
Abstract
We report results of DNA analysis with next generation sequencing (NGS) of 21 consecutive Italian patients from 17 unrelated families with clinical diagnosis of Usher syndrome (4 USH1 and 17 USH2) searching for mutations in 11 genes: MYO7A, CDH23, PCDH15, USH1C, USH1G, USH2A, ADGVR1, DFNB31, CLRN1, PDZD7, HARS. Likely causative mutations were found in all patients: 25 pathogenic variants, 18 previously reported and 7 novel, were identified in three genes (USH2A, MYO7A, ADGRV1). All USH1 presented biallelic MYO7A mutations, one USH2 exhibited ADGRV1 mutations, whereas 16 USH2 displayed USH2A mutations. USH1 patients experienced hearing problems very early in life, followed by visual impairment at 1, 4 and 6 years. Visual symptoms were noticed at age 20 in a patient with homozygous novel MYO7A missense mutation c.849G > A. USH2 patients’ auditory symptoms, instead, arose between 11 months and 14 years, while visual impairment occurred later on. A homozygous c.5933_5940del;5950_5960dup in USH2A was detected in one patient with early deafness. One patient with homozygous deletion from exon 23 to 32 in USH2A suffered early visual symptoms. Therefore, the type of mutation in USH2A and MYO7A genes seems to affect the age at which both auditory and visual impairment occur in patients with USH.
Similar content being viewed by others


Introduction
Usher Syndrome (USH) is a syndromic inherited retinal dystrophy (IRD). It is a clinically and genetically heterogeneous autosomal recessive disorder, characterized by retinitis pigmentosa (RP)1,2,3 and bilateral sensorineural deafness, with or without vestibular dysfunction.
USH has a prevalence of 3.2 to 6.2 cases per 100000, thus representing the leading genetic cause of combined hearing and vision loss4,5. Three different clinical subtypes have been so far identified based on hearing, vestibular and visual symptoms6. Type 1 (USH1) is the most severe form of USH, characterized by severe congenital deafness, vestibular dysfunction and prepubertal RP onset7. Deafness is less severe in USH type 2 (USH2), for which the vestibular function remains normal, while RP symptoms generally occur during puberty7. Finally, USH type 3 (USH3) is characterized by post lingual hearing loss, while the age of RP occurrence is offset between the second to fourth decade and can be subject to variable vestibular dysfunctions6. Sixteen loci and thirteen disease-causing genes associated with USH have been so far identified (https://sph.uth.edu/retnet/), thus demonstrating the wide genetic heterogeneity of this syndrome. Such heterogeneity makes molecular diagnosis with Sanger sequencing of single genes rather challenging, since this particular diagnostic strategy, although accurate, requires a lot of time and resources6. Microarray technology for the simultaneous detection of known mutations in Usher related genes, instead, has proven to perform poorly, proving diagnostic efficiency as low as 12%, when applied on the Italian population8. Nevertheless, thanks to the recent availability of next generation sequencing (NGS) in routine genetic testing, it is now possible to simultaneously screen an increasing number of selected genes (targeted NGS), as well as the whole exome or the entire genome. So far, NGS has proven to be a rapid and cost-effective diagnostic method, which is also a useful tool for the identification of novel disease-causing genes9. Targeted NGS is being currently applied on patients affected by hereditary diseases with high gene heterogeneity, such as Usher syndrome and, both syndromic and non-syndromic, inherited retinal dystrophies10,11,12,13.
Deeper understanding of the genetic basis allows further analysis of possible genotype-phenotype correlations. Besides being useful for genetic counselling, such knowledge base is fundamental to an accurate clinical diagnosis and prognosis of individual patients and thus represents an invaluable tool for the development of personalized treatments.
The report describes the phenotypes associated with the mutations detected during genetic testing with targeted NGS. The tests were carried out on a group of 21 consecutive Italian patients from 17 unrelated families affected by Usher syndrome who were seeking genetic counselling.
Methods
Patients
This is a retrospective study. The study comprises 21 consecutive Italian patients (15 males and 6 females) from 17 unrelated families with a clinical diagnosis of Usher syndrome who were seeking genetic counselling at the Mauriziano Hospital in Torino, Italy. The pedigrees of the 17 families are reported in Fig. 1.

Pedigrees of the families included in the study.
The data collection complies with the Italian law. The study was conducted in accordance with the provisions stated in the Declaration of Helsinki (59th World Medical Association General Assembly; Seoul, Korea; October 2008).
All the patients and their relatives were duly informed about the advantages and limitations of the test and were required to sign informed consent. Detailed medical, personal and family history was obtained from the patients and their relatives, specifically recording the age of onset of deafness, visual deficiency and equilibrium impairment.
In most cases, the available ophthalmological clinical data were composed by slit-lamp anterior segment and fundus examinations, best corrected visual acuity, Goldmann applanation tonometry, electroretinogram (ERG), visual evoked potentials (VEP), visual field test, optic coherence tomography (OCT). A few patients had also sustained colour vision tests, fundus autofluorescence and fluorescein angiography.
DNA analysis
Within the framework of the Italian rare diseases registry, the diagnostic genetic unit of AOU Careggi in Firenze offers National Health System patients free genetic testing for IRD. Genetic testing is mainly requested to confirm clinical diagnosis, for genetic counselling for patients and their families and to allow participation in gene therapy trials.
Genomic DNA was isolated from peripheral leukocytes, using the QiaSymphony DNA Blood Midi kit on the QIAsymphony SP workstation (Qiagen), according to the manufacturer’s protocol.
A custom Haloplex panel was designed using Agilent’s online SureDesign tool (https://earray.chem.agilent.com/suredesign/index.htm), Targeted NGS of coding regions and exon-intron junctions of a panel of 11 genes were performed: MYO7A (MIM 276903), CDH23 (MIM 605516), PCDH15 (MIM 605514), USH1C (MIM 605242), USH1G (MIM 607696), USH2A (MIM 608400), ADGVR1 (MIM 602851), DFNB31 (MIM 607928), CLRN1 (MIM 606397), PDZD7 (MIM 612971), HARS (MIM 142810). Genes ABDH12 (MIM 613599,612674), CEP250 (MIM 609689) and CIB2 (MIM 614869) were not included in the panel because mutations in these genes have only been recorded in a few non-Caucasian families. PDZD7 gene was included because it has been suggested to be a modifier gene in subjects with USH2A mutations and potentially involved with ADGRV1 in digenic USH214.
The target regions were captured using the Agilent HaloPlex Target Enrichment System Kits for Illumina Sequencing following Agilent protocols. The captured target libraries were amplified by PCR, quality controlled and quantified using the BioAnalyzer 2100 (Agilent Technologies, Inc. Santa Clare, CA). Equimolar amounts of differentially indexed samples were pooled before pair-ended sequencing at 300 cycles on the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA). In addition, the deep intronic variant (c.7595-2144A > G) in intron 40 of USH2A gene was searched15.
The criteria used to distinguish new mutations from polymorphisms is ExAC frequency. We filtered variants with a MAF < 0.05. All the new mutations reported in this study are not validated at RNA and protein levels.
The new mutations reported were investigated in 48 healthy subjects (20 females, 28 males) with Sanger sequencing.
For the new point mutations leading to aminoacid substitution, pathogenicity predictions from Bioinformatic tools SIFT, PhyloP, AGVGD, MutationTaster and Polyphen2 were compared.
For intronic mutations, although our laboratory cannot validate that the mutations observed indeed affect the splicing process, the bioinformatics tools available predicted all of them to be pathogenic; furthermore the variants were at the exon-intron junction and such variants both at RNA level and in classical genetics are reported to affect the splicing process.
The presence of all pathogenic and likely pathogenic variants detected was confirmed with Sanger sequencing, processed with the automated Core System (Beckman Coulter, Fullerton, CA). After purification, amplicons were sequenced on the 3730 DNA Analyzer (ABI, Foster City, CA). The sequences were assembled and analyzed using SeqScape software (ABI). Variants of unknown pathogenicity were interpreted with Alamut 2.6 (Interactive Biosoftware, Rouen, France), a decision-support software application for medical molecular genetics. The software relies on web-based prediction software, such as Align-GVGD, SIFT, PolyPhen, Mutation Taster (hosted by Interactive Biosoftware). Note that Alamut 2.6 scoring systems provide a predictive evaluation only for missense variants. In selected patients multiplex ligation-dependent probe amplification (MLPA) was also performed. The MLPA reaction (P361-A1/P362-A2 SALSA MLPA kit; MRC Holland, Amsterdam, The Netherlands) was performed according to the manufacturer’s recommendations. One microliter of each reaction product was separated on a POP-7 polymer with capillary electrophoresis using the 3730 DNA Analyzer (ABI). Freely available software provided by MRC Holland was used to analyze the MLPA data (Coffalyser; MRC Holland).
When relatives were available (14 families), segregation analysis was performed.
Results
DNA analysis
DNA analysis results were in accordance with the diagnosis for all patients clinically diagnosed with USH. The average of sequencing depth was about >99.9%. We obtained about 70 variants for each sample. We filtered for function and frequency according to ACMG guidelines16. We obtained about 1–3 variants for sample that were validated by Sanger sequencing.
Likely causative mutations were identified in three Usher related genes: USH2A, MYO7A, ADGRV1. For the causative mutations found in this study, reported and novel, there aren’t neither in vivo functional experiments showing that the mutations cause the USH phenotype, nor in vitro experiments showing that the mutations will cause genetic dysfunction.
The results are reported in Table 1. Further heterozygous mutations in Usher related genes were observed in nine patients (Table 2). In Table 3 the frequency in our normal population of the new mutation and the pathogenecity prediction from Bioinformatic tools SIFT, PhyloP, AGVGD, MutationTaster and Poluphen2 for the new point mutations leading to aminoacid substitution, are reported. In Fig. 1 the pedigree of the 17 families and the genotypes of the patients and of their relatives are reported.
Phenotype
Details of the phenotypes are reported in Table 4.
Discussion
This report retrospectively analyses the clinical and genetic data of 21 consecutive Italian patients from 17 unrelated families affected by Usher syndrome undergoing genetic analysis by targeted NGS of 11 genes (MYO7A, CDH23, PCDH15, USH1C, USH1G, USH2A, ADGVR1, DFNB31, CLRN1, PDZD7, HARS). USH2 syndrome was the most frequent clinical diagnosis, accounting for 81% of the patients, while USH1 was diagnosed in 19%. None of the patients were affected by USH3.
24 likely pathogenic variants - 17 previously reported and 7 novel - were identified in three genes (USH2A, MYO7A, ADGRV1). Five mutations were detected in MYO7A, two of which were novel; 18 mutations were identified in USH2A (4 of which novel) and one novel homozygous mutation was identified in ADGRV1. Additional variants with uncertain pathogenic significance in USH2A, MYO7A, ADGRV1, CDH23 genes were further identified in 9 patients. In these patients, the additional variants were not considered to be the main causative mutations because two other causative variants were present (reported or novel but with a strong impact at the protein level). We cannot exclude a modifier role for these uncertain variants.
Genetic testing result was in accordance with previous clinical diagnosis or clinical suspicion for all patients. All subjects with USH1 or suspected USH1 displayed biallelic MYO7A mutations and all subjects with USH2 presented at least two USH2A or two ADGRV1 mutations. Such results are in line with previous studies that report MYO7A and USH2A gene mutations being among the most frequent causes of USH1 and USH217. The frameshift mutation c.2299delG (p.Glu767Serfs*21) in the USH2A gene was the most frequent in our cohort, since it was detected in 6 patients from four unrelated families. This mutation is quite frequent (0.16 to 0.44) in several cohorts of patients18,19,20,21. According to current literature, this is the most common mutation in European patients, accounting for approximately 30% of all European cases of USH2A 22. The high frequency of such mutation has been reported in several different populations and was proven to be the result of an ancestral mutation that has then spread throughout Europe and other continents due to migratory movements23.
Segregation analysis revealed three patients (TO15, TO18 and TO19) with a USH2A allele carrying more than one mutation. This finding underlines the importance of performing segregation analysis on patients suffering from recessive disorders to identify the exact genotype. Although such thoroughness adds up to the costs, it is essential for genetic counselling and reliable family risk evaluation.
All subjects with USH1 and biallelic MYO7A mutations were diagnosed with deafness and vestibular function impairment within their first 18 months of life. This lead to an initial clinical diagnosis of myopathy with neurodevelopmental delay in one patient (TO17), while the brother (TO16) was diagnosed with a pervasive developmental disorder. For all patients, deafness was initially attributed to a possible prenatal or postnatal infection. Three patients were tested in early childhood for GJB2 gene mutations, since this gene is deemed to be the leading cause for hereditary deafness within the European population24. At the time of diagnosis, the absence of pathogenic variants in GJB2 gene had, in fact, misled the physician in reinforcing the hypothesis of an infective aetiology of deafness. The parents of two of these patients, having been reassured about a low risk of recurrence, had a second affected child. This further stresses the fundamental importance of providing an exact diagnosis to children affected by deafness. Such diagnosis can be accomplished by offering targeted NGS for syndromic and non-syndromic deafness-related genes whenever prenatal or postnatal infective aetiology is not documented.
Visual symptoms have also proven to occur at an early age, ranging from 1 to 6 years, hence well before the age of 10, as generally observed in USH1 subjects25. Only patient TO11 had a late onset of visual symptoms, which went unnoticed until he was 19. This patient has a novel homozygous missense mutation in MYO7A gene that might have a milder effect on retinal function.
A recent study on the Italian population confirms that hearing and visual impairment generally occur at an earlier age in patients carrying MYO7A mutations compared to those carrying USH2A mutations. The mean age for hearing loss and visual symptoms is generally between 5 ± 1 months and 16 ± 3 years respectively26.
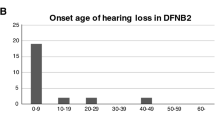
Patients with USH2 carrying USH2A or ADGRV1 mutations were diagnosed deaf and displayed visual symptoms at an older age compared to subjects with MYO7A mutations. Deafness usually occurred between 11 months and 14 years (mean age 5 years) within the reported range 8 months – 31 years27, whereas visual impairment generally onsets later, among 10 to 38 year old, (mean 15 ± 8.4 years) again within the reported age range 8–76 (average age 35.5)27. Two homozygous c.5933_5940del and 5950_5960dup mutations in USH2A were detected in patient TO19, whose deafness onset was recorded when he was 11 months old and whose visual symptoms were noticed only later when he was 17 years old.
The age at which the patients with USH2 in our cohort lamented symptoms was generally earlier than what a recent study on Italian patients with USH17 reports. The study comprises 36 patients (three USH1 and 33 USH2) and reports the average age of visual symptoms onset to be 17.5 ± 8.8 years. OCT revealed macular oedema in 29% of the patients in our study, a comparable percentage of cystoid macular lesions (from 28 to 49% in different studies) were similarly reported in studies on patients with RP28,29,30.
In conclusion, patients with USH exhibited clinical severity, which appears to be related to the mutated gene and to the specific type of mutation. Homozygosity for deletion from exon 23 to 32 and homozygosity for c.5933_5940del and c.5950_5960dup in USH2A were associated with a severe phenotype. It is known that mutations in USH2A can lead either to USH2 or to non-syndromic RP. Mutations carried by USH2 patients, tend to be more severe than those found in non-syndromic RP patients31,32. Therefore, there is evidence that, even within the USH2 phenotype, there ought be a severity gradient depending on the specific mutation33,34. We also observed a MYO7A biallelic mutation that was generating a phenotype with vestibular dysfunction, though it entailing milder hearing and visual symptoms. The role of additional heterozygous mutations in other related Usher genes remains to be further investigated.
These results, thus, provide useful data not only for tailored genetic counselling but they also provide additional clues for early clinical diagnosis of patients with Usher syndrome.
Nevertheless, this study present some limits. The heterogeneity of the clinical information available, in fact, did not allow for statistical analysis.
References
Keats, B. J. & Corey, D. P. The Usher syndromes. Am J Med Genet. 89, 158–166 (1999).
Saihan, Z., Webster, A. R., Luxon, L. & Bitner-Glindzicz, M. Update on Usher syndrome. Curr Opin Neurol. 22, 19–27 (2009).
Bonnet, C. & El-Amraoui, A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 25, 42–49 (2012).
Kimberling, W. J. et al. Frequency of Usher syndrome in two paediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet. Med. 12, 512–516 (2010).
Lenarduzzi, S. et al. Usher syndrome: an effective sequencing approach to establish a genetic and clinical diagnosis. Hear Res. 320, 18–23 (2015).
Mathur, P. & Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 1852, 406–420 (2015).
Lentz, J., Keats, B. J. Usher syndrome type II, in GeneReviews® (ed Pagon, R. A) (Seattle,1993–2017).
Vozzi, D. et al. Molecular epidemiology of Usher syndrome in Italy. Mol Vis. 17, 1662–1668 (2011).
Bras, J. et al. Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci. 13, 453–464 (2012).
Huang, X. F. et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One. 8, e63832 (2013).
Wang, G., Liu, Y., Zhu, D., Klau, G. W. & Feng, W. Bioinformatics Methods and Biological Interpretation for Next-Generation Sequencing Data. Biomed Res Int. 2015, 690873 (2015).
van El, C. G. et al. Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. Eur J Hum Genet. 21, 580–584 (2013).
Bonnet, C. et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur J Hum Genet. 24, 1730–1738 (2016).
Ebermann, I. et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest. 120, 1812–1823 (2010).
Vaché, C. et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum Mutat. 33, 104–108 (2012).
Richards, S. et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 5, 405–424 (2015).
Sodi, A. et al. MYO7A and USH2A gene sequence variants in Italian patients with Usher syndrome. Mol Vis. 20, 1717–1731 (2014).
Beneyto, M. M. et al. Prevalence of 2314delG mutation in Spanish patients with Usher syndrome type II (USH2). Ophthalmic Genet. 21, 123–128 (2000).
Dreyer, B. et al. Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet 8, 500–506 (2000).
Liu, X. Z. et al. A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet. 64, 1221–1225 (1999).
Weston, M. D. et al. Genomic structure and identification of novel mutations in Usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 66, 1199–1210 (2000).
Aller, E. et al. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur J Hum Genet. 12, 407–410 (2004).
Dreyer, B. et al. A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am J Hum Genet. 69, 228–234 (2001).
Smith, R.J.H., Jones, M.K.N. Nonsyndromic Hearing Loss and Deafness, DFNB1. In GeneReviews® (Pagon, R.A. et al.) (Seattle,1993–2017).
Gorlin, R. J., Tilsner, T. J., Feinstein, S. & Duvall, A. J. Usher’s syndrome type III. Arch Otolaryng. 105, 353–354 (1979).
Testa, F. et al. Clinical presentation and disease course of usher syndrome because of mutations in MYO7A or USH2A. Retina. (2016). [Epub ahead of print].
Abadie, C. et al. Audiological findings in 100 USH2 patients. Clin Genet. 82, 433–438 (2012).
Ozdemir, H., Karacorlu, M. & Karacorlu, S. Intravitreal triamcinolone acetonide for treatment of cystoid macular oedema in patients with retinitis pigmentosa. Acta Ophthalmol Scand. 83, 248–51 (2005).
Hajali, M., Fishman, G. A. & Anderson, R. J. The prevalence of cystoids macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br J Ophthalmol. 92, 1065–1068 (2008).
Lai, Y. H., Capasso, J. E., Kaiser, R. & Levin, A. V. Intraretinal cystoid spaces in a patient with retinitis pigmentosa due to mutation in the MAK gene. Ophthalmic Genet. 37, 424–426 (2016).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 133, 331–345 (2014).
Jiang, L. et al. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet J Rare Dis. 10, 110 (2015).
Yang, J. et al. Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 6, e1000955 (2010).
Pan, L. & Zhang, M. Structures of usher syndrome 1 proteins and their complexes. Physiology (Bethesda). 27, 25–42 (2012).
Author information
Authors and Affiliations
Contributions
C.M.E. and C.M. conceived the study, A.M., I.P. and F.T. conducted the genetic analysis, M.V., R.S., M.P.M. and C.A. analyzed the results. C.M.E., L.D., C.M. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eandi, C.M., Dallorto, L., Spinetta, R. et al. Targeted next generation sequencing in Italian patients with Usher syndrome: phenotype-genotype correlations. Sci Rep 7, 15681 (2017). https://doi.org/10.1038/s41598-017-16014-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16014-z
This article is cited by
-
Spectrum of variants associated with inherited retinal dystrophies in Northeast Mexico
BMC Ophthalmology (2024)
-
Diagnostic yield of panel-based genetic testing in syndromic inherited retinal disease
European Journal of Human Genetics (2020)
-
Novel Usher syndrome pathogenic variants identified in cases with hearing and vision loss
BMC Medical Genetics (2019)
-
High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
