
Abstract
As a kind of important biocatalysts, Pseudomonas lipases are commonly applied in various industrial fields. Pflip1, a new extracellular lipase gene from Pseudomonas. fluorescens Pf0–1, was first cloned and respectively expressed in Escherichia coli BL21(DE3) and Pichia pastoris KM71, the recombinant proteins Pflip1a and Pflip1b were later purified separately. Then Pflip1a was further characterized. The optimum pH of Pflip1a was 8.0 and its optimal temperature was 70 °C. After incubation at 70 °C for 12 h, Pflip1a could retain over 95% of its original activity. It showed the highest activity towards p-nitrophenyl caprylate. Moreover, its activity was profoundly affected by metal ion, ionic surfactants and organic solvents. Furthermore, the two obtained recombinant lipases were immobilized on the magnetic nanoparticles for biodiesel preparation. The GC analysis showed that for the immobilized lipases Pflip1b and Pflip1a, the biodiesel yield within 24 h respectively attained 68.5% and 80.5% at 70 °C. The activities of the two immobilized lipases still remained 70% and 82% after 10 cycles of operations in non-solvent system. These characteristics and transesterification capacity of the recombinant protein indicated its great potential for organic synthesis, especially for biodiesel production.
Similar content being viewed by others

Introduction
Lipases, also known as triacylglycerol acylhydrolase (EC 3.1.1.3), are a big kind of hydrolases which act on carboxylic ester bonds, and completely hydrolyze triglycerides into fatty acids and glycerol. Additionally, they can catalyze esterification and transesterification reactions in non-aqueous media. These features make lipases play a significant role in biotechnological industries1,2,3,4. Lipases can be easily isolated from animals, plants and microorganisms, while bacteria, yeast and fungi are the main sources of the commercial lipases5. Bacterial lipases exhibit higher thermostability and tolerance to denaturing reagents and/or organic solvents in comparison with fungal and yeast lipases, and they are also more amenable to genetic manipulation6. So, they are widely applied in industry and academic researches. Among various bacterial lipases, those from Pseudomonas and Burkholderia (formerly Pseudomonas) genera are the commonest members applied in the field of biotechnological applications7. Particularly, it has been proved that lipase(s) from Pseudomonas fluorescens possess the enzymatic merits in chiral resolution of racemic mixtures and biodiesel production8,9,10.
In general, lipases from Pseudomonas have be classified into two categories. Lipases derived from P. aeruginosa and B. glumae belonging to one category. The functional expression of such enzymes requires a lipase-specific foldase (Lif). These enzymes provide classic signal peptide to complete their secretion through cell membrane. And lipases from P. fluorescens and Serratia marcescens were as the members of the other category. This sorts of lipases do not contain typical N-terminal signal peptide while containing C-terminal-targeting signal. Their secretion is completed via ATP-binding cassette (ABC) exporters which consist of an inner membrane ATP-binding cassette, a membrane fusion protein and an outer membrane protein11,12.
Because of their function in the degradation of fats and synthesis of esters, lipases have extensive applications, for instance, processing of fats, detergents, food processing, synthesis of fine chemicals, paper manufacturing, production of maquillage, and medicines13. As known, efficiently expression of recombinant enzymes has become a precondition for commercial applications. To achieve the consistency of the internal circumstances, lipase genes from P. fluorescens and P. alcaligenes were usually expressed in homological hosts, while their expression levels are fairly low. However, some former studies indicated that expression of them in Escherichia coli might provide another option to achieve high expression level14,15,16.
On the other hand, Pichia pastoris is a popular yeast species which is frequently applied to express recombinant proteins both in the fields of scientific researches and industries. Generally, foreign gene can be integrated into genome stably through single crossover which happens in the region of His4 or AOXl with the purpose of generating recombinants with Mut+ phenotype (among which methanol is commonly used). Additionally, it may also be used to substitute for the AOX1 locus between the end sequence of 5-AOX1 and 3-AOX1 so as to produce MutS phenotype (among which methanol is gradually used). So far, over 350 recombinant proteins from human, bacteria, yeasts and filamentous fungi have been successfully expressed in P. pastoris systems, with protein production of more than 10 g/L in some cases17,18,19,20,21,22,23,24. There also are a few reports on the expression of Pseudomonas lipases in yeast18,25,26.
Moreover, fossil diesel fuel is expected to be partly substituted by biodiesel which is produced via transesterification of oil with short-chain alcohol. To date, chemical methods have been the mainstream of producing commercial biodiesel, while the requirement of excessive energy and environmental pollution appear to be its major disadvantages. Compared with chemical methods, enzymatic catalyzed methods by lipases have many advantages, such as mild reaction conditions, simplified procedure, and little pollution. But the key problem of enzymatic catalyzed methods is that lipases are expensive and easy inactivation. Thus, an efficient conversion of biodiesel depends on lipase characteristics and its high productivity, which can effectively reduce production cost to a great extent27.
Therefore, in this research, a new lipase gene, Pflip1 from P. fluorescens Pf0–1 was successfully expressed in heterologous hosts of E. coli and P. pastoris, respectively. The recombinant lipases were characterized with reference to biodiesel preparation. Then, they were further immobilized on the magnetic nanoparticles and used for transesterification of soybean oil into biodiesel to assess potential feasibility.
Results
Cloning and overexpression of pflip1
The Pflip1 gene (891 bp) was obtained by touchdown PCR using primers Pflip1F and Pflip1R with P. fluorescens Pf0–1 chromosomal DNA as template. Then, the gene Pflip1 was respectively expressed in E. coli and in P. pastoris to obtain the recombinant proteins. In E. coli, under the influence of strong T7 promoter, the Pflip1 gene was inserted in pET28a and in translational fusion with His-tag. Upon the induction of IPTG, E. coli BL21(DE3) haboring the recombinant plasmid encoded a polypeptide of 296 amino acid, whose molecular mass was 32.1 kDa. After the break, the supernatant of the recombinant Pflip1a showed a moderate activity at 217 U/ml (438 U/mg) under its optimum conditions: pH 8.0; temperature 70 °C, with p-NP, p-nitrophenyl caprylate as substrates. Figure 1 showed the result of SDS-PAGE analysis. The molecular mass of the recombinant protein was ca 32 kDa, which coincides with the actual one.

The result of SDS-PAGE gel showing the purified recombinant protein Pflip1a. Lane 1: Protein molecular weight marker (Fermentas, SM0431). Lane 2: supernatants of recombinant E. coli BL21 (DE3) cells lysates harbouring Pflip1a. Lane 3: flow-through buffer. Lane 4-Lane 8: washing buffer with an imidazole concentration gradient (30, 60, 100, 200, and 500 mM, respctly). The recombinant Pflip1a lipase is marked by an arrow in the figure.
Similarly, in P. pastoris, the Pflip1 gene was inserted in pPIC9K and in translational fusion with His-tag. Then, it was transformed into the P. pastoris strain KM71 cells. After the cells were spread in MD plate, a single clone was selected to grow in tributyrin plate, colonies with a clear hydrolysis holas were selected to grow in BMMY medium broth (Fig. 2A). The highest activity of the supernatant of fermentation in BMMY medium broth was measured to be 105 U/ml (336 U/mg). The purified recombinant protein, Pflip1b was analyzed via SDS-PAGE, and the result was shown in Fig. 2B.

Enzyme activity identification and SDS-PAGE analysis of recombinant protein. (A) Colonies had some hydrolysis circles in the middle of the tributyrin plate, which indicated that recombinant P. pastoris cells were with enzyme activity. (B) The result of SDS-PAGE gel showing the purified recombinant protein Pflip1b.
Molecular structure modeling
The Pflip1 gene encoded a 296 amino acid sequence with a theoretical molecular mass of 32.09 kDa. Further BLASTP analysis proved that Pflip1 shown high similarity to AAC15585, a lipase from P. fluorescens (identity: 92.6%); AEV60646, a triacylglycerol lipase from P. fluorescens F113 (identity: 82.1%); WP_015093259, an lipase from Pseudomonas sp. UW4 (identity: 85.5%); CAA32193, a unnamed protein product from P. fragi (identity: 43.9%); CAC07191, a lipase from P. fragi (identity: 47.1%), which indicates that Pflip1 is a bacterial I.1 subfamily lipase (Fig. 3). To classify Pflip1, a phylogenetic tree was built for the lipolytic enzymes which represent five different lipase subfamilies. As shown in Fig. 3, Pflip1 is really in subfamily I.1.

Phylogenetic tree of Pflip1 and other closely related lipolytic enzymes. The phylogenetic analysis was accomplished with the neighbor-joining method of MEGA 5.0. The values at nodes indicated the bootstrap percentage of 1,000 replicates, the lengths of the branches shown the relative divergence among the reference lipase amino acid sequences, and the scale bar indicated the amino acid substitutions per position. Database accession numbers were shown in brackets after each enzyme.
Through searching in the RCSB PDB Protein Database, it was discovered that the crystal structure of the lipase (with accession number of IEX9) from P. aeruginosa had the largest identity of amino acid sequence (46.8%) with Pflip1. Therefore, it is deemed as the most appropriate template for homology modeling28. According to validation completed by online PROCHECK (http://nihserver.mbi. ucla.edu/SAVES/), the most preferred regions contains 86.2% of the residues in the modeled structure, and only 3 out of 296 amino acids are located in the disallowed regions, which indicates that the model is reliable. The 3D structure showed a variant of the α/β hydrolase fold, and Ser83, Asp241, and His263 as the catalytic active center. The first two β-strands and one α-helix (αE) are present compared with the “canonical” α/β hydrolase fold. The formation of a stabilizing intramolecular disulfide bridge derived from the absence of helix αE. The octahedrally-coordinated calcium ion is used to stabilize the His263-containing loop (Figs 4 and 5).

Multiple sequence alignment between Pflip1 and other closely related lipolytic enzymes: IEX9, a lipase from Pseudomonas aeruginosa; AAC15585, a lipase from P. fluorescens; AEV60646, a triacylglycerol lipase from P. fluorescens F113; WP_015093259, an lipase from Pseudomonas sp. UW4; CAA32193, a unnamed protein product from P. fragi; CAC07191, a lipase from P. fragi. Cluster 1.83 and ESpript 2.0 were used to analyze multiple sequence alignment. The alpha helix, beta sheet, random coil, and beta turn are identical to α, β, η, and T, respectively.

3D model of Pflip1. The α-helix, β-sheet, random coil and beta turn are shown in the cartoon in red, yellow and green, respectively. The catalytic triads (Ser83, Asp241 and His263) are shown as spheres in magenta, blue and orange, respectively. “N” and “C” denote the N and C termini, respectively.
Characterization of recombinant lipase Pflip1a
The relative molecular mass of recombinant protein Pflip1a is ca 32 kDa, and Fig. 1 shows the SDS-PAGE analysis of Pflip1. The recombinant protein was active at a pH range from 5.0 to 10.0, and the optimum pH was 8.0 (Fig. 6A). The recombinant protein retained more than 75% of its maximal activity at a pH value range of 7.5 to 9.0. And the recombinant protein was active between 40 °C and 100 °C, especially between 70 °C and 90 °C, it retained at least 97.3% of its maximal activity at temperature range from 70 °C to 90 °C. Its optimal temperature was 70 °C (Fig. 6B).

Characterization of Pflip1a. (A) Effect of pH on the activity of Pflip1a. The enzyme activity in Tris-HCl (50 mM, pH 8.0) was taken as 100%. (citrate-phosphate buffer (pH 5.0–6.5), Tris-HCl buffer (pH 7.0–8.5), Glycine-NaOH buffer (pH 9.0–10.0), and final concentration all were 50 mM). (B) Effect of temperature on the activity of Pflip1a. (40 °C–100 °C). (C) Effect of p-NP esters of different chain lengths on the activity of Pflip1a. (C2, C4, C8, C10, C12, C14, and C16). (D) Thermal stability of Pflip1a. The enzyme was incubated at 70 °C (■), 90 °C (▲), 80 °C (●).
The purified recombinant lipase could hydrolyze p-NP esters, it was active in p-NP esters of acyl fatty acid chain lengths ranging from C2 to C16, and showed the optimum activity in C8. Compared with C8, the relative activities of Pflip1a in C10 and C12 were 71.3% and 63.9%, respectively. In contrast, it has better activity towards the esters of short-chain and medium-chain fatty acids compared with those of long-chain fatty acids (Fig. 6C).
The thermostability of Pflip1a was examined in Tris-HCl (pH 8.0) at 70 °C, 80 °C, and 90 °C for 12 h (Fig. 6D). More than 70% of its maximal activity remained after incubation at 80 °C for 10 h, and about 70% of residual activity was retained after exposure at 90 °C for 7 h. The results indicate that Pflip1a has very good thermostability (Fig. 6D).
The effects of various metal ions and EDTA on the activities of Pflip1a were presented in Fig. 7A. The results showed that Ca2+, Mg2+, Ba2+ significantly enhanced its activity by 30.2%, 24.0% and 25.6%, respectively. On the contrary, Cu2+, Zn2+, Fe3+, Al3+ and EDTA markedly decreased its activity by 19.4%, 20.6%, 34.5%, 11.9% and 24.8%, whereas, Na+, K+, Mn2+ had no remarkable effect on its activity.

Effects of additives on Pflip1a activity. (A) Effects of metal ions and EDTA on the activity of Pflip1a. 1, control; 2, Na+; 3, K+; 4, Ca2+; 5, Mg2+; 6, Cu2+; 7, Mn2+; 8, Ba2+; 9, Zn2+; 10, Fe3+; 11, Al3+; 12, EDTA. (B) Effects of surfactants on the activity of Pflip1a. 1, control; 2, SDS; 3, CTAB; 4, NP-40; 5, Triton X-100; 6, Tween 20; 7, Tween 40; 8, Tween 80. (C) Effects of organic solvents on the activity of Pflip1a. 1, control; 2, methanol; 3, ethanol; 4, tert-butanol; 5, glycerol; 6, acetone; 7, chloroform; 8, n-hexane.
The effects of various surfactants on the activities of the Pflip1a were given in Fig. 7B. The results indicated that ionic surfactants SDS and CTAB remarkably inhibited its activity by 11.9% and 37.3%, whereas, non-ionic surfactants NP-40, Triton X-100, Tween 20, Tween 40, and Tween 80 significantly increased its activity by 8.4%, 11.3%, 31.3%, 29.0%, and 17.4%, respectively.
The activities of the Pflip1a in various organic solvents were also tested (Fig. 7C). It was only inhibited in the presence of chloroform (78.1%), whereas, other organic solvents stimulated its activity to some extent, and the maximum activity was exhibited in the presence of n-hexane (134.4%), followed by acetone (130.3%), tert-butanol (130.2%), ethanol (118.1%), glycerol (119.4%), and methanol (112.3%).
Biodiesel preparation using the immobilized lipase Pflip1b and Pflip1a and their recyclability
In the present study, immobilization treatment strategy was employed for lipases Pflip1b and Pflip1a to catalyze the transesterification of soybean oil into biodiesel. The two lipases were covalently attached on magnetic nano-particles. The reaction conditions were given in Section ‘Materials and Methods’. The immobilization efficiencies of immobilized Pflip1a and Pflip1b were 84.2% and 76.4%. The activity recoveries of immobilized Pflip1a and Pflip1b were 182.5% and 166.7%, respectively. The activities have been increased 1.8 and 1.7-fold for Pflip1a and Pflip1b after immobilization. The GC analysis (Fig. 8A) showed the biodiesel conversion for the immobilized lipases Pflip1b and Pflip1a. It can be seen that the substrate conversion rates after 24 h reaction at 70 °C were 68.5% and 80.5%, respectively. However, the conversion rates were significantly different between lipase Pflip1b and Pflip1a.

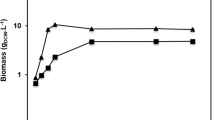
Conversion of soybean oil to biodiesel. (A) Detection of esterification reaction products by GC; (B) Reusability of the immobilized lipases Pflip1b and Pflip1a.
Reusability is one of the most important factors for the industrial applications of enzyme. The operational stability of the immobilized Pflip1b and Pflip1a were respectively presented in Fig. 8B. It can be seen from Fig. 8B that the two immobilized lipases showed high operational stability. The conversion rate of the two immobilized lipases had no obvious loss and still remained 70% and 82% of its original activity after running for 10 cycles in the solvent-free system.
Discussion
P. fluorescens species was widely existed in the environment. These bacteria could be easily separated, so they were an excellent source of lipase gene resources. In this study, a new P. fluorescens lipase was first cloned and respectively overexpressed in E. coli and P. pastoris, the enzymatic property of Pflip1a was characterized, and both recombinant enzymes were further applied in biodiesel production. There were many lipase genes that had been cloned from genus Pseudomonas. Nevertheless, there were many unsolved problems in expression of the proteins encoded by these genes in E. coli. In addition, many recombinant proteins, including SIK W1 lipase isolated from P. fluorescens 28, LipM from P. moraviensis M929, and LipR from Pseudomonas sp. R0–1430, form inclusion bodies because of incorrect folding or excessively rapid expression and accumulation of protein31. In this research, when Pflip1 of P. fluorescens Pf0–1 was transformed into E. coli BL21 (DE3) induced at low temperature, Pflip1a was overexpressed in a soluble and catalytically active form. This result indicates that the thermostable lipase genes may be expressed in psychrophilic and mesophilic organisms, and E. coli can slow down expression level of heterologous protein at low temperature, so it is expressed in soluble form. So far, many lipase genes from bacteria, yeasts and filamentous fungi have been functionally expressed. However, there were few reports on the expression of Pseudomonas lipases in yeast in comparison with effective expression of lipase genes in other organisms. Due to the differences of prokaryotic and eukaryotic expression systems, it is generally difficult for bacterial lipase to be expressed in active form in yeast. Nevertheless, in this study, we tried to express the bacterial a new lipase gene, P. fluorescens lipase gene in P. pastrois cells. The purified enzyme Pflipb was obtained. The catalytic activity of Pflip1b was much weaker than Pflip1a from E. coli, it shows that bacterial lipases are more suitable to express in prokaryotic expression system.
Among the members of I.1 lipase subfamily discussed above, Pflip1a showed its unique thermophilic property. The optimal hydrolyzing temperature for C8 was 70 °C, significantly higher than other homologous lipases. And high activity can be maintained continuously at high temperature of 70 °C. The lipase KB700A derived from Pseudomonas sp. displayed an optimal temperature of 35 °C32, and that of SIK W1 lipase came from P. fluorescens displayed a range of 45–55 °C28. This indicates that Pflip1a has great potential for various high temperature industrial applications.
It was reported that a majority of heavy metal ions deactivated lipases from Pseudomonas 31. But in this study, Pflip1a was enhanced by Ca2+, Mg2+, Ba2+, and other metal ions inhibited its activity. LipC12 from Yersinia enterocolitica subsp. palearctica Y11 had a preference for binding Ca2+, its maximum activity was achieved with addition of Ca2+, there may be a calcium ion hole for LipC12 dependent33. Similar to LipC12, the activity of Pflip1a was also heavily dependent on calcium ion. The activation by Ca2+ proved that the calcium-binding domain was important for the activity of Pflip1a. The fact that some divalent metal ions, including Ca2+, Mg2+ and Ba2+ could activate Pflip1a indicates that metal ions appear to be replaceable. Generally, non-ionic surfactants, such as Tween 20, Tween 40, Tween 60, Tween 80, and NP-40, and ionic surfactants, including SDS and CTAB, usually inhibited the activity of lipases34. But Pflip1a had good tolerance to these surfactants. Only SDS and CTAB inhibited its activity, other surfactants could promote. In particular, Tween 20 had a significant promoting effect on the improvement of enzyme activity. As known, lipases could be interfacial-activated due to the oil–water interface27. As a class of surfactants, Tween 20 may greatly promote the activation of lipase interface. The industrial applications of lipases could be broadened and enhanced greatly by using them in micro- or non-aqueous systems. Thus, high stability and activity of lipases in the presence of organic solvents were a prerequisite and desirable for biotransformation31. In view of this, the activities of Pflip1a in various organic solvents were also investigated. Pflip1a had an excellent tolerance to organic solvents, it was only inhibited in the presence of chloroform, and other organic solvents could stimulate its activity. These results show that Pflip1a has a potential for application in organic synthesis and transesterification.
The preparation of lipase-catalyzed biodiesel was generally recognized as an environmental-friendly technique. An efficient enzyme for biodiesel conversion is popularly welcomed. Its high productivity could effectively reduce the utilization cost of lipases to a great extent. Lipases originated from different biological sources usually have their own special characteristics. Some previous researches have proved that lipases from the same species of P. fluorescens exhibited different transesterification ability35,36,37. In our study, the biodiesel conversion rate of P. fluorescens Pflip1 reached 83.8% and that of Pflip1a is obviously higher than Pflip1b. The probable reason is attributable to the modifications of post translation in P. pastoris affecting the catalytic activity of the recombinant bacterial lipases. This to say, too much modification is not necessary for the proper folding of the lipase. Although most of the current used lipases are expressed in fungal systems, sometimes expression of bacterial lipase in prokaryotic system is more suitable.
Materials and Methods
Bacterial Strains, Plasmids and Chemicals
P. fluorescens Pf0–1 was preserved in our laboratory, E. coli TOP10 (Novagen, Darmstadt, Germany) and E. coli BL21(DE3) (Novagen, Darmstadt, Germany) were grown at 37 °C in LB medium or on agar plates. P. pastoris strain KM71 was grown in YPD broth (2% tryptone, 1% yeast extract, 2% dextrose) at 28 °C, or BMMY broth (1% yeast extract, 1.34% YNB, 4 × 10−5% biotin, 2% dextrose, 100 mM potassium buffer, 0.5% methanol) at 28 °C, or MD broth plates (2% dextrose, 4 × 10−5% biotin, 1.34% YNB and 2% agar) at 28 °C for screening yeast recombinants, or tributyrin agar plate (2% tryptone, 1% yeast extract, 1.34% YNB, 100 mM potassium buffer, 4 × 10−5% biotin, 1% glycerol at 16 °C, 1% tributyrin and 2% agar) at 28 °C. pMD19-T cloning vector (Takara, Otsu, Japan), pET-28a (Novagen, Darmstadt, Germany) and the plasmid pPIC9K (Invitrogen, Carlsbad, USA) were applied for constructing the expression plasmids. The antibiotics ampicillin (Ap; 100 μg/ml) and kanamycin (Km; 50 μg/ml) were used for screening in this study.
All strains were saved in 15% (v/v) glycerin solution and frozen at −20 °C and −80 °C. DNA sequencing has been completed by Invitrogen Biotechnology Company (Guangzhou, china). Taq polymerase, Restriction enzyme and T4 ligase were obtained from Takara (Otsu, Japan). Substrate p-nitrophenyl (p-NP) esters were purchased from Sigma-Aldrich (St.Louis, USA). And other chemicals of analytical grade were purchased from Sinopharm (Beijing, China). Primers used in the experiments are listed in Table 1.
DNA manipulations
Genomic DNA of Pf0–1 was extracted by Bacteria Genomic DNA Kit (CWBIO, Beijing, China) for genome extraction. It was obtained via Touchdown PCR with degenerate primers Pflip1F and Pflip1R.
Functional expression and purification of recombinant lipase Pflip1a in E. coli
After amplified with primers Pflip1aF and Pflip1aR, the PCR product was inserted into pET-28a by digested with Nde I and Hind III, such that the recombinant protein would have a carboxyl terminal of 6-his-tag. Then, it was transformed into the expression strain E. coli BL21(DE3) and grown overnight at 37 °C in a 5 ml LB medium. Later, the mixture was diluted with 1:100 into 1 L of LB broth containing ampicillin (100 μg/ml) and grown. After the optical density of the mixture at OD600 was between 0.4 and 0.5, the recombinant Pflip1a was induced with 0.1 mM IPTG at 16 °C for 20 h.
After centrifugation at 7,000 × g for 20 min at 4 °C and resuspension in lysis buffer (0.5 M NaCl, 20 mM Tris-HCl, pH 8.0), 6 L of culture was harvested and then disrupted by a One Shot Cell Disrupter (Constant Systems, Daventry, UK). Insoluble material was isolated by centrifugation at 12,000 × g for 20 min at 4 °C and supernatant was then passed through a 0.45 µm syringe-end filter before loading on His-Tag column. After that, the supernatants were transferred to a Ni-NTA affinity chromatography column (GE Healthcare, Pittsburgh, PA, USA). After the column balanced with lysis buffer, the recombinant protein was added into it and then eluted using washing buffer with an imidazole concentration gradient of 0, 30, 60, 100, 200, and 500 mM. Finally, 30 ml of the washing buffer containing the target recombinant enzymes was dialyzed in 20 mM Tris-HCl buffer (pH 8.0).
Then, SDS-PAGE was used to analyze the recombinant lipases. Unstained and pre-stained protein markers (Fermentas, SM0431 and SM0671 Waltham, USA) were used as references.
Functional expression and purification of recombinant lipase Pflip1b in P. pastoris
After amplified with primers Pflip1bF and Pflip1bR, the PCR product was cloned into pPIC9K by digested with EcoR I and Not I and ligated. It had a carboxyl terminal of 6-his-tag. Then, it was transformed into P. pastoris strain KM71 cells. Transformants grew on MD plates at 28 °C for 2–3 days. Subsequently, 25 colonies with large hydrolyzing halos were picked up and grown overnight in the YPD at 28 °C. These colonies was diluted with 1:100 into 1 L of BMMY broth and grown for 7 days.
After centrifuged at 7,000 × g for 20 min at 4 °C and passed through a 0.45 µm syringe in a concentration tube, 6 L of the supernatants was concentrated and collected. Then, the concentrated solution (concentrated for 20 times) was transferred to a Ni-NTA affinity chromatography column. The other purification procedure of the recombinant protein is similar to the above-mentioned Pflip1a. The analysis method is similar to the above stated.
Biochemical characterization of Pflip1a
The optimal substrate was examined using a standard assay except for different p-nitrophenyl esters as substrates, they were p-nitrophenyl (p-NP) acetate (C2), butyrate (C4), p-NP hexanoate (C6), p-NP caprylate (C8), p-NP decanoate (C10), laurate (C12), p-NP myristate (C14), and p-NP palmitate (C16), respectively. The optimum pH was analyzed through measuring the enzyme activity at 70 °C with C8 as substrate and in different buffers with pH ranging from 5.0 to 10.0 (citrate-phosphate buffer,pH 5.0–6.5; Tris-HCl buffer, pH 7.0–8.5; glycine-NaOH buffer, pH 9.0–10.0). The temperature effect was tested using a standard assay except for varied temperatures (40 °C-100 °C). In a standard assay, the total reaction system of 1 ml contains 940 μl of Tris–HCl buffer (50 mM, pH 8.0), 10 μl of p-NP ester (100 mM, soluble in acetonitrile), 40 μl of ethanol and 10 μl of the diluted enzyme solution. The blank contained the same components without addition of enzyme solution38. Unless otherwise described, lipase activity was measured in this standard assay at 70 °C for 10 min. All experiments were performed in triplicates. The mixtures were centrifuged, and the absorbance of the resulting supernatants was measured at 410 nm by use of UV-1600 spectrophotometer (MAPADA, Shanghai, China)39,40. One unit of lipase activity (U) was defined as the amount of enzyme that released 1 μmol p-NP per minute under the assay conditions.
The effects of metal ions (Na+, K+, Mn2+, Ca2+, Mg2+, Ba2+, Cu2+, Zn2+, Fe3+, and Al3+) and the chelating agent EDTA on enzyme activity were determined by adding 10 mM of each into the reaction system and then followed by a lipase activity assay. The influences of detergents (SDS, CTAB, Tween-20, NP-40, Triton X-100, Tween 20, Tween 40, and Tween 80) on enzyme activity were investigated by measuring the retaining activities through addition of 1% v/v of various detergents into the reaction system. After 1 h treatment, the residual activities were again measured in the standard assay conditions. For the organic solvent tolerance of the purified enzymes, aliquots of the recombinant enzymes were incubated in 30% of organic solvents such as chloroform, methanol, acetone, ethanol, tert-butanol, glycerol and n-hexane. After 2 h treatment, the residual activities were measured in the standard assay conditions as stated above.
Sequence analysis
The Pflip1 accession numbers of nucleotide sequence and amino acid sequence in the NCBI (http://www.ncbi.nlm.nih.gov/) were Pfl01_0571 and ABA72315.1, respectively.
The BlastP program of NCBI (http://blast.ncbi.nlm.nih.gov/) were applied to search for protein sequence similarity. ClustalW (version 1.83) and ESPript 2.2 (http://espript.ibcp.fr/ESPript/ESPript/) were employed to analyze multiple sequence alignments41. Phylogenetic tree analysis was accomplished with the neighbor-joining method of MEGA 5.042. A bootstrap analysis with 1000 replicates was applied to estimate the reliability of the tree. The ExPASy proteomics server (http://us.expasy.org/tools/protparam.html) was adopted for the analysis of protein physicochemical parameters (ProtParam tool).
Immobilization of the two recombinant lipases on magnetic nanoparticles and enzymatic properties assay
The purified recombinant lipases were separately immobilized on magnetic nanoparticles (self-made in our lab) in aqueous phase as follows27: 0.5 ml Pflip1a lipase solution (0.19 mg-protein/ml) and 0.5 ml Pflip1b lipase solution (0.17 mg-protein/ml) were respectively added in 4 ml 0.05 M Tris-HCl buffer (pH 8.0) containing 0.5 ml magnetic nano-particles activated by glutaraldehyde solution (i.e. MNP-G). The mixture was shaken at 30 °C for 2 h. Subsequently, the immobilized lipases were easily separated with a magnet and washed for several times with buffer. The enzymatic activities of the free lipase and the immobilized lipase were assayed by adopting the method obtained previously27. A brief description is29: 1 ml reaction system contained 940 µl of Tris-HCl buffer (50 mM, pH 8.0), 10 µl of p-NP caprylate (C8, 100 mM), 40 µl of ethanol, and a certain amount of the free or immobilized lipase. The mixture was pre-incubated at 70 °C for 5 min before the reaction, and then incubated at 70 °C for 10 min. After doing so, 2 ml of acetone was added to quench the reaction. Then, the absorbance of p-nitrophenol released was recorded at OD410. A unit is defined as the amount of enzyme liberating 1 µmol of fatty acid per minute. Protein concentrations were measured by Bradford assay43. The blank contained all the components except for the dilution of the enzyme solution. All experiments were performed for three times.
Transesterification of soybean oil into biodiesel and recyclability assay
The total reaction system containing 2.19 g soybean oil, 10 wt % immobilized enzyme, 5 wt % buffer (K2HPO4-KH2PO4, 0.1 mol/L, pH = 8.0) was conducted in a 50 mL shaking flask at 60 °C with a shaking speed of 200 rpm for 42 h27,44,45. The molar ratio of oil/ethanol was 1:4 and ethanol was added in three steps at the same interval of 6 h. Unless otherwise specified, all the dosage percentages were calculated on the basis of oil weight. The recyclability of the two immobilized lipases was further studied. By the end of the reaction, the immobilized lipases were separated from the mixtures with a magnet, washed with buffer, then under the same conditions reused for the new batch and calculated the biodiesel yield of each batch. The fatty acid alkyl esters (biodiesel) were measured by a GC-9790 gas chromatograph (Agilent HP-INNOWAX capillary column 30 m × 0.25 mm × 0.25 μm, J&W Scientific, Folsom, CA), and the operating conditions were subject to those described by Yan et al.46.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
References
Gupta, R., Gupta, N. & Rathi, P. Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol. 64, 763–781 (2004).
Houde, A., Kademi, A. & Leblanc, D. Lipases and Their Industrial Applications. Appl Biochem Biotechnol. 118, 155–170 (2004).
Casas-Godoy, L. et al. Lipases: An overview, in Lipases and phospholipases, methods and protocols. Humana Press. 861, 3–30 (2012).
Faber, K. Biotransformations in Organic Chemistry. Berlin: Springer-Verlag (2011).
Rapp, P. & Backhaus, S. Formation of extracellular lipases by filamentous fungi, yeasts, and bacteria. Enzyme Microb Technol. 14, 938–943 (1992).
Nagarajan, S. New Tools for Exploring “Old Friends—Microbial Lipases”. Appl Biochem Biotechnol. 168, 1163–1196 (2012).
Shao, H., Xu, L. & Yan, Y. J. Thermostable lipases from extremely radioresistant bacterium Deinococcus radiodurans: cloning, expression, and biochemical characterization. J Basic Microbiol. 54, 984–995 (2014).
Saxena, R. K., Sheoran, A., Giri, B. & Davidson, W. S. Purification strategies for microbial lipases. J Microbiol Methods. 52, 1–18 (2003).
Iso, M. et al. Production of biodiesel fuel from triglycerides and alcohol using immobilized lipase. Journal of Molecular Catalysis B Enzymatic, (2001).
Luo, Y., Zheng, Y., Jiang, Z., Ma, Y. & Wei, D. A novel psychrophilic lipase from Pseudomonas fluorescens with unique property in chiral resolution and biodiesel production via transesterification. Appl Microbiol Biotechnol. 73, 349–355 (2006).
Zhao, X. et al. An Organic Soluble Lipase for Water-Free Synthesis of Biodiesel. Appl Biochem Biotechnol. 143, 236–243 (2007).
Rosenau, F. & Jaeger, K. Bacterial lipases from Pseudomonas: Regulation of gene expression and mechanisms of secretion. Biochimie. 82, 1023–1032 (2000).
Arpigny, J. L. & Jaeger, K. E. Jaeger, Bacterial lipolytic enzymes: classification and properties. Biochem J. 343, 177–83 (1999).
Sharma, R., Chisti, Y. & Banerjee, U. C. Production, purification, characterization, and applications of lipases. Biotechnol Adv. 19, 627–62 (2001).
Gerritse, G., Hommes, R. W. & Quax, W. J. Development of a Lipase Fermentation Process That Uses a Recombinant Pseudomonas alcaligenes Strain. Appl Environ Microbiol. 64, 2644–2651 (1998).
Ahn, J. H., Pan, J. G. & Rhee, J. S. Homologous Expression of the Lipase and ABC Transporter Gene Cluster, tliDEFA, Enhances Lipase Secretion in Pseudomonas spp. Appl Environ Microbiol. 67, 6606–5511 (2001).
Shu, Z., Jiang, H., Lin, R. & Jiang, Y. Technical methods to improve yield, activity and stability in the development of microbial lipases. J Mol Catal B: Enzym. 62, 1–8 (2010).
Jiang, Z. et al. Cloning and Expression of a Novel Lipase Gene From Pseudomonas fluorescens B52. Mol Biotechnol. 31, 1073–6085 (2005).
Aloulou, A. et al. Constitutive expression of human pancreatic lipase-related protein 1 in Pichia pastoris. Protein Expr Purif. 47, 415–421 (2006).
Yu, X. W., Wang, L. L. & Xu, Y. Rhizopus chinensis lipase: Gene cloning, expression in Pichia pastoris and properties. J Mol Catal B: Enzym. 57, 304–311 (2009).
Pfeffer, J. et al. High yield expression of Lipase A from Candida antarctica in the methylotrophic yeast Pichia pastoris and its purification and characterisation. Appl Microbiol Biotechnol. 72, 931–938 (2006).
Fernández, L. et al. High-level expression and characterization of Galactomyces geotrichum (BT107) lipase I in Pichia pastoris. Protein Expr Purif. 49, 256–264 (2006).
Zhao, W., Wang, J., Deng, R. & Wang, X. Scale-up fermentation of recombinant Candida rugosa lipase expressed in Pichia pastoris using the GAP promoter. J Ind Microbiol Biotechnol. 35, 189–195 (2008).
Shen, Q. et al. The effect of gene copy number and co-expression of chaperone on production of albumin fusion proteins in Pichia pastoris. Appl Microbiol Biotechnol. 96, 763–772 (2012).
Yang, J., Zhang, B. & Yan, Y. Cloning and Expression of Pseudomonas fluorescens 26-2 Lipase Gene in Pichia pastoris and Characterizing for Transesterification. Appl Biochem Biotechnol. 159, 355–365 (2009).
Wang, C. H. et al. Cloning and Sequence Analysis of a Novel Cold-Adapted Lipase Gene from Strainlip35 (Pseudomonas sp.). Agric. Sci. China. 10, 1216–1221 (2008).
Liu, T. et al. A novel oriented immobilized lipase on magnetic nanoparticles in reverse micelles system and its application in the enrichment of polyunsaturated fatty acids. Bioresour Technol. 132, 99–102 (2013).
Lee, Y. P., Chung, G. H. & Rhee, J. S. Purification and characterization of Pseudomonas fluorescens SIK W1 lipase expressed in Escherichia coli. Biochim Biophys Acta. 1169, 156–164 (1993).
Yang, W. et al. A novel eurythermic and thermostale lipase LipM from Pseudomonas moraviensis M9 and its application in the partial hydrolysis of algal oil. BMC Biotechnol. 10, https://doi.org/10.1186/s12896-015-0212-2 (2015).
Yang, W., Xu, L., Zhang, H. & Yan, Y. Characterizing LipR from Pseudomonas sp. R0-14 and Applying in Enrichment of Polyunsaturated Fatty Acids from Algal Oil. J Microbiol Biotechnol. 25, 1880–1893 (2015).
Peng, R., Lin, J. & Wei, D. Purification and Characterization of an Organic Solvent-Tolerant Lipase from Pseudomonas aeruginosa CS-2. Appl Biochem Biotechnol. 162, 733–743 (2010).
Rashid, N. et al. Low-Temperature Lipase from Psychrotrophic Pseudomonas sp. Strain KB700A. Appl Environ Microbiol. 67, 4064–4069 (2001).
Meier, R. et al. A Calcium-gated Lid and a Large beta-Roll Sandwich Are Revealed by the Crystal Structure of Extracellular Lipase from Serratia marcescens. J Biol Chem. 282, 31477–31483 (2007).
Shao, H., Xu, L. & Yan, Y. Thermostable lipases from extremely radioresistant bacterium Deinococcus radiodurans: Cloning, expression, and biochemical characterization. J Basic Microbiol. 54, 984–995 (2013).
Panizza, P. et al. Acidic lipase Lip I.3 from a Pseudomonas fluorescens-like strain displays unusual properties and shows activity on secondary alcohols. J Appl Microbiol. 114, 722–32 (2013).
Lima, L. N. et al. Immobilization of Pseudomonas fluorescens lipase on hydrophobic supports and application in biodiesel synthesis by transesterification of vegetable oils in solvent-free systems. J Ind Microbiol Biotechnol. 42, 523–535 (2015).
Xun, E. N. et al. Immobilization of Pseudomonas fluorescens Lipase onto Magnetic Nanoparticles for Resolution of 2-Octanol. Appl Biochem Biotechnol. 168, 697–707 (2012).
Zheng, X. et al. A novel cold-adapted lipase from Acinetobacter sp. XMZ-26: gene cloning and characterisation. Appl Microbiol Biotechnol. 90, 971–980 (2011).
Luo, Y. et al. A novel psychrophilic lipase from Pseudomonas fluorescens with unique property in chiral resolution and biodiesel production via transesterification. Appl Microbiol Biotechnol. 73, 349–355 (2006).
Kumar, S. et al. Production, purification, and characterization of lipase from thermophilic and alkaliphilic Bacillus coagulans BTS-3. Protein Expression Purif. 41, 38–44 (2005).
Gouet, P., Courcelle, E. & Stuart, D. I. ESPRIPT: analysis of multiple sequence alignments in PostScript. Bioinformatics. 15, 305–8 (1999).
Tamura, K. et al. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Molecular Biology and Evolution. 24, 1596–1599 (2007).
Kruger, N. J. The Bradford Method For Protein Quantitation. The Protein Protocols Handbook-Part of the series Springer Protocols Handbooks, Wiley-VCH Verlag, Weinheim. pp. 17-24 (2009).
Kaieda, M. et al. Biodiesel fuel production from plant oil catalyzed by Rhizopus oryzae lipase in a water-containing system without an oganic solvent. J. Biosci. Bioeng. 88, 627–631 (1999).
Jin, X., Li, J. & Huang, P. et al. Immobilized protease on the magnetic nanoparticles used for the hydrolysis of rapeseed meals. J. Magn. Magn. Mater. 322, 2031–2037 (2010).
Yan, J. et al. Enzymatic enrichment of polyunsaturated fatty acids using novel lipase preparations modified by combination of immobilization and fish oil treatment. Bioresour Technol. 102, 7154–7158 (2011).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31170078), the National High Technology Research and Development Program of China (Nos. 2011AA02A204 and 2013AA065805), the Natural Science Foundation of Hubei Province (No. 2015CFA085), and the Fundamental Research Funds for HUST (Nos. 2014NY007 and 2014QN119). The authors thank Ms. Chen Hong, from the Centre of Analysis and Test, Huazhong University of Science and Technology for biodiesel analysis.
Author information
Authors and Affiliations
Contributions
Wu Liu designed the experimental scheme and did the most of the preparation and characterizations. Menggang Li contributed to the measurement of G.C.-M.S. and H.P.L.C. analysis. Wu Liu and Yunjun Yan wrote and revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, W., Li, M. & Yan, Y. Heterologous expression and characterization of a new lipase from Pseudomonas fluorescens Pf0–1 and used for biodiesel production. Sci Rep 7, 15711 (2017). https://doi.org/10.1038/s41598-017-16036-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16036-7
This article is cited by
-
A Review on Lipases: Sources, Assays, Immobilization Techniques on Nanomaterials and Applications
BioNanoScience (2024)
-
Recent insight into the advances and prospects of microbial lipases and their potential applications in industry
International Microbiology (2024)
-
Haloalkaline Lipase from Bacillus flexus PU2 Efficiently Inhibits Biofilm Formation of Aquatic Pathogen Vibrio parahaemolyticus
Probiotics and Antimicrobial Proteins (2022)
-
Artificial consortia of Bacillus amyloliquefaciens HM618 and Bacillus subtilis for utilizing food waste to synthetize iturin A
Environmental Science and Pollution Research (2022)
-
Optimization of lipase production using fungal isolates from oily residues
BMC Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
