
Abstract
Granulocyte-macrophage colony-stimulating factor (GM-CSF) produced by T helper 17 (Th17) cells plays an essential role in autoimmune diseases. Transcriptional regulation of Th17 cell differentiation has been extensively studied, but post-transcriptional regulation of Th17 cell differentiation has remained less well characterized. The RNA-binding protein HuR functions to promote the stability of target mRNAs via binding the AU-rich elements of the 3′ untranslated region (3′UTR) of numerous pro-inflammatory cytokines including IL-4, IL-13, IL-17 and TNF-α. However, whether HuR regulates GM-CSF expression in Th17 cells has not been fully investigated. Here we showed that HuR conditional knockout (KO) Th17 cells have decreased GM-CSF mRNA in comparison with wild-type (WT) Th17 cells, and that HuR binds directly to GM-CSF mRNA 3′UTR. Interestingly, HuR deficiency increased the levels of certain microRNA expression in Th17 cells; for example, miR-466i functioned to mediate GM-CSF and IL-17 mRNA decay, which was confirmed by in vitro luciferase assay. Furthermore, we found that HuR promoted Mxi1 expression to inhibit certain miRNA expression. Taken together, these findings indicate that interaction of HuR and miR-466i orchestrates GM-CSF expression in Th17 cells.
Similar content being viewed by others
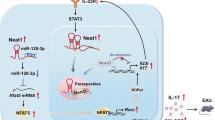


Introduction
Naïve CD4+ T cell differentiate into different subsets of T helper cells after antigen stimulation that are crucial for orchestrating adaptive immune responses. Apart from the well characterized Th1 and Th2 cells that have been known for more than three decades ago, a new subset of Th17 cells has been identified in the past ten years1,2,3,4,5,6. Because of the essential and non-redundant role of Th17 cells in a number of autoimmune diseases including experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS), rheumatoid arthritis, and psoriasis7,8,9, regulation of Th17 cell differentiation has been extensively studied10,11,12,13. The signature cytokine for Th17 cells is interleukin 17 (IL-17); later researchers found that GM-CSF is also produced by Th17 cells7,14. Two research groups have independently demonstrated that GM-CSF is essential for Th17 cells to induce autoimmune neuroinflammation7,14. Further study showed that the responsiveness of bone marrow-derived monocytes to GM-CSF plays a pivotal role in EAE6. Thus, administration of recombinant GM-CSF resulted in more severe EAE15, and mice with transgenic overexpression of GM-CSF in T cells spontaneously developed autoimmune neuroinflammation16. Interestingly, elevated concentrations of GM-CSF have been reported in the cerebrospinal fluid of patients with relapsing-remitting MS17,18, suggesting that GM-CSF may play a similar pathogenic role in human MS. Thus, targeting GM-CSF by antibody has been tested in numerous clinical trials. Some trials have shown promising results in treatment of rheumatoid arthritis, and others are still in progress for treating MS patients19. A recent Phase I clinical trial showed that targeting human GM-CSF by MOR103 is safe20. Given the importance of Th17 cells and their cytokines in numerous types of autoimmune inflammation7,10,14,21,22, it is imperative to further study regulation of GM-CSF production in order to develop novel approaches for targeting it.
Gene expression is post-transcriptionally regulated by RNA-binding proteins (RBPs)23,24. Although most RBPs destabilize target mRNAs, a few of these, including HuR, bind to 3′UTR of proinflammatory cytokines to stabilize them24,25,26. In particular, HuR binds to adenylate-uridylate-rich elements (AREs) located in the 3′UTR of unstable genes to selectively mediate mRNA stabilization23,27,28. We previously showed that HuR post-transcriptionally stabilizes IL-17 and CCR6 mRNA and promotes their expression in autoimmune neuroinflammation29,30. However, it remains unclear whether HuR modulates GM-CSF expression in Th17 cells.
It is well known that interaction of HuR and microRNAs (miRNAs) controls target mRNA expression in response to environmental stimuli31,32. MiRNAs are small, non-coding RNAs with approximately 21 to 24 nucleotides (nt)33, which regulate the expression of numerous target genes by mediating their mRNA decay and/or repressing their translation33,34,35. The most common motif is perfect pairing between nucleotides 2 and 7 at the 5′ end of the miRNA, which is called the ‘seed’ region. It is thought that most miRNA-mRNA interactions involve the seed region at the 5′ end of miRNA. The small size of miRNAs provides a limited amount of information for specificity. Furthermore, as partial pairing between a miRNA and a target site is often sufficient for miRNA function, which means that a single miRNA can regulate multiple mRNAs but which also makes target predictions complicated36. Although cells contain hundreds of miRNAs, only a limited number of miRNAs have been validated functionally. Given the importance of miRNAs in regulating expression of numerous genes, further characterizing specific miRNA function will improve our understanding of gene regulation. Here we provide evidence that HuR post-transcriptionally modulates GM-CSF expression in Th17 cells. The level of miR-466i is increased in HuR KO Th17 cells compared with WT Th17 cells. The 3′UTRs of GM-CSF and IL-17 mRNA are potential targets of miR-466i, as shown by Targetscan analysis. In vitro luciferase transfection assay demonstrated that miR-466i could target GM-CSF and IL-17 mRNA 3′UTRs for decay, suggesting that miR-466i has potential as a novel reagent for therapeutic intervention in autoimmune inflammation.
Results
Knockout of HuR reduces GM-CSF expression in Th17 cells
Our previous studies showed that the levels of HuR mRNA and protein correlate with IL-17 mRNA and protein expression in Th17 cells and that knockout of HuR reduces IL-17 mRNA and protein levels in Th17 cells29. Considering that GM-CSF plays a non-redundant role in Th17 cell induction of autoimmune demyelination7,14, we were interested in determining if HuR regulates GM-CSF mRNA expression in Th17 cells. To address this question, we used HuR conditional KO (OX40-Cre HuRf/f) mice29. Naive CD4+ T cells do not express OX40, but activated T cells do36, thus deleting HuR. An advantage of using this approach is that conditional knockout of HuR does not affect early development of T cells. Naive CD4+ T cells were isolated from wild type (WT) and KO mouse splenocytes, then were cultured under Th0 and Th17 cells-polarizing conditions. Western blot assays were performed to quantify HuR levels. Endogenous HuR was gradually degraded after cell activation in KO CD4+ T cells from day 3 to 5 of culture, and HuR was barely detected at day 529. Quantitative real-time PCR (qRT-PCR) data showed that KO Th0 and Th17 cells had significantly less GM-CSF mRNA as compared with WT counterparts on day 4 of culture (Fig. 1a,b). Flow cytometry analysis also showed that the GM-CSF protein was also reduced in HuR KO Th0 and Th17 cells in comparison with WT cells (Fig. 1c). Results were confirmed by ELISA assay (Fig. 1d,e). These results demonstrated that knockout of HuR reduced GM-CSF mRNA and protein in Th0 and Th17 cells.

Knockout of HuR reduces GM-CSF mRNA in CD4+ T cells. Naive WT and HuR KO CD4+ T cells were isolated as described in Materials and Methods and cultured with anti-CD3 and anti-CD28 stimulation in Th0 and Th17 cell polarizing conditions. Expression of GM-CSF mRNA (a,b) and protein (c–e) in Th0 and Th17 cells was measured by qRT-PCR, flow cytometry, and ELISA, respectively. Knockout of HuR reduced the GM-CSF mRNA and protein levels in Th0 and Th17 cells compared with WT counterparts. Results in (a,b,d and e) represent the summary of three independent experiments. The representative of flow analysis was shown in (c). **p < 0.01, ***p < 0.001.
HuR binds to GM-CSF mRNA 3′UTR
The regulation of GM-CSF mRNA expression by HuR suggested that GM-CSF mRNA might be a direct target of HuR. To study the function of HuR in regulation of GM-CSF in Th17 cells, we did RNA immunoprecipitation (RIP) assay to test if HuR protein binds to GM-CSF mRNA. The association of GM-CSF mRNA with HuR was tested by isolating RNA from the ribonucleoprotein (RNP) complexes with anti-HuR Ab (Fig. 2a). Our results showed a remarkable GM-CSF mRNA enrichment in the anti-HuR immunoprecipitation (IP) sample compared with an isotype-matched IgG control (Fig. 2b). IL-23R mRNA was not enriched in the HuR pulldown sample as a negative control (Fig. 2b). To confirm HuR interaction with GM-CSF mRNA, we utilized biotin pulldown, a second independent method. Two biotinylated transcripts that are complementary to sequences in GM-CSF mRNA ORF (open reading frame) and 3′ UTR were generated according to a protocol described in Materials and Methods (Fig. 2c). The biotinylated transcripts were incubated with cytoplasmic protein lysates from NIH/3T3 cells, which contain large amounts of HuR protein. The result of western blot showed that HuR was clearly detected in the samples precipitated by the biotinylated transcript of GM-CSF mRNA 3′ UTR (Fig. 2c–e). HuR protein could not be detected in the GAPDH 3′ UTR biotinylated probe pulldown complex (Fig. 2e), and this transcript has no putative HuR binding sites29. Taken together, these results indicated that HuR directly binds to 3′ UTR of GM-CSF mRNA, suggesting a regulatory relationship. Thus, HuR binds to and stabilizes GM-CSF mRNA, resulting in increased mRNA accumulation in Th17 cells.

HuR regulates GM-CSF mRNA by binding and stabilizing its 3′UTR in Th17 cells. (a) Schematic diagram of RIP analysis. (b) RIP analysis was performed to detect GM-CSF mRNA enrichment after IP using anti-HuR (3A2) or isotype–matched antibody (IgG1) from Th17 cell cytoplasmic extracts. IL-23R mRNA was assayed as a negative control. (c–e) Biotin pulldown was used to determine whether HuR directly binds to 3′UTR of GM-CSF mRNA. (c,d) Schematic diagrams of biotin probe corresponding to GM-CSF and 3′UTR and biotin-pull down assay, respectively. (e) HuR protein could be detected by Western blot in biotinylated transcripts spanning GM-CSF 3′UTR, but not in GM-CSF mRNA ORF biotinylated transcripts. GAPDH biotinylated transcripts were unreactive (e). One of two independent experiments is shown in (e). **p < 0.01.
HuR inhibits miR-466i expression in Th17 cells
Since miRNAs play an important role in mediating target mRNA decay, decreasing mRNA levels by miRNAs represents a better approach to disrupt their protein function. We used several miRNA analysis programs to explore miRNAs that have the potential to target GM-CSF and IL-17 mRNAs. Interestingly, by Targetscan analysis, we found many miRNAs that could potentially bind to GM-CSF and IL-17 mRNAs, respectively (Supplementary Table 1). Among these miRNAs, it is only miR-466i that has potential binding sites in both GM-CSF and IL-17 mRNAs (Fig. 3a–d). Previous work showed that overexpression of miR-446i upregulates IL-10 expression in macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation, and miR-466i inhibits antiviral innate immune response by targeting interferon-α37,38. A recent study showed that miR-466i plays a bipolar role in inflammation, promoting an acute inflammatory response initially and enhancing resolution during the late stage of inflammation39. However, whether miR-466i mediates GM-CSF and IL-17 mRNA degradation remains unknown, thus, making this an important question to address.

MiR-466i has multiple potential binding sites in GM-CSF and IL-17A mRNA 3′UTR. (a) There are nine miR-466i potential binding sites in 3′UTR of GM-CSF mRNA. There are three miR-466i potential binding sites listed in (b). Three miR-466i potential binding sites exist in 3′UTR of IL-17A mRNA which were listed in (c) and (d).
Interaction of RNA-binding protein HuR with miRNAs post-transcriptionally regulates gene expression32. Our previous studies showed that HuR post-transcriptionally modulates IL-17 mRNA expression and promotes autoimmune neuroinflammation29. Knockout of HuR reduces IL-17 mRNA half-life and destabilizes it. Here, we found that HuR directly regulated GM-CSF production by binding to 3′UTR of GM-CSF mRNA (Fig. 2b and e). We therefore performed qRT-PCR to examine miR-466i expression in WT and HuR KO Th17 cells. Interestingly, miR-466i was highly expressed in HuR KO Th17 cells in comparison with WT Th17 cells, but there was no significant change in miR-155 expression (Fig. 4a), suggesting that miR-466i may play an active role in mediating GM-CSF and IL-17 mRNA decay in the absence of HuR, and that HuR inhibits expression of certain miRNAs27.

HuR inhibits miR466i but promotes Mxi1 expression in Th17 cells. Naïve CD4+ T cells were isolated from spleen of WT and HuR knockout mice and polarized as described in Materials and Methods. (a) Th17 cells were harvested after 5 days of cell culture. Expression of miRNAs in Th17 cells was analyzed by qRT-PCR and normalized by level of U6 snRNA. Expression of Mxi1 mRNA (b) and protein (c and d) was measured by qRT-PCR and Western blot, respectively. (e) Knockdown of Mxi1 in WT Th17 cells by Mxi1 siRNA transfection increased expression of miR-466i compared with the counterparts treated with scramble siRNA. (f) Knockdown of Mxi1 in WT Th17 cells also significantly reduced GM-CSF mRNA expression compared with WT Th17 cells transfected with scramble siRNA. Data in a, b, d, e and f summarize three individual experiments. The representative Western blots are shown in (c).
To understand how HuR negatively regulates expression of certain miRNAs, we checked the expression of some molecules related to miRNA expression in HuR KO and WT Th17 cells. It is well known that Drosha, Dicer, Ago1/2 and c-Myc are actively involved in miRNA expression40, but their expression showed no significant change in the presence or absence of HuR (data not shown). However, expression of Mxi1 mRNA and protein, a repressor of c-Myc41, was significantly reduced in the absence of HuR compared with WT Th17 cells (Fig. 4b–d). Thus, it is possible that HuR promotes Mxi1 expression, which suppresses c-Myc function, resulting in its indirectly inhibiting expression of certain miRNAs. In line with this notion, knockdown of Mxi1 in WT Th17 cells by Mxi1 siRNA transfection increased miR-466i expression but decreased GM-CF mRNA compared with counterparts treated with scramble siRNA (Fig. 4e,f).
MiR-466i-mediates GM-CSF and IL-17 mRNA decay by interacting with their 3′UTRs
To further investigate whether miR-466i modulates GM-CSF and IL-17 expression by degrading it through interacting with their 3′UTRs, a reporter vector with firefly and Renilla Dual-Luciferase (RDL) containing GM-CSF 3′UTR was co-transfected with miR-466i mimics into HeLa cells (Fig. 5a). Overexpression of miR-466i decreased luciferase activity of the reporter construct by analysis of firefly luciferase activity which was normalized by Renilla luciferase activity, but overexpression of scramble miRNAs did not (Fig. 5a), suggesting that miR-466i functions to degrade GM-CSF mRNA through binding to 3′UTR. Similarly, overexpression of miR-466i in HeLa cells transfected with RDL vector containing IL-17 mRNA 3′UTR also reduced luciferase activity (Fig. 5b). These results thus suggest that miR-466i mediates GM-CSF and IL-17 mRNA decay through binding to their 3′UTRs. In line with these results, overexpression of miR-466i in WT Th17 cells by transfection reduced expression of GM-CSF and IL-17 mRNA compared to that with scramble miRNA (Fig. 5c).

MiR-466i directly targets GM-CSF and IL-17 3′UTR. (a) HeLa cells were transfected with reporter containing GM-CSF 3′UTR, and overexpression of miR-466i decreased the luciferase activity of vector containing GM-CSF 3′UTR. (b) Similar results were obtained for the luciferase activity of vector containing IL-17 3′UTR. (c) Overexpression of miR-466i by transfection in Th17 cells reduced the expression of GM-CSF and IL-17 mRNA by qRT-PCR assay. Results in (a–c) are a summary of at least three independent experiments. EV (empty vector). *p < 0.05, **p < 0.01, ***p < 0.001.
MiR-466i regulates GM-CSF protein expression in macrophages
To further confirm the result that miR-466i modulates GM-CSF expression, macrophages were transfected with miR-466i and scramble miRNAs. GM-CSF mRNA and protein levels were examined by qRT-PCR and flow cytometry, respectively. The data showed that expression of GM-CSF mRNA was remarkably decreased when macrophages were transfected with miR-466i compared with scramble miRNA (Fig. 6a); however, the IL-6 mRNA level was moderately increased in macrophages transfected with miR-466i, which is consistent with a previous report39. The GM-CSF protein level was also decreased in macrophages transfected with miR-466i mimics compared with scramble miRNA (Fig. 6b,c). This result indicated that miR-466i also regulates GM-CSF expression in macrophages.

MiR-466i overexpression reduced GM-CSF expression in activated macrophages. RAW264.7 cells were transfected with miR-466i mimics or scramble control at a final concentration of 10 nM, and 24 hours later after LPS stimulation (100 ng/ml), the cells were harvested. (a) Expression of GM-CSF and IL-6 mRNA levels were measured by qRT-PCR analysis and normalized by GAPDH. (b,c) GM-CSF protein expression levels were measured by flow cytometry. Data in (a) are a summary of three individual experiments. The representative flow cytometric analysis is shown in (b) and (c).
Taken together, these results suggest that HuR and miR-466i interaction modulates GM-CSF expression in Th17 cells. MiR-466i targets GM-CSF and IL-17 mRNA decay by binding to their 3′UTRs, a finding that may lead to development of miR-466i as a novel therapeutic intervention for autoimmune inflammation.
Discussion
MiRNAs are small (approximately 22 bases) non-protein-coding RNAs that recognize target sequences of imperfect complementarity in cognate mRNA and either destabilize them or inhibit protein translation. In principle, gene expression is post-transcriptionally regulated under the control of RNA-binding protein and miRNA interaction32. Our current study shows that HuR binds to GM-CSF and IL-17 mRNA by inhibiting miR-466i expression and by preventing miR-466i-mediated targeting of GM-CSF and IL-17 mRNA decay.
In this study, we first used OX40-Cre HuRf/f mice to demonstrate that HuR promotes GM-CSF mRNA expression. Ablation of HuR reduced GM-CSF mRNA and protein expression in Th0 and Th17 cells. We previously reported that knockout of HuR impacted IL-17 expression29. GM-CSF and IL-17 in Th17 cells are thus similarly post-transcriptionally regulated by HuR.
Here we found that HuR functions to bind to GM-CSF mRNA 3′UTR. HuR ablation therefore reduced GM-CSF mRNA expression, which is consistent with previous reports showing that GM-CSF can be controlled by a post-transcriptional mechanism through ARE in its 3′UTR and stabilized by HuR42,43,44,45. However, the mechanism by which GM-CSF mRNA was degraded in the absence of HuR had not been completely characterized in previous reports. Thus, our current study extended previous findings by showing that HuR deficiency in T cells using conditional knockout mice deceased GM-CSF production and increased miR-466i expression. In vitro analysis suggested that miR-466i has the capacity to mediate GM-CSF and IL-17 mRNA decay. The fact that knockout of HuR increased miR-466i, miR-409 and miR-335 expression30 is consistent with a previous report showing that overexpression of HuR reduces miR-16 expression, and that HuR inhibits miR-16 targeting of Cox-246. To understand why knockout of HuR increased certain miRNA expression, we also performed anti-HuR immunoprecipitation (IP) western blots to determine whether HuR and Ago2 are physically associated with each other. Not surprisingly, Ago2 could not be detected in the anti-HuR IP complex (data not shown). It has been reported that c-Myc is a transcription factor with the capacity to promote certain miRNA expression47,48, and that HuR recruits miRNA let-7 to mediate c-Myc RNA decay49. Mxi1 is a transcription repressor that negatively regulates c-Myc function41. Although knockout of HuR did not impact the expression of c-Myc mRNA (data not shown), Mxi1 mRNA and protein expression was significantly reduced in the absence of HuR (Fig. 4). Therefore, knockout of HuR reduced Mxi1 expression, which may alleviate its repressive effect on the function of c-Myc, leading to an increase in expression of certain miRNAs. Based on this idea, we speculated that knockdown of Mxi1 in WT Th17 cells would result in increased miR-466i expression. Interestingly, we obtained new data which support this notion (Fig. 4). However, how HuR modulates miRNA biogenesis via Mxi1 only and/or other factors need to be further studied.
By in vitro luciferase assay, we demonstrated that overexpression of miR-466i reduced activity of the vector containing GM-CSF or IL-17 mRNA 3′UTR in transfected Hela cells, suggesting that miR-466i has the potential to target GM-CSF and IL-17 mRNA. Indeed, overexpression of miR-466i in WT Th17 cells and macrophages decreased GM-CSF mRNA and its protein production, respectively (Fig. 6). A previous report showed that miR-466i increased anti-inflammatory cytokine IL-10 production by blocking RNA-binding protein tristetraprolin, which destabilized IL-10 mRNA37. A recent report demonstrated that miR-466i could target MyD88 mRNA 3′UTR for decay to downregulate IL-12 production and increase IL-10 production50. miR-466i might therefore have the therapeutic potential to target GM-CSF-mediated inflammation and to increase IL-10 production for promoting resolution of inflammation.
Since HuR binds to GM-CSF 3′UTR (Fig. 2), and GM-CSF 3′UTR contains nine potential miR-466i binding sites (Supplementary Table 1), it is likely that HuR and miR-466i might competitively bind to the same target32. However, we cannot rule out the possibility that with HuR binding, the space-structure of GM-CSF mRNA 3′UTR changes, which prevents miR-466i from binding to it32. In any case, HuR inhibition of miR-466i biogenesis also contributes to the effect of HuR on promoting GM-CSF expression.
Overall, RNA-binding protein HuR post-transcriptionally regulates GM-CSF and IL-17 mRNA expression by binding to target mRNAs. HuR promotes Mxi1 expression to inhibit the expression of certain miRNAs. HuR and miR-466i interaction orchestrates GM-CSF and IL-17 mRNA fate. Taking advantage of the many newly developed small molecule inhibitors for HuR in anti-tumor research51,52, and based on our current study on HuR regulation of GM-CSF production, it would be worthwhile to test the effects of these newly developed HuR inhibitors on anti-autoimmune inflammation.
Materials and Methods
Cell culture
Mouse naive CD4+ T cells were isolated from splenocytes of WT and HuR conditional knockout (HuR KO) mice29 using CD4 negative selection kits (StemCell Tech., Vancouver, Canada) following the manufacturer’s protocol. Cells were differentiated into Th17 cells as previously described29,53. Isolated naive CD4+ T cells were activated with plate-bound anti-CD3 (10 µg/ml) and soluble anti-CD28 (3 µg/ml) in the presence of TGF-β (3 ng/ml), IL-6 (20 ng/ml), IL-23 (20 ng/ml), anti-IFN-γ (10 µg/ml) and anti-IL-4 (10 µg/ml). Naïve CD4+ T cells stimulated with anti-CD3 and anti-CD28 and without other polarizing cytokines were designated as Th0 cells. All cytokines were purchased from R&D Systems (Minneapolis, MN) and Peprotech (Rocky Hill, NJ), and antibodies were purchased from eBiosciences (San Diego, CA) and Biolegend (San Diego, CA).
RAW 264.7 macrophages (ATCC) were cultured in DMEM with 10% FBS plus penicillin (100 U/ml) and streptomycin (100 μg/ml) in 5% CO2 atmosphere at 37 °C.
All mice used are on the C57BL/6 background and were breed at the animal facility of Arkansas Bioscience Institute at Arkansas State University and/or the Thomas Jefferson University. Animal experiments were approved by the Arkansas State University (ASTATE) and Thomas Jefferson University (TJU) IACUC committee. We followed the regulation of NIH, ASTATE and TJU according to federal and institutional guidelines.
Flow cytometry
Cells obtained from in vitro 5 day culture were stained for surface markers with FITC-conjugated anti-CD4, PE-conjugated anti-GM-CSF, and allophycocyanin-conjugated anti-IL-17 (Biolegend, San Diego, CA). Acquisitions were made with a BD FACSAria II (BD Biosciences, San Diego, CA). Flowjo software was used for data analysis.
RNA isolation and Quantitative real time-PCR (qRT-PCR)
Cultured cells were collected and total RNA was extracted using TRIzol (Invitrogen, Life Technologies Corp., Thermo Fisher Scientific). Five hundred ng of RNA was reverse transcribed into cDNA using SuperScript III Kit (Life Technologies, Fisher Thermos Scientific Inc.) according to the manufacturer’s protocols. The cDNA was subjected to qRT-PCR analysis using the CFX96 Real-Time PCR Detection system (Bio-Rad, Hercules, CA) with SYBR Green reagent Kit (Invitrogen, Life Technologies Corp., Thermo Fisher Scientific) according to the manufacturer’s protocols. The levels of test mRNAs were normalized to the levels of Gapdh mRNA for each sample. Forward and reverse primers for specific murine target genes are listed in Table 1.
RNA Immunoprecipitation (RIP)
RIP was performed according to published protocols29,54. Th17 polarized cells at day 4 to 5 culture were lysed using polysome lysis buffer supplemented with RNase inhibitors and protease inhibitors55. Lysates were pre-cleared by adding 30 µg of IgG1 (BD Bioscience) and 50 µl of Protein-A/G Sepharose beads swollen in NT2 buffer with 5% BSA. Beads were coated by adding either IgG1 (BD Biosciences, San Diego, CA) as control or anti-HuR antibody 3A2, and incubated overnight at 4 °C. After extensive washes of pre-coated Protein-A/G sepharose beads, the pre-cleared lysate was added and incubated for 4 h at 4 °C, and then 30 µg of proteinase K was added to digest protein by incubation at 55 °C for 30 min. The extracted RNA was reverse transcribed into cDNA, and qRT-PCR was preformed to detect the presence of specific target mRNAs.
Biotin pulldown assay
Biotinylated transcripts were synthesized as previously described29. Forward primers that contained the T7 RNA polymerase promoter sequence (5′-CCAAGCTTCTAATACGACTCACTATAGGGAGA-3′ [T7]) and reverse primers used to generate cDNA are listed as following: GM-CSF ORF forward plus T7 5′-GCTTCTAATACGACTCACTATAGGATGTGGCTGCAGAA-3′; GM-CSF ORF Reverse: 5′-TCATTTTTGGCCTGGTTTTTTGCATTCAAAGGGG-3′; GM-CSF 3′UTR Forward plus T7: 5′-GCT TCTAATACGACTCACTATAGGGGAAGCCCAGGCCAG-3′; GM-CSF 3′UTR Reverse: 5′-CTG GTAAGACATTCTCAATAAATAGA-3′. PCR-amplified products were purified and used as templates for the synthesis of biotinylated RNA with T7 RNA polymerase and biotin-conjugated UTP for murine RNAs. Biotinylated transcripts were incubated with NIH/3T3 cell lysate at room temperature. The RIP complexes were isolated with paramagnetic streptavidin-conjugated Dynabeads (Invitrogen, Life Technologies Corp., Thermo Fisher Scientific). Bound HuR protein in the pulldown pellet was analyzed by western blots to evaluate whether HuR directly binds to 3′ UTR of GM-CSF mRNA.
Transfection and luciferase assay
HeLa cells (ATCC) were cultured in DMEM containing 10% FBS, penicillin (100U/ml), streptomycin (100 μg/ml), 2 mM L-glutamine and non-essential amino acids (Invitrogen, Thermo Fisher Scientific) in 24-well plates and incubated overnight. Lipofectamine 2000 (Invitrogen, Life Technologies Corp., Thermo Fisher Scientific) was used to transfect Hela cells with pRL-CMV-renilla-luciferase plasmid and firefly-luciferase plasmid DNA containing GM-CSF or IL-17 mRNA 3′UTR. Hela cells were co-transfected with 40–80 nmol miR-466i and scramble miRNAs (Life Technologies, Thermo Fisher Scientific). Twenty-four hours later, cells were lysed and luciferase activities were measured using a Dual-Luciferase Reporter Assay System according to the manufacturer’s instructions (Promega, Madison, WI). Firefly luciferase activity was normalized to renilla luciferase in each sample.
For transfection of siRNA, naïve WT CD4+ T cells were transfected with Mxi1 siRNA and scramble siRNA (Life Technologies Corp., Thermo Fisher Scientific) using Lonza 4D-nucleofector according to the manufacturer’s instructions.
Statistics
Bar graphs in figures represent average values ± SEM unless indicated otherwise. Statistical significance between experimental groups was calculated using a two-tailed unpaired Student t test and is shown in the graphs as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
References
Park, H. et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature immunology 6, 1133–1141, https://doi.org/10.1038/ni1261 (2005).
Harrington, L. E. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature immunology 6, 1123–1132, https://doi.org/10.1038/ni1254 (2005).
Mangan, P. R. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234, https://doi.org/10.1038/nature04754 (2006).
Dong, C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nature reviews. Immunology 8, 337–348, https://doi.org/10.1038/nri2295 (2008).
Molinero, L. L., Cubre, A., Mora-Solano, C., Wang, Y. & Alegre, M. L. T cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proceedings of the National Academy of Sciences of the United States of America 109, 18529–18534, https://doi.org/10.1073/pnas.1204557109 (2012).
Croxford, A. L. et al. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43, 502–514, https://doi.org/10.1016/j.immuni.2015.08.010 (2015).
El-Behi, M. et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nature immunology 12, 568–575, https://doi.org/10.1038/ni.2031 (2011).
Hirota, K. et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. The Journal of experimental medicine 204, 2803–2812, https://doi.org/10.1084/jem.20071397 (2007).
Hedrick, M. N. et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. The Journal of clinical investigation 119, 2317–2329 (2009).
Korn, T., Bettelli, E., Oukka, M. & Kuchroo, V. K. IL-17 and Th17 Cells. Annual review of immunology 27, 485–517, https://doi.org/10.1146/annurev.immunol.021908.132710 (2009).
Ivanov, I. I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133, https://doi.org/10.1016/j.cell.2006.07.035 (2006).
Zhou, L. & Littman, D. R. Transcriptional regulatory networks in Th17 cell differentiation. Current opinion in immunology 21, 146–152, https://doi.org/10.1016/j.coi.2009.03.001 (2009).
Ichiyama, K. et al. The MicroRNA-183-96-182 Cluster Promotes T Helper 17 Cell Pathogenicity by Negatively Regulating Transcription Factor Foxo1 Expression. Immunity 44, 1284–1298, https://doi.org/10.1016/j.immuni.2016.05.015 (2016).
Codarri, L. et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature immunology 12, 560–567, https://doi.org/10.1038/ni.2027 (2011).
McQualter, J. L. et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. The Journal of experimental medicine 194, 873–882 (2001).
Spath, S. et al. Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity 46, 245–260, https://doi.org/10.1016/j.immuni.2017.01.007 (2017).
Carrieri, P. B. et al. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacology and immunotoxicology 20, 373–382, https://doi.org/10.3109/08923979809034820 (1998).
Perrella, O., Carrieri, P. B., De Mercato, R. & Buscaino, G. A. Markers of activated T lymphocytes and T cell receptor gamma/delta + in patients with multiple sclerosis. European neurology 33, 152–155 (1993).
Shiomi, A. & Usui, T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators of inflammation 2015, 568543, https://doi.org/10.1155/2015/568543 (2015).
Constantinescu, C. S. et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurology(R) neuroimmunology & neuroinflammation 2, e117, https://doi.org/10.1212/nxi.0000000000000117 (2015).
Maddur, M. S., Miossec, P., Kaveri, S. V. & Bayry, J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. The American journal of pathology 181, 8–18, https://doi.org/10.1016/j.ajpath.2012.03.044 (2012).
Ponomarev, E. D. et al. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. Journal of immunology (Baltimore, Md.: 1950) 178, 39–48 (2007).
Kafasla, P., Skliris, A. & Kontoyiannis, D. L. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nature immunology 15, 492–502, https://doi.org/10.1038/ni.2884 (2014).
Anderson, P. Post-transcriptional control of cytokine production. Nature immunology 9, 353–359, https://doi.org/10.1038/ni1584 (2008).
Tiedje, C. et al. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic acids research 44, 7418–7440, https://doi.org/10.1093/nar/gkw474 (2016).
Lopez de Silanes, I., Zhan, M., Lal, A., Yang, X. & Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proceedings of the National Academy of Sciences of the United States of America 101, 2987–2992, https://doi.org/10.1073/pnas.0306453101 (2004).
Techasintana, P., Davis, J. W., Gubin, M. M., Magee, J. D. & Atasoy, U. Transcriptomic-Wide Discovery of Direct and Indirect HuR RNA Targets in Activated CD4+ T Cells. PloS one 10, e0129321, https://doi.org/10.1371/journal.pone.0129321 (2015).
Casolaro, V. et al. Posttranscriptional regulation of IL-13 in T cells: role of the RNA-binding protein HuR. The Journal of allergy and clinical immunology 121, 853–859.e854, https://doi.org/10.1016/j.jaci.2007.12.1166 (2008).
Chen, J. et al. Posttranscriptional gene regulation of IL-17 by the RNA-binding protein HuR is required for initiation of experimental autoimmune encephalomyelitis. Journal of immunology (Baltimore, Md.: 1950) 191, 5441–5450, https://doi.org/10.4049/jimmunol.1301188 (2013).
Chen, J. et al. The RNA-binding protein HuR contributes to neuroinflammation by promoting C-C chemokine receptor 6 (CCR6) expression on Th17 cells. The Journal of biological chemistry, doi:10.1074/jbc.M117.782771 (2017).
Ahuja, D., Goyal, A. & Ray, P. S. Interplay between RNA-binding Protein HuR and microRNA-125b Regulates p53 mRNA Translation in Response to Genotoxic Stress. RNA biology, 0, https://doi.org/10.1080/15476286.2016.1229734 (2016).
Srikantan, S., Tominaga, K. & Gorospe, M. Functional interplay between RNA-binding protein HuR and microRNAs. Current protein & peptide science 13, 372–379 (2012).
Xiao, C. & Rajewsky, K. MicroRNA control in the immune system: basic principles. Cell 136, 26–36, https://doi.org/10.1016/j.cell.2008.12.027 (2009).
Jonas, S. & Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nature reviews. Genetics 16, 421–433, https://doi.org/10.1038/nrg3965 (2015).
Iwakawa, H. O. & Tomari, Y. The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends in cell biology 25, 651–665, https://doi.org/10.1016/j.tcb.2015.07.011 (2015).
Croft, M., So, T., Duan, W. & Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological reviews 229, 173–191, https://doi.org/10.1111/j.1600-065X.2009.00766.x (2009).
Ma, F. et al. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. Journal of immunology (Baltimore, Md.: 1950) 184, 6053–6059, https://doi.org/10.4049/jimmunol.0902308 (2010).
Li, Y. et al. MicroRNA-466l inhibits antiviral innate immune response by targeting interferon-alpha. Cellular & molecular immunology 9, 497–502, https://doi.org/10.1038/cmi.2012.35 (2012).
Li, Y. et al. Plasticity of leukocytic exudates in resolving acute inflammation is regulated by MicroRNA and proresolving mediators. Immunity 39, 885–898, https://doi.org/10.1016/j.immuni.2013.10.011 (2013).
Ha, M. & Kim, V. N. Regulation of microRNA biogenesis. Nature reviews. Molecular cell biology 15, 509–524, https://doi.org/10.1038/nrm3838 (2014).
Zervos, A. S., Gyuris, J. & Brent, R. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell 72, 223–232 (1993).
Hachiya, M., Suzuki, G., Koeffler, H. P. & Akashi, M. Irradiation increases expression of GM-CSF in human fibroblasts by transcriptional and post-transcriptional regulation. Experimental cell research 214, 343–350, https://doi.org/10.1006/excr.1994.1266 (1994).
Migliaccio, A. R. et al. Transcriptional and posttranscriptional regulation of the expression of the erythropoietin receptor gene in human erythropoietin-responsive cell lines. Blood 82, 3760–3769 (1993).
Shaw, G. & Kamen, R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46, 659–667 (1986).
Wang, J. G. et al. LFA-1-dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. Journal of immunology (Baltimore, Md.: 1950) 176, 2105–2113 (2006).
Young, L. E., Moore, A. E., Sokol, L., Meisner-Kober, N. & Dixon, D. A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Molecular cancer research: MCR 10, 167–180, https://doi.org/10.1158/1541-7786.mcr-11-0337 (2012).
Wang, X., Zhao, X., Gao, P. & Wu, M. c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Scientific reports 3, 1942, https://doi.org/10.1038/srep01942 (2013).
Dews, M. et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nature genetics 38, 1060–1065, https://doi.org/10.1038/ng1855 (2006).
Kim, H. H. et al. HuR recruits let-7/RISC to repress c-Myc expression. Genes & development 23, 1743–1748, https://doi.org/10.1101/gad.1812509 (2009).
Mukherjee, B. et al. Antimony-Resistant Leishmania donovani Exploits miR-466i To Deactivate Host MyD88 for Regulating IL-10/IL-12 Levels during Early Hours of Infection. Journal of immunology (Baltimore, Md.: 1950) 195, 2731–2742, https://doi.org/10.4049/jimmunol.1402585 (2015).
Lang, M. et al. HuR Small-Molecule Inhibitor Elicits Differential Effects in Adenomatosis Polyposis and Colorectal Carcinogenesis. Cancer research 77, 2424–2438, https://doi.org/10.1158/0008-5472.can-15-1726 (2017).
Wu, X. et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS chemical biology 10, 1476–1484, https://doi.org/10.1021/cb500851u (2015).
Xie, L. et al. The immunomodulator AS101 suppresses production of inflammatory cytokines and ameliorates the pathogenesis of experimental autoimmune encephalomyelitis. Journal of neuroimmunology 273, 31–41, https://doi.org/10.1016/j.jneuroim.2014.05.015 (2014).
Stellato, C. et al. Coordinate regulation of GATA-3 and Th2 cytokine gene expression by the RNA-binding protein HuR. Journal of immunology (Baltimore, Md.: 1950) 187, 441–449, https://doi.org/10.4049/jimmunol.1001881 (2011).
Atasoy, U., Watson, J., Patel, D. & Keene, J. D. ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is upregulated during serum stimulation and T cell activation. Journal of cell science 111(Pt 21), 3145–3156 (1998).
Acknowledgements
This work was supported by NIH grant (R01AI119135) and NIH Grant Number p20GM103429 from the IDeA Networks of Biomedical Research Excellence (INBRE) Program of the National Center for Research Resources.
Author information
Authors and Affiliations
Contributions
J.C. and S.Y. designed and performed most of the experiments; W.A. did some flow cytometry analysis and mouse genotyping; G.H. did some luciferase transfection assay; J.C., U.A., A.R., and S.Y. analyzed the data; J.C. and S.Y. wrote the manuscript. All authors reviewed the results and approved the final version of manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, J., Adamiak, W., Huang, G. et al. Interaction of RNA-binding protein HuR and miR-466i regulates GM-CSF expression. Sci Rep 7, 17233 (2017). https://doi.org/10.1038/s41598-017-17371-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17371-5
This article is cited by
-
Glycolytic enzymes in non-glycolytic web: functional analysis of the key players
Cell Biochemistry and Biophysics (2024)
-
RBP–RNA interactions in the control of autoimmunity and autoinflammation
Cell Research (2023)
-
Development of a novel and synthetic HematoMiR technology that broadly modulates quiescence of stem cells and enhances HSC expansion
Cellular and Molecular Life Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
