
Abstract
Deep-sea sediments contain high concentrations of rare earth element (REE) which have been regarded as a huge potential resource. Understanding the marine REE cycle is important to reveal the mechanism of REE enrichment. In order to determine the geochemistry characteristics and migration processes of REE, seawater, porewater and sediment samples were systematically collected from the western Pacific for REE analysis. The results show a relatively flat REE pattern and the HREE (Heavy REE) enrichment in surface and deep seawater respectively. The HREE enrichment distribution patterns, low concentrations of Mn and Fe and negative Ce anomaly occur in the porewater, and high Mn/Al ratios and low U concentrations were observed in sediment, indicating oxic condition. LREE (Light REE) and MREE (Middle REE) enrichment in upper layer and depletion of MREE in deeper layer were shown in porewater profile. This study suggests that porewater flux in the western Pacific basin is a minor source of REEs to seawater, and abundant REEs are enriched in sediments, which is mainly caused by the extensive oxic condition, low sedimentation rate and strong adsorption capacity of sediments. Hence, the removal of REEs of porewater may result in widespread REE-rich sediments in the western Pacific basin.
Similar content being viewed by others


Introduction
The rare earth element (REE) is important not only in world industrial demand1, but also in tracing various geochemical processes2. REE is widely used for geochemical studies of estuaries, oceans and hydrothermal processes3,4,5,6,7,8,9,10,11,12,13,14,15,16,17. The main sources of REEs to seawater are the riverine flux18,19,20 and the eolian flux21. The fact that removal of REEs tend to occur rapidly at the low salinity end of the mixing zone, reflecting REE scavenging by salt-induced coagulation and flocculation of Fe-rich, organic colloids22. The order of removal is LREE > MREE > HREE2. In this case, LREE and MREE with high mobility are preferentially absorbed onto particulates and removal from seawater23. REE mainly exist as carbonate complexation with positive or negative charge24,25, and the intensity of complexes improve following the increase of the atomic number25,26. At higher salinities the particles with strong REEs enrichment release the REE back to solution10,22.
The seawater REE cycle, however, has not been fully understood yet27,28. Johannesson et al.29 proposed that groundwater from seafloor would input abundant REEs into seawater, representing a “new” REEs inputs to seawater. There are also a lot of investigations on the REE behaviors in porewater of marine sediment2,21,28. The records of REE pattern can be used to demonstrate processes of REE cycling in oceans28, and the study of REE fractionation and migration between seawater and porewater play a key role for understanding early diagenesis and REE cycling mechanisms21,28,30,31,32.
Porewater which offer a link between bottom water and sediments is a carrier of the chemical components, and act as sensitive indicators of diagenesis33. There are few works about porewater profiles for REE due partially to analytical difficulties30,31,33,34. Existing REE data of porewater are mainly from shallow sea region2,31,33,35,36,37, and margin and slope facies2,28. However, there are still few investigations for REE cycling in deep-sea basin, and in the porewater of pelagic sediment. In addition, deep-sea mud in the Pacific Ocean has been considered as an important potential resource for REE1. Researchers of REE-rich sediments mainly focused on enrichment mechanism38,39,40,41 notwithstanding the fact that investigating REE cycle in deep ocean basin may be also important for uncovering the origin of REE-rich sediments.
In this paper, high-resolution REE profiles in both seawater and porewater are presented for 2 sites in the western Pacific basin. The object is to achieve an improved understanding of (1) features of REE migration and fractionation, and (2) the REE cycling mode in deep-sea area in the western Pacific.
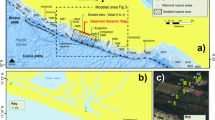
A comfortable succession of marine sediments was deposited in Pigafetta basin of the western Pacific surrounded by seamounts, with the depth of water ranging from 5000 m to 6500 m (Fig. 1). The seawater oxygen minimum zone (OMZ) ranges from 600 m to 1000 m with oxic bottom water, and the nutrients (SiO3 2−, NO3 − and PO4 3−) show the low values at surface water followed by a steep rise at OMZ and deep seawater42. The Pigafetta basin is typical of pelagic sediment having no abundant riverine input and low values of terrigenous, organic contents and sedimentation rate43 compared to the margin and slope sediments2,28. These sediments are dominated by zeolitic brown clay in the depth of 0–40 m (equivalent to the Late Cretaceous to the Pliocene)43, and show REE enrichment characteristics1 with the highest concentration exceeding 6500 ppm near the area38. Seawater samples collected at CTD1 site (water depth ~5688 m) and aliquots of 32-cm-thick sediments were collected by box core (MABC25) at water depth ~5322 m in July 2015. The upper part of site (shallow sediments, ~0–8 cm) contains sandy clay with gravel, with high moisture content, low mobility and low viscosity. Whereas the lower part (deep sediments, ~8–32 cm) are dominated by pelagic clay with low moisture content and relatively high viscosity.

Location of the research area in the western Pacific region.
Results and Discussion
Vertical profiles of REE, Mn, Fe and sulfate in seawater and porewater
The seawater, porewater and sediment results are presented in the Tables 1, 2 and 3, and Fig. 2. The contents of REE (ΣREE) are low in seawater (31.8–168 pmol/L) and display an increasing trend with depth (Fig. 2), similar to previously published REE data28,44,45,46,47,48,49 (Fig. 3). The increasing trend of REE concentrations in seawater may be caused by many factors. REEs in upper seawater column are removed by adsorption on downward settling particles followed by dissolution of REEs from these particles upon settling into deep seawater44. Alternatively, this REEs concentrations gradients may be produced by the REEs-rich upward flux from porewater into bottom water3.

Seawater profile of REE and porewater profile of REE, Mn, Fe and SO4 2−, Western Pacific.

The distributions of REE concentrations from seawater, and variations of REE, Mn and Fe values in porewater. Data source: North Pacific, South Pacific, North Atlantic, South Atlantic, Northwest Pacific, San Francisco Bay from refs.44,45,46,47,48,49 respectively, Oregon Slope and Buzzards Bay from ref.28 and ref.33 respectively, California Margin and Nazca Ridge off Peru from ref.2.
For comparison, La, Nd, Sm and Yb seawater column profiles from previously researches are shown in Fig. 3. As can be seen, the content variations revealed in our results are comparable to the North Pacific, particularly for La and Nd. In the upper seawater, nutrients are assimilated by phytoplankton, which is remineralized and released REE back into solution, resulting in low concentrations at the surface water but a rapidly increase in the oxygen minimum zone (OMZ)50. The OMZ (~600–1000 m) in study area shows significant enrichment of nutrients (nitrate, phosphate and silicate)42 and REEs (Fig. 3), and thus suggests that REEs are influenced by biogeochemical processes in shallow water. However, advective transport and water mass mixing may be the dominant process controlling REEs in the intermediate and deep ocean47. It was established that REEs concentrations increase in deep water from North Atlantic to North Pacific45 (Fig. 3). Data in this study show relatively high values of Sm and low concentrations of Yb in deep water compared to North Pacific (Fig. 3), and this observation might probably have resulted from different water mass. The most obvious influence on REE in the western Pacific is the northward spreading Antarctic bottom water (AABW)44. Kawabe et al.51 inferred that these water mass is mixed with North Pacific Deep Water (NPDW) in study area. Therefore, the striking similarity in REE concentrations between deep water in Northwest Pacific nearby research area and the deep-water samples of CTD1 are observed (Fig. 3).
REE concentrations in porewater are relatively high, with the peak value at sediment-water interface (SWI). Total concentrations of REE range from 120 pmol/L to 8531 pmol/L with relatively high abundances in upper part of the core (Fig. 2).
REE contents (except the top sample) show relatively low values similar to Nazca Ridge off Peru (Fig. 3) which exhibits no Fe increase in porewater2. It was suggested that high concentrations of REEs in porewater are derived from reduction of oxides in reducing condition2,28,33, which is supported by the strong increase of dissolved Mn and Fe at the upper part of sediments at each site (Fig. 3). However, there is extremely low Fe concentrations (below detection limit) and weak enrichment of Mn in this study coinciding with California Margin site (without Fe present) (Fig. 3), which indicates that the studied interval of the MABC25 profile is oxic or suboxic. This interpretation is also supported by the sulfate concentrations that are close to typical seawater value of ~28 mmol/L52 for all porewater samples (Fig. 2) and no obvious excursion in Ba for sediment samples (Table 3). Mn/Al ratios in bulk sediment are used as a sensitive indicator of suboxic condition, due to manganese is soluble under suboxic condition in porewater and transport upward until oxic zone53. Consequently, sediments under suboxic condition exhibit low Mn/Al value of ~0.0152. The Mn/Al ratios in MABC25 sediments range from 0.062 to 0.099, thus showing much higher values than the value of ~0.01 which is suggestive of oxic sedimentation throughout. This interpretation is also evidenced by extremely low U concentrations of bulk rock (2.31~2.61 ppm)52. The dissolved Mn of porewater values correspond to results of ocean drilling program which show seawater-like value 22 m below seafloor (mbsf) and peak value (~86 μmol/L) at 232 mbsf in research area43.
There are obvious REE enrichment at SWI (discussed below) and weak enrichment of dissolved Mn at a depth of ~0–2 cm below the seafloor (cmbsf). The characteristics of dissolved Mn may be in contradiction to oxic condition in porewater. Reconciling this conflict, Kim et al.54 and Machida et al.55 observed that the geochemistry of ferromanganese nodule on deep-sea sediment is similar to that of the adjacent Fe-Mn crust on the seamounts nearby the research area, thus indicating a high flux of detrital components of Fe-Mn crust from seamounts to the study site. The increase of dissolved Mn at top sediment is probably caused by interference of partial dissolution of these detrital materials. The detrital grains, remarkable peak of dissolved Al concentration, which mainly comes from detrital components and positive correlations between dissolved Al and trace elements, as observed in upmost sediments of MABC25 site42, supporting this interpretation.
Comparison between seawater and porewater REE distribution patterns
The PAAS-normalized patterns of seawater samples display the typical characteristics of seawater3, with a progressive enrichment toward heavy REE, a depletion of Ce and a slight enrichment of Gd (Fig. 4). Specifically, the intermediate and deep seawater column (~1000–5663 m) exhibits a stronger enrichment of HREE, and a stronger depletion of Ce (with an average Ce/Ce* value of 0.19) than those of the upper seawater column (~0–1000 m) (Fig. 4).

PAAS shale-normalized REE distribution spectra of seawater and porewater.
The “MREE bulge” pattern was observed in porewater with anoxic condition2. Porewater REE patterns in this study, however, show a slight decline to left and negative Ce anomalies (Fig. 4), also reflecting oxic condition in the sedimentary profile of the western Pacific, which is in consistent with the central Pacific56 and the eastern Pacific area57. The REE patterns of porewater can be divided into two patterns. Porewater REE patterns of the upper part of core (shallow porewater, ~0–8 cm) show a flatter pattern with weak negative Ce anomalies and the enrichments of LREE and MREE relative to bottom seawater. The lower part of the porewater (~8–32 cm), however, shows stronger positive Eu anomalies and negative Ce anomalies (Fig. 4).
Migration and fractionation of REE
The ratios between LREE (La-Nd), MREE (Sm-Dy) and HREE (Ho-Lu) are expressed as LaN/YbN, LaN/SmN and SmN/YbN respectively. REE contents in the surface seawater is low and LaN/YbN, LaN/SmN and SmN/YbN values are 0.59, 0.71 and 0.83 respectively (Fig. 5), indicating a relative HREE enrichment. The surface seawater also shows a flat REE pattern and a relatively weak Ce depletion (Ce/Ce* = 0.38). When the main contribution of REE is river or eolian input, the PAAS-normalised REE would exhibit a shale-like flat pattern with LaN ≈ CeN ≈ PrN ≈ LuN 10,31,58,59. Organic matter, Fe and Mn particle scavenged abundant REEs from seawater with adsorption capacity of LREE > MREE > HREE2. Therefore, surface seawater usually shows a HREE enriched pattern and a relatively flat distribution in comparison with deep seawater2.

Variation characteristics of REE in seawater and porewater.
The values of LaN/YbN and SmN/YbN show obvious decline and LaN/SmN ratios rise at depth of OMZ42 (~600–1000 m), followed by nearly constant values of these ratios in deep water (~1000–5663 m), reflecting significant enrichment of HREE and depletion of LREE and MREE at intermediate and deep seawater (Fig. 5). These results are consistent with data from the northern Atlantic3, the South Atlantic47, the South Pacific45 and the North Pacific44. The variation of REE fractionation may be produced by adsorption and the strength of complexation. Deep seawater displayed a significantly HREE-enriched pattern due to preferential scavenging of LREE and MREE by particles, and stronger compelxation of HREE in seawater23,60. The deep-water samples which are influenced by biochemical processes and water mass mixing47 show constant REE contents and distribution patterns (Figs 4 and 5), indicating a stable state. The upward porewater flux can lead to REE enrichment in deep water3. The ΣREE of bottom seawater sample, however, exhibit low value relative to surface porewater and even lower than the seawater sample at depth of ~4000 m, indicating that the diffusion of flux from sediments is low. This result is consistent with the suggestion of Piepgras and Jacobsen44. They also observed no significant upward REEs flux from the oxic sediments. Cerium (Ce) exists as trivalent or tetravalent forms depending on the redox conditions2,13,15,61,62. Ce/Ce* values decrease with water depth, suggesting that surface seawater is influenced by REEs sources with LREE enrichment and higher Ce/Ce* values. Deep seawater, however, may be influenced by oxic AABW44, with low Ce/Ce* values due to Ce scavenging by non-dissolve oxide.
Porewater at SWI (~0–1 cm depth of sediment) shows a significant REE enrichment, with concentrations almost hundred times higher than surface seawater as a result of particles degradation during early diagenesis2,28,33. There are abundant particles in the surface water but few particles in deep seawater in research area (unpublished data). Although, Müller et al.57 inferred that only 1% particles could reach seafloor, these should not be ignored in REE cycling. It is believed that the particles are dominantly in particulate coatings composed of Mn- or Fe-Mn oxides and organic matter, and only Fe-oxides and organic matter coatings are the main carriers of REEs2. The dissolved Fe is not present in MABC25 (Fig. 2), thus the REEs may be released by organic matter. Most of organic matter would be expected to degrade within the upper ~1–2 cm63, particularly in oxic sediments which shows a markedly faster degradation rate of organic matter relative to anoxic sediments2. This interpretation is also evidenced by the low values of total organic carbon (~0.2–0.4 wt%) of shallow sediments in North Pacific64 and the extremely low value (0.03 wt%) of sediments in research area43. It was established that the organic matter in surface sediments shows non-negligible REE concentrations with an average value of ~39 ppm, and displays a slight decline to right pattern with enrichment of LREE and MREE and no or slight positive Ce anomalies65. These results are in agreement with substantial LREE, MREE and Ce scavenging by organic matter in the upper water column21. The organic matter mainly originated from plankton in euphotic zone63, but εNd values suggesting that there is a detrital provenance in Northwest Pacific65. The ΣREE of porewater at SWI show much higher concentrations compared to the published data from oxic surface sediments (Fig. 3), indicating another source of REEs in surface porewater besides organic matter. It is possible that detrital components may play a key role for REEs enrichment. Freslon et al.65 infer that partial dissolution of easily dissolvable volcanogenic particles could occur in Northwest Pacific. Lots of seamounts around the MABC25 (Fig. 1) and abundant enrichment of dissolved Al at SWI42, indicating that dissolution of detrital components is also a reason for the significant increment in ΣREE in surface porewater. The LaN/YbN and SmN/YbN values increase and LaN/SmN (Fig. 5) obviously decrease, representing LREE and MREE enrichments. These characteristics are similar to organic matter65 and detrital components2, indicating that the LREE and MREE carried by organic matter and detrial components have been released into porewater during burial in the sediment surface. The increasing of Ce/Ce* values may also be generated by these processes (Fig. 5).
The shallow porewater (~1–8 cm) show a rapid decline of ΣREE (i.e. mean value of ~731 pmol/L) and the decrease of LaN/YbN values (Fig. 5), which may reflect the decrease of REE release from organic matter and detrial components, or the increase of REE removal. The slightly increased MREE/MREE* values may reflect lower activity and thus gradually released of MREE from particles. Similarly, the decreased LaN/SmN ratios and increased SmN/YbN ratios at depths of >3 cm show MREE enrichment of porewater.
REEs of porewater in deeper sediments (~8–32 cm) show very low contents, i.e., ΣREE less than bottom seawater. The decreasing ΣREE values may be influenced by early diagenetic reaction33. The REEs may be removed by precipitation of REE-rich mineral or the absorption by sediments21. The steep drop of MREE/MREE* and SmN/YbN, and obvious increase of LaN/SmN values indicate a severe removal of MREE (Fig. 5). It is possible due to the exhaust of particles and absorption of sediments. The uptake of MREE is further evidenced by increase of MREE/MREE* in sediment samples with depth from 0.35 to 0.38 (Table 3). MREE may be influenced by surface charges of Fe- and Mn- oxyhydroxides. Specifically, Mn- oxyhydroxides tend to display negative charges that attract LREE, but Fe- oxyhydroxides develop positive charges that attract HREE66, leading to a preferential scavenging of MREE in porewater by Fe-Mn particles. Alternatively, recent works observed that REEs exhibited a strongly positive correlation with phosphorus38,39,40,41,67,68, and phosphate minerals in sediments could also uptake abundant MREE69,70,71, resulting in depletion of MREE in porewater. The sequestering of REE has been observed by investigating the partitioning of REE between different reservoirs in research area41. Wang et al.41 calculated that the apatite occupies above 70% of the total REE budget and Fe-Mn micro-nodules and zeolite only occupy 2% and 3% respectively. Thus, REE mainly uptake by apatite in sediments. Ce/Ce* values exhibited gradual decreasing trend in the interval (Fig. 5) with oxic condition. The most reasonable explanation is the preferentially removal of Ce in deeper porewater33.
As an element with multiple valences, europium (Eu) exist as trivalent in seawater and would produce positive Eu anomalies under hydrothermal input or strongly reducing conditions61. There are no obvious hydrothermal activity and reducing conditions in research site. Thus, the positive Eu anomalies in porewater may result from abundantly uptake of other MREEs by sediments, or interference of Ba related to ICP-MS analytical techniques72. However, the seawater-like concentration of sulfate in porewater and low values of Ba in sediments point to the fact that there are low dissolved Ba concentrations in porewater. The low values of Ba interference may reflect the original signature of Eu62.
From the discussions above, we can conclude that: (1) surface seawater column displays a relatively flat REE distributions pattern due to influence of the input flux carried by wind or ocean current, (2) deep seawater shows HREE enrichment, and LREE and MREE depletion because of biogeochemical processes and water mass mixing (3) porewater at SWI exhibits REE enrichment (especially LREE and MREE) resulting from degradation of organic matter and dissolution of detrital components, (4) particles begin to release MREE in shallow sediments due to a slow regeneration rate, and (5) porewater in deeper sediments presents a steep fall of REE concentrations and MREE depletion as a result of absorption of sediments.
Deep-sea REE cycling and their indicative significance
Historically, riverine inputs were considered as the primary source of REEs to seawater18,19,20 and could be transported to marine via particles, solution and colloids with a total concentration of 39 × 106 mol/y73. Similarly, the flux of REEs from eolian or atmosphere input also adds abundant REEs into ocean. Hydrothermal with low REE contents are poorly quantified21 but likely play a limited role in REE cycle. Recent works found that groundwater could discharge lots of REEs to seawater29, and porewater is also thought to be a significant source of REEs to seawater28,31,32,33. Chen et al.21 predicted that coastal and shelf sediments would diffuse REEs to seawater 10 times higher than river inputs. Deep sea area (i.e. continental margin and slope) also show the same characteristics with Nd from porewater to seawater up to ~110 × 106 mol/y28.
There are HREE depleted pattern in shallow porewater, and MREE and HREE enriched pattern (similar to the pattern of deep seawater) in deeper porewater (Fig. 6) at near-shore margin site of the Pacific2. This observation is not unique28. Abbott et al.28 observed the same porewater REE patterns in the sediments of margin and slope sites (Fig. 6) and interpreted that HREE depletion patterns for shallow porewater may have a primary diagenetic source. Moreover, the HREE enrichment patterns for deep porewater which displayed a similar REE pattern with bottom seawater, suggesting that porewater in shore margin or slope sediments are the main REE source to seawater28. However, our data shows difference REE distribution patterns between bottom seawater and porewater at shallow or deep sediments in the western Pacific basin (Figs 4 and 6). The REE concentration of the surface porewater are much higher than that of the bottom ocean water, suggesting a REE diffusion flux from sediments to seawater. However, their REE patterns are somewhat different, which may be generated by relatively high mixing rate of deep seawater with mean speed values of ~2.8 cm/s detected by current meter in a year (unpublished data) and/or the limited REEs flux from sediments to seawater. This is also corroborated by the obviously different LaN/YbN, LaN/SmN and SmN/YbN values between bottom water and porewater (Fig. 5). This feature is apparently different with the shore margin or slope model (Fig. 6).

Possible reasons for REE enrichment of deep water sediments in study area are different characteristics of sediments, sedimentation rates and redox condition compared with shallow water regions. The shallow sediments (~0–8 cm) dominated by sandy clay with high moisture content and permeability, and the deeper sediments (~8–32 cm) contains brown pelagic clay with low moisture content and permeability. The sandy clay in shallow sediments mainly influenced by detrital components from seamounts and organic matter input show substantial release of REEs from sediment into porewater and the high permeability contribute to REEs migrate to deep sediments. The deeper pelagic clays characterized by low permeability and superior adsorption abilities for REEs74, largely absorbed REEs from shallow porewater. Meanwhile, the low sedimentation rate is favorable for REE enrichment because the ΣREE of sediments is mainly controlled by REE scavenging processes from seawater to sediments75. The age of sediments should be no older than Cenozoic, and the sedimentation rate is lower than 0.5 m/m.y. in pelagic clays in research area43. The extremely low sedimentation rate results in the great the uptake of REEs during sedimentation. In addition, redox condition also plays a key role for REE enrichment67. The shore margin or slope sediments generally display a relative narrow oxic condition, e.g., only 1mm-thick of sediment in Buzzard bay30. Under anoxic condition, there is re-mineralization of oxides in sediments and discharge of abundant REEs2. Thus, REEs flux from sediments to seawater primarily comes from the re-mineralization flux21. In this study, oxic condition are extensive in porewater (discussed above), which indicated low chances of re-mineralization of oxides. Meanwhile, there are likely extensive re-mineralization of organic matter and dissolution of detrital components. These REEs could be absorbed by sediments which exhibit excellent absorbency.
Arsouze et al.76 presented Nd oceanic model. This study continues the modeling work and adds the porewater characteristics to establish a possible REE migration and cycle model in the western Pacific basin, which is further compared with REE characteristics of shore margin and slope area (Fig. 7). Two significant REE sources have been proposed (river source is indistinctive in research area), in which eolian sources (LREE ≈ MREE ≈ HREE) input REEs into surface seawater, and re-mineralization porewater flux from shore margin or slope sediments input abundant REEs into bottom seawater (Fig. 7). The particles scavenge REEs from upper water column followed by part re-mineralization in deep seawater. With the settlement of these particles on the seafloor, abundant REEs are released (LREE > MREE > HREE) to porewater at SWI (Fig. 7). The obvious enrichment of REEs in porewater of surface sediments likely leads to part of REEs diffusion to bottom water. However, it may be also reasonable to suppose that abundant REEs are absorbed by the deeper sediments. Preferential uptake of MREE is observed in sediments probably because of existence of apatite. Finally, REEs from upper seawater column are enriched in sediment through a series of migration and fractionation process (Fig. 7).

REE cycle model in western Pacific basin.
This model can also account for REE enrichment mechanism. Under anoxia condition, oxides which show enrichment of REEs tend to be regenerated and thus release abundant REEs into seawater. However, sediments with extensively oxic condition in the study area offer a favorable site for oxides preservation. Also, strong adsorption capacity and low sedimentation rate play a key role for REE enrichment. It is worth to remark that pelagic clay in basin displays beneficial preservation condition, low sedimentation rate and excellent absorbency. Thus, we infer that this REE cycle model in deep sea basin is the main reason for widespread enrichment of REEs in sediments of the western Pacific Ocean.
Conclusions
Surface seawater displays relatively flat patterns due to the eolian sources. The intermediate and deep seawater column showed HREE enrichment patterns because of particles (i.e. Fe-Mn or organic matters) scavenging (LREE > MREE > HREE) and strong carbonate complexation of HREE. The deep water is also influenced by the water mass mixing. The generation of REE-rich particles on the seafloor release abundant REEs (LREE > MREE > HREE) to porewater at the SWI, leading to strong REE enrichment at uppermost porewater with a flat REE pattern. Decomposition present in decreasing trend with depth and part of REEs likely diffuse to bottom ocean water. However, we infer that lots of REEs are absorbed by the deeper sediments. The REE enrichment in basin may be generated by strong adsorption capacities, low sedimentation rate and widespread oxic conditions. We conclude that this mechanism may provide a valuable insight into the REE cycling in the deep sea.
Methods
Sampling
The seawater and sediment samples were collected in the ship. All bottles, samplers, tubing and filters were cleaned by acid and ultrapure water before use. Seawater was collected by Sea-Bird Electronics 917 Plus CTD (Conductance, Temperature and Depth) device. Niskin sampling bottles were deployed on an epoxy-coated CTD rosette attached to a non-metallic line in order to minimize chemical contamination, and were programmed to trip at determined depths (bottom water 25 m above seafloor). About 1 L seawater per sample was filtered (0.4 μm) and transferred into polyethylene bottle immediately after sampling. These samples were acidified to pH of 2 with high purity HCl.
Sediment samples were obtained using box core during the Haiyang-6 Cruise by the Guangzhou Marine Geological Survey (GMGS). Within two hours after sediment collection, high-resolution porewater samples were extracted from sediments via Rhizon CSS (Rhizosphere Research Products, Netherlands) moisture sampler which was composed of core solution sampler and syringe. This sampler made of a hydrophilic porous polymer tube, with pore diameter of 0.1 μm, extended with a polyvinyl chloride tube. The outer diameter of a Rhizon is 2.4 mm, and the filter section has a length of 5 cm. Rhizons can minimize sampling artifacts and obtain good results of pH, alkalinity, sulfate, and various metal ions in porewater77. Immediately following sampling, the porewater samples were acidified to pH 2 with ultra pure HCl and subsequently transferred into the polyethylene tubes. The seawater, porewater and sediment samples were stored in seal and cool condition (4 °C).
Analytical methods
Dissolved REE concentrations were measured following the main procedures described in Akagi et al.78 and Freslon et al.79. To increase the accuracy of these measurements, a condensation procedure via the co-precipitation was applied to the seawater and porewater samples. Briefly, 25 ml of sample was mixed in a pre-cleaned Teflon centrifuge tube with 0.5 ml of 20 mg/L Ga standard solution. The mixture was shaken and then allowed to equilibrate for 15 min. The solution was increased pH to ~9.0 using 2 M NaOH, which led to co-precipitation of magnesium hydroxides with gallium. The sample was centrifugated for 5 min at 3000 rpm and the supernatant discarded. Following the process of centrifugation was conducted three times, the residue of precipitate (the pre-concentration factor of up to ~200)78 was washed by ultrapure water and then redissolved in 4 ml 5% ultrapure diluted HNO3 in cleaned Teflon centrifuge tube. Rare earth elements were measured via ICAP-Q Inductively Coupled Plasma Mass Spectrometry (ICP-MS) (Thermo Fisher Scientific, MA, USA) at Third Institute of Oceanography, State Oceanic Administration, China.
For the procedural blanks, the reagent blanks caused by the whole procedure were determined by carrying out the same pre-concentration procedure without use of a seawater or porewater sample. The blanks varied between 0.041 (for Tm) and 3.60 (for La) pmol/L and the procedural detection limit (three times the standard deviation of 10 blank measurements) range from 0.012 (for Tm) to 0.98 (for La) pmol/L (Table 4). Due to no certified reference materials (CRM) for porewater available, seawater CRM (NASS-6) were used to assess the accuracy (ten repeated analyses). The analytical results for every REE agreed with literature values80 (Table 4). The external accuracy was assessed via repeat analyses of a porewater sample. The relative standard deviation (RSD) for every REE in samples was ≤10% (seven repeated analyses MABC25-5) (Table 4). The recovery from the column was evaluated using 100 ng/L REE multi-element standard solutions and the sample mean recoveries range from 93% to 102%.
The concentrations of SO4 2− were measured by using a 790 Personal IC (Metrohm, Herisau, Switzerland) ion chromatograph equipped with a conductivity detector and an anion exchange column (Metrosep A Supp 4). Sulfate concentrations were determined on 500 folds diluted with ultrapure water. A bicarbonate solution (i.e. 1.8 mmol/L Na2CO3 + 1.7 mmol/L NaHCO3) was used as the eluent and the analytical precision estimated to be <1%. Dissolved Fe and Mn of porewater were determined on 50 folds diluted with ultrapure water followed by the method of Hu et al.81 and analyzed using the ELEMENT XR High Resolution ICP-MS (Thermo Fisher Scientific, MA, USA). The analytical reproducibility and precision was better than 10%. The SO4 2−, Fe and Mn of porewater were analyzed in the Center for Marine Geochemistry Research at the Nanjing University.
Sediment samples were dried, crushed and pulverized (200 mesh). Major element, trace element and REE analyses of sediment followed the method of Chen et al.82. Mn and Al were analyzed using AXIOSX X-ray fluorescence spectrometry (PANalytical B. V., Almelo, The Netherlands) at the Key Laboratory of Marine Mineral Resources, Ministry of Land and Resources, China. Overall analytical precision and accuracy were better than 3%. For the REE and trace element analyses, the samples were dissolved using an HCl, HF, and HClO4 acid mix, and rhodium was added as an internal standard for concentrations calculation. The REE and U were measured using a X2 ICP-MS (Thermo Fisher Scientific, MA, USA), and Ba was determined by using Optima 4300DV, Inductively Coupled Plasma Optical Emission Spectrometer (PerkinElmer, MA, USA) at the Key Laboratory of Marine Mineral Resources, Ministry of Land and Resources, China. Analytical precision for elemental concentrations was generally better than 5%.
REE concentrations were normalized to PAAS83. Ce/Ce* = CeN/(1/2LaN + 1/2PrN), Eu/Eu* = EuN/(1/2SmN + 1/2GdN) were calculated50 (Bau and Dulski, 1996) and MREE/MREE* = 2*average (MREE)/average (LREE) + average (HREE) was calculated according to Chen et al.21.
Data Availability
The datasets analysed during the current study are available from the corresponding author upon reasonable request.
References
Kato, Y. et al. Deep-sea mud in the Pacific Ocean as a potential resource for rare-earth elements. Nat. Geosci. 4, 535–539 (2011).
Haley, B. A., Klinkhammer, G. P. & McManus, J. Rare earth elements in pore waters of marine sediments. Geochim. Cosmochim. Acta 68, 1265–1279 (2004).
Elderfield, H. & Greaves, M. J. The rare earth elements in seawater. Nature 296, 214–219 (1982).
Klinkhammer, G., Elderfield, H. & Hudson, A. Rare earth elements in seawater near hydrothermal vents. Nature 305, 185–188 (1983).
Murphy, K. & Dymond, J. Rare earth element fluxes and geochemical budget in the eastern equatorial Pacific. Nature 307, 444–447 (1984).
Elderfield, H., Upstill-Goddard, R. & Sholkovitz, E. R. The rare earth elements in rivers, estuaries, and coastal seas and their significance to the composition of ocean waters. Geochim. Cosmochim. Acta 54, 971–991 (1990).
Bertram, C. J. & Elderfield, H. The geochemical balance of the rare earth elements and neodymium isotopes in the oceans. Geochim. Cosmochim. Acta 57, 1957–1986 (1993).
German, C. R., Masuzawa, T., Greaves, M. J., Elderfield, H. & Edmond, J. M. Dissolved rare earth elements in the Southern Ocean: Cerium oxidation and the influence of hydrography. Geochim. Cosmochim. Acta 59, 1551–1558 (1995).
Sherrell, R. M., Field, M. P. & Ravizza, G. Uptake and fractionation of rare earth elements on hydrothermal plume particles at 9°25’N, east Pacific Rise. Geochim. Cosmochim. Acta 63, 1709–1722 (1999).
Sholkovitz, E. & Szymczak, R. The estuarine chemistry of rare earth elements: Comparison of the Amazon, Fly, Sepik and the Gulf of Papua systems. Earth Planet. Sci. Lett. 179, 299–309 (2000).
Lacan, F. & Jeandel, C. Tracing Papua New Guinea imprint on the central equatorial Pacific Ocean using neodymium isotopic compositions and rare earth element patterns. Earth Planet. Sci. Lett. 186, 497–512 (2001).
Wang, Z. L. & Yamada, M. Geochemistry of dissolved rare earth elements in the Equatorial Pacific Ocean. Environ. Geol. 52, 779–787 (2007).
He, G. W., Sun, X. M., Yang, S. X., Zhu, K. C. & Song, C. B. A comparison of REE geochemistry between polymetallic nodules and cobalt-rich crusts in the Pacific Ocean. Geology in China 38(2), 462–472 (2011).
Ling, H. F. et al. Cerium anomaly variations in Ediacaran–earliest Cambrian carbonates from the Yangtze Gorges area, South China: Implications for oxygenation of coeval shallow seawater. Precambrian Res. 225, 110–127 (2013).
Deng, Y. N., Guo, Q. J., Zhu, M. Y. & Zhang, J. M. REE Geochemistry of Kerogen from Early Cambrian Black Rock Series in Western Hunan. Earth Science (Journal of China University of Geosciences) 39(3), 283–292 (2014).
Zhao, M. Y. & Zheng, Y. F. Marine carbonate records of terrigenous input into Paleotethyan seawater: geochemical constraints from Carboniferous limestones. Geochim. Cosmochim. Acta 141, 508–531 (2014).
Zhao, M. Y. & Zheng, Y. F. A geochemical framework for retrieving the linked depositional and diagenetic histories of marine carbonates. Earth Planet. Sci. Lett. 460, 213–221 (2017).
Martin, J. M., Hodahl, O. & Philippot, J. C. Rare earth element supply to the ocean. J. Geophys. Res. 81, 3119–3124 (1976).
Greaves, M. J., Statham, P. J. & Elderfield, H. Rare earth element mobilization from marine atmospheric dust into seawater. Mar. Chem. 46, 255–260 (1994).
Sholkovitz, E. R., Elderfield, H., Szymczak, R. & Casey, K. Island weathering: River sources of rare earth elements to the Western Pacific Ocean. Mar. Chem. 68, 39–57 (1999).
Chen, J. B. et al. Diagenetic uptake of rare earth elements by bioapatite, with an example from Lower Triassic conodonts of South China. Earth-Sci. Rev. 149, 181–202 (2015).
Johannesson, K. H. et al. Rare earth element behavior during groundwater–seawater mixing along the Kona Coast of Hawaii. Geochim. Cosmochim. Acta 198, 229–258 (2017).
Nozaki, Y. & Alibo, D. S. Importance of vertical geochemical processes in controlling the oceanic profiles of dissolved rare earth elements in the northeastern Indian Ocean. Earth Planet. Sci. Lett. 205, 155–172 (2003).
Ohta, A. & Kawabe, I. Theoretical study of tetrad effect observed in REE distribution coefficients between marine Fe–Mn deposit and deep seawater, and in REE(III)-carbonate complexation constants. Geochem. J. 34, 455–473 (2000).
Tang, J. & Johannesson, K. H. Ligand extraction of rare earth elements from aquifer sediments: implications for rare earth element complexation with organic matter in natural waters. Geochim. Cosmochim. Acta 74, 6690–6705 (2010).
Luo, Y. & Byrne, R. H. Carbonate complexation of yttrium and the rare earth elements in natural waters. Geochim. Cosmochim. Acta 68, 691–699 (2004).
Van de Flierdt, T. et al. New constraints on the sources and behavior of neodymium and hafnium in seawater from Pacific Ocean ferromanganese crusts. Geochim. Cosmochim. Acta 68, 3827–3843 (2004).
Abbott, A. N., Haley, B. A., McManus, J. & Reimers, C. E. The sedimentary flux of dissolved rare earth elements to the ocean. Geochim. Cosmochim. Acta 154, 186–200 (2015).
Johannesson, K. H. et al. Submarine groundwater discharge is an important net source of light and middle REEs to coastal waters of the Indian River Lagoon, Florida, USA. Geochim. Cosmochim. Acta 75, 825–843 (2011).
Elderfield, H. & Scholkovitz, E. Rare earth elements in the pore waters of reducing nearshore sediments. Earth Planet. Sci. Lett. 82, 280–288 (1987).
Sholkovitz, E. R. Chemical evolution of rare earth elements: Fractionation between colloidal and solution phases of filtered river water. Earth Planet. Sci. Lett. 114, 77–84 (1992).
Haley, B. A. & Klinkhammer, G. P. Complete separation of rare earth elements from small volume seawater samples by automated ion chromatography: Method development and application to benthic flux. Mar. Chem. 82, 197–220 (2003).
Sholkovitz, E. R., Piepgras, D. J. & Jacobsen, S. B. The porewater chemistry of rare earth elements in Buzzards Bay sediments. Geochim. Cosmochim. Acta 53, 2847–2856 (1989).
Piper, D. Z. Rare earth elements in the sedimentary cycle: A summary. Chem. Geol. 14, 285–304 (1974).
Bayon, G. et al. Evidence for intense REE scavenging at cold seeps from the Niger Delta margin. Earth Planet. Sci. Lett. 312, 443–452 (2011).
Himmler, T. et al. Rare earth element geochemistry in cold-seep pore waters of Hydrate Ridge, northeast Pacific Ocean. Geo-Mar. Lett. 33, 369–379 (2013).
Soyol-Erdene, T. & Huh, Y. Rare earth element cycling in the pore waters of the Bering Sea Slope (IODP Exp. 323). Chem. Geol. 358, 75–89 (2013).
Kon, Y. et al. Geochemical characteristics of apatite in heavy REE-rich deep-sea mud from Minamitorishima area, southeastern Japan. Resour. Geol. 64(1), 47–57 (2014).
Ren, J. B. et al. Enrichment mechanism of rare earth elements and yttrium in deep-sea mud of Clarion-Clipperton Region. Earth Science Frontiers 22(4), 200–211 (2015).
Zhu, K. C., Ren, J. B., Wang, H. F. & Lu, H. F. Enrichment mechanism of REY and Geochemical characteristics of REY-rich pelagic clay from the central Pacific. Earth Science (Journal of China University of Geosciences) 40(6), 1052–1060 (2015).
Wang, F. L., He, G. W., Sun, X. M., Yang, Y. & Zhao, T. P. The host of REE+ Y elements in deep-sea sediments from the Pacific Ocean. Acta Petrologica Sinica 32(7), 2057–2068 (2016).
Deng, Y. et al. Trace elements geochemistry characteristics of seawater and porewater in basin, western Pacific. Earth Science (in press).
Lancelot, Y. et al. Proc. ODP, Init. Repts. Vol. 129, 1–57 (Ocean Drilling Program, 1990).
Piepgras, D. J. & Jacobsen, S. B. The behavior of rare earth elements in seawater: Precise determination of variations in the North Pacific water column. Geochim. Cosmochim. Acta 56, 1851–1862 (1992).
Zhang, J. & Nozaki, Y. Rare earth element and yttrium in seawater: ICP-MS determination in the East Caroline and South Fiji basins of the western South Pacific Ocean. Geochim. Cosmochim. Acta 23, 4631–4644 (1996).
Mawji, E. et al. The GEOTRACES Intermediate Data Product 2014. Mar. Chem. 177, 1–8 (2015).
Zheng, X. Y., Plancherel, Y., Saito, M. A., Scott, P. M. & Henderson, G. M. Rare earth elements (REEs) in the tropical South Atlantic and quantitative deconvolution of their non-conservative behavior. Geochim. Cosmochim. Acta 177, 217–237 (2016).
Tanaka, M., Shimizu, H. & Masuda, A. Features of the heavy rare-earth elements in seawater. Geochem. J. 24, 39–46 (1990).
Hatje, V., Bruland, K. W. & Flegal, A. R. Determination of rare earth elements after pre-concentration using NOBIAS-chelate PA-1®resin: Method development and application in the San Francisco Bay plume. Mar. Chem. 160, 34–41 (2014).
Schijf, J., Christenson, E. A. & Byrne, R. H. YREE scavenging in seawater: A new look at an old model. Mar. Chem. 177, 460–471 (2015).
Kawabe, M., Fujio, S. & Yanagimoto, D. Deep-water circulation at low latitudes in the western North Pacific. Deep-Sea Res. I 50, 631–656 (2003).
Borowski, W. S., Paull, C. K. & Ussler, W. III Global and local variations of interstitial sulfate gradients in deep-water, continental margin sediments: Sensitivity to underlying methane and gas hydrates. Mar. Geo. 159, 131–154 (1999).
Osborne, A. H. et al. The potential of sedimentary foraminiferal rare earth element patterns to trace water masses in the past. Geochem. Geophhys. Geosyst. 18, 1550–1568 (2017).
Kim, J. et al. Southward Shift of the Intertropical Convergence Zone in the Western Pacific During the Late Tertiary: Evidence from Ferromanganese Crusts on Seamounts West of The Marshall Islands. Paleoceanography 21, 247–257 (2006).
Machida, S. et al. Geology and Geochemistry of Ferromanganese Nodules in the Japanese Exclusive Economic Zone around Minamitorishima Island. Geochem. J. 50, 1–17 (2016).
Klinkhammer, G. P., Heggie, D. T. & Graham, D. W. Metal Diagenesis in Oxic Marine Sediments. Earth Planet. Sci. Lett. 61, 211–219 (1982).
Müller, P. J., Hartmann, M. & Suess, E. The Chemical Environment of Pelagic Sediments. In: Halbach, P., Friedrich, G. & Stackelberg, U. V. (Eds). The Manganese Nodule Belt of the Pacific Ocean Geological Environment. Ferdinand Enke, Stuttgart, pp. 70–90 (1988).
Hoyle, J., Elderfield, H., Gledhill, A. & Greaves, M. The behaviour of the rare earth elements during mixing of river and sea waters. Geochim. Cosmochim. Acta 48, 143–149 (1984).
Sholkovitz, E. R. The geochemistry of rare earth elements in the Amazon River estuary. Geochim. Cosmochim. Acta 57, 2181–2190 (1993).
Sholkovitz, E. R., Landing, W. M. & Lewis, B. L. Ocean particle chemistry: The fractionation of rare earth elements between suspended particles and seawater. Geochim. Cosmochim. Acta 58, 1567–1579 (1994).
Shields, G. & Stille, P. Diagenetic constrains on the use of cerium anomalies as palaeoseawater proxies: an isotopic and REE study of Cambrian phosphorites. Chem. Geol. 175, 29–48 (2001).
Guo, Q. J. et al. REE and trace element patterns from organic-rich rocks of the Ediacaran-Cambrian transitional interval. Gondwana Res. 36, 81–93 (2016).
Hammond, D. E., Mcmanus, J., Berelson, W. M., Kilgore, T. E. & Pope, R. H. Early diagenesis of organic material in equatorial Pacific sediments: stoichiometry and kinetics. Deep-Sea Res. II 43, 1365–1412 (1996).
Muller, P. J. & Suess, E. Productivity, sedimentation rate, and sedimentary organic matter in the oceans—i. organic carbon preservation. Deep Sea Res. 26, 1347–1362 (1979).
Freslon, N. et al. Rare earth elements and neodymium isotopes in sedimentary organic matter. Geochim. Cosmochim. Acta 140, 177–198 (2014).
Prakash, L. S. et al. Distribution of REEs and yttrium among major geochemical phases of marine Fe–Mn-oxides: comparative study between hydrogenous and hydrothermal deposits. Chem. Geol. 312–313, 127–137 (2012).
Emsbo, P., McLaughlin, P. I., Breit, G. N., Bray, E. A. & Koenig, A. E. Rare earth elements in sedimentary phosphate deposits: Solution to the global REE crisis? Gondwana Res. 27, 776–785 (2015).
Yasukawa, K. et al. Geochemistry and mineralogy of REY-rich mud in the eastern Indian Ocean. J. Asian Earth Sci. 93, 25–36 (2014).
Byrne, R. H., Liu, X. & Schijf, J. The influence of phosphate coprecipitation on rare earth distributions in natural waters. Geochim. Cosmochim. Acta 60, 3341–3346 (1996).
Rasmussen, B., Buick, R. & Taylor, W. R. Removal of oceanic REE by authigenic precipitation of phosphatic mineral. Earth Planet. Sci. Lett. 164, 135–149 (1998).
Hannigan, R. E. & Sholkovitz, E. R. The development of middle rare earth element enrichments in freshwaters: weathering of phosphate minerals. Chem. Geol. 175, 495–508 (2001).
Dulski, P. Interferences of oxide, hydroxide and chloride analyte species in the determination of rare earth elements in geological samples by inductively coupled plasma-mass spectrometry. Fresen. J. Anal. Chem. 350, 194–203 (1994).
Nozaki, Y. Rare earth element and their isotopes in the ocean. In: Steele, J. H., Thorpe, S. A. & Turekian, K. K. (Eds). Encyclopedia of Ocean Sciences. Academic Press, London, pp. 2354–2366 (2001).
Liu, J. H., Zhang, L. J. & Liang, H. F. The REE geochemistry of sediments in core CC48 from the East Pacific Ocean. Oceanologia Et Limnologia Sinica 25(1), 15–22 (1994).
Menendez, A., James, R. H., Roberts, S., Peel, K. & Connelly, D. Controls on the distribution of rare earth elements in deep-sea sediments in the North Atlantic Ocean. Ore Geol. Rev. 87, 100–113 (2017).
Arsouze1, T., Dutay, J. C., Lacan, F. & Jeandel, C. Reconstructing the Nd oceanic cycle using a coupled dynamical–biogeochemical model. Biogeosciences 6, 2829–2846 (2009).
Seeberg-Elverfeldt, J., Schlüter, M., Feseker, T. & Kölling, M. Rhizon sampling of porewaters near the sediment-water interface of aquatic systems. Limnol. & Oceanogr.: Methods 3, 361–371 (2005).
Akagi, T., Fuwa, K. & Haraguchi, H. Simultaneous multi-element determination of trace metals in sea water by inductively-coupled plasma atomic emission spectrometry after coprecipitation with gallium. Anal. Chim. Acta 177, 139–151 (1985).
Freslon, N., Bayon, G., Birot, D., Bollinger, C. & Alix, B. J. Determination of rare earth elements and other trace elements (Y, Mn, Co, Cr) in seawater using Tm addition and Mg(OH)2 co-precipitation. Talanta 85, 582–587 (2011).
Raso, M., Censi, P. & Saiano, F. Simultaneous determinations of zirconium, hafnium, yttrium and lanthanides in seawater according to a co-precipitation technique onto iron-hydroxide. Talanta 116, 1085–1090 (2013).
Hu, Y. et al. Impact of anaerobic oxidation of methane on the geochemical cycle of redox-sensitive elements at cold-seep sites of the northern South China Sea. Deep-Sea Res. II 122, 84–94 (2015).
Chen, F. et al. Evidence of Intense Methane Seepages from Molybdenum Enrichments in Gas Hydrate-Bearing Sediments of the Northern South China Sea. Chem. Geol. 443, 173–181 (2016).
McLeannan, S. M. Rare earth elements in sedimentary rocks: influence of provenance and sedimentary processes. Rev. Mineral. 21, 169–200 (1989).
Acknowledgements
Thanks to Xin Zhang, Yanmei Wang, Piaoer Fu, Zhe Yu and the staffs in Haiyangliuhao ship for their assistances in sampling and laboratory works. We deeply appreciate Dr. Peter Chukwunonso Okoli and Dr. Mingyu Zhao for revision suggestions of English expression. YN acknowledges financial support from Key Laboratory of Marine Mineral Resources, Ministry of Land and Resources (No. KLMMR-2015-A-03, KLMMR-2013-A-24), China Geological Survey Program (GZH201100303) and NSFC (Nos. 41625006).
Author information
Authors and Affiliations
Contributions
Y.D. performed sampling, chemical analysis and then wrote the main manuscript text; J.R. prepared the figures in this article; Q.G. conceived the main experimental scheme and manuscript review; J.C., H.W., and C.L. were responsible for sampling and revision suggestion for the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, Y., Ren, J., Guo, Q. et al. Rare earth element geochemistry characteristics of seawater and porewater from deep sea in western Pacific. Sci Rep 7, 16539 (2017). https://doi.org/10.1038/s41598-017-16379-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16379-1
This article is cited by
-
Distribution of REY-Rich Mud in Deep-Sea Sediments from the Magellan Seamount Trail Zone in the Northwestern Pacific
Ocean Science Journal (2024)
-
Elemental Geochemical Distribution of the Jurassic Yan'an Formation in the Ningdong Coalfield and its Controlling Effect on Coal Reservoir Properties
Natural Resources Research (2023)
-
Rare earth element signatures of Paleoproterozoic Sallopat phosphorites of Aravalli Basin, India: implications for diagenetic effects and depositional environment
Acta Geochimica (2023)
-
Differentiating between hydrothermal and diagenetic carbonate using rare earth element and yttrium (REE+Y) geochemistry: a case study from the Paleoproterozoic George Fisher massive sulfide Zn deposit, Mount Isa, Australia
Mineralium Deposita (2022)
-
Rare earth elements in surface waters and sediments of the Mgoua watershed, south western Cameroon
Arabian Journal of Geosciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
