
Abstract
One of the major challenges of the current Zika virus (ZIKV) epidemic is to prevent congenital foetal abnormalities, including microcephaly, following ZIKV infection of pregnant women. Given the urgent need for ZIKV prophylaxis and treatment, repurposing of approved drugs appears to be a viable and immediate solution. We demonstrate that the common anti-malaria drug chloroquine (CQ) extends the lifespan of ZIKV-infected interferon signalling-deficient AG129 mice. However, the severity of ZIKV infection in these mice precludes the study of foetal (vertical) viral transmission. Here, we show that interferon signalling-competent SJL mice support chronic ZIKV infection. Infected dams and sires are both able to transmit ZIKV to the offspring, making this an ideal model for in vivo validation of compounds shown to suppress ZIKV in cell culture. Administration of CQ to ZIKV-infected pregnant SJL mice during mid-late gestation significantly attenuated vertical transmission, reducing the ZIKV load in the foetal brain more than 20-fold. Given the limited side effects of CQ, its lack of contraindications in pregnant women, and its worldwide availability and low cost, we suggest that CQ could be considered for the treatment and prophylaxis of ZIKV.
Similar content being viewed by others
Introduction
The recent re-emergence of Zika virus (ZIKV) represents a public health emergency1. ZIKV is a member of the Flaviviridae family of viruses, which are transmitted to humans by mosquitoes and ticks and are responsible for millions of infections annually. ZIKV has historically been restricted to tropical/sub-tropical regions but it has now reached the Southern USA2. Recent studies have demonstrated that ZIKV infects and predominantly damage human neural precursors; however, the cellular and molecular mechanisms of ZIKV pathogenesis remain poorly understood3,4,5,6.
ZIKV infection causes moderately severe disease in about 20% of infected adults and is normally accompanied by mild symptoms such as headache, low fever, malaise, skin rashes, conjunctivitis, and muscle and joint pain7. More alarming is the evidence linking ZIKV infection in pregnant women to severe microcephaly in the foetus. The number of birth defects in children born to ZIKV-infected mothers is about 20-fold higher than normal8. In addition, ZIKV infection of adults can cause neuronal pathology such as Guillain-Barré syndrome, a rapid-onset acute polyneuropathy caused by an autoimmune response to the peripheral nervous system9,10.
In the absence of established anti-ZIKV treatments, repurposing of available drugs approved for use in pregnant women is likely to be the most effective way to reduce ZIKV infection in adults and to limit birth defects in newborns. Chloroquine (CQ) phosphate/CQ hydrochloride (Aralen) has a long and successful history as an oral anti-malarial chemotherapy, and there are no substantiated reports that CQ used for malarial prophylaxis11 causes foetal harm. CQ is also prescribed for the treatment of rheumatoid arthritis and other autoimmune disorders. Recently, CQ was suggested to inhibit ZIKV infection in several cell models, including human neural precursor cells (NPCs) derived from induced pluripotent stem cells (iPSCs)12. CQ was also shown to inhibit autophagy and ZIKV propagation in interferon signalling-deficient pregnant mice13. Here, we demonstrate that administration of CQ significantly extended the lifespan of interferon (IFN) type I and II receptor-deficient AG129 mice infected with Brazilian strain ZIKV (ZIKVBR, Brazil-ZKV2015), a common preclinical model for ZIKV research. However, the severity of disease precludes the use of AG129 mice for the investigation of vertical ZIKV transmission. To develop a suitable model for this purpose, we used SJL mice, which have a normal IFN signalling response and have previously been used for the study of ZIKV pathogenesis4. We found not only that SJL mice support chronic ZIKVBR infection but also that the virus can be transmitted vertically, making this a more relevant model of ZIKV infection in humans14. Notably, administration of CQ to pregnant SJL mice during mid-late gestation markedly reduced ZIKVBR infection in the foetal brain. Collectively, our data suggest that CQ could be effectively and readily employed for the treatment and prophylaxis of ZIKV infection in humans.
Results
CQ protects human neural progenitors from ZIKV infection
Human foetal NPCs are the major target of ZIKV in the developing brain5,15. To examine the effect of CQ in vitro, we infected monolayer cultures of primary human foetal NPCs with ZIKVBR (Brazilian strain ZKV2015) and cultured them in the absence or presence of up to 40 μM CQ. Consistent with work by others12, we found that CQ efficiently (90% inhibition at 6 µM) reduced ZIKVBR infection of primary human foetal NPCs. To mimic ZIKVBR infection in the context of the 3-dimensional architecture of the developing human brain, we examined neurospheres derived from human iPSCs (Fig. 1a). We found that CQ treatment reduced both the percentage of ZIKVBR-positive cells (Fig. 1b) and the level of apoptosis in the neurospheres with an IC50 of ~10 μM (Fig. 1c and d).

CQ inhibits ZIKV infection and apoptosis in human neurospheres. (a–d) Human iPSC-derived neurospheres were infected with ZIKVBR (multiplicity of infection = 1) and immediately treated with CQ at 5 µM, 20 µM, or 40 µM. MOCK = uninfected neurospheres. DMSO = DMSO treated neurospheres. Quantification of (a) neurosphere size at 96 h post-infection, (b) ZIKV-positive cells, (c) cleaved caspase 3 (CC3)-positive cells, and (d) CC3 and ZIKV double-positive cells. Data are the mean ± SEM of triplicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with “ZIKV” one-way ANOVA with Dunnett’s multiple comparisons test.
CQ attenuates acute ZIKV-induced mortality in AG129 mice
To corroborate the in vitro findings, we first examined AG129 mice, which lack receptors for type I (α/β) and type II (γ) IFNs and have previously been used to model ZIKV infection16,17. To test the prophylactic effects of CQ, mice were administered 50 mg/kg/day CQ in drinking water for 2 days and then infected with ZIKVBR (2 × 103 PFU retro-orbitally). CQ treatment was continued at the same dose for 5 days and then at 5 mg/kg/day until the end of the experiment. Control mice received drinking water alone.
We observed that CQ extended the average lifespan of ZIKV-infected AG129 mice to 15 days (p < 0.01, log-rank Mantel–Cox test; Fig. 2a) and significantly attenuated ZIKV-induced weight loss (p < 0.01, unpaired t-test with Welch’s correction; Fig. 2b). Overall animal health was assessed using a modified 6-point scoring system15, which showed that CQ-treated mice remained in good health and survived for longer than the vehicle-treated mice (80% vs 0% of animals alive on day 13, respectively) (Fig. 2c and d). These results indicate that CQ attenuated disease severity in ZIKV-infected AG129 mice, which is considered the most severe model of ZIKV infection16.

CQ attenuates ZIKV infection in AG129 mice. (a) Survival curves for control and CQ- treated mice (n = 6 and 5, respectively). Note that 60% of the CQ-treated mice were euthanized on day 15. p = 0.0075 by log-rank Mantel–Cox test. (b) Weight loss following ZIKV infection in control and CQ-treated mice. Data are the mean ± SEM. *p < 0.05, **p < 0.01 compared with controls by unpaired t-test with Welch’s correction. (c,d) Visual appearance health scores in ZIKV-infected control (c) and CQ-treated (d) mice. Note that by day 13, all control mice had died but 60% of CQ-treated mice were alive.
SJL mice support chronic infection with ZIKV
Mice deficient in IFN response genes, such as single knockout (Ifnar1) A129, double knockout (Ifnar1, Ifnar2) AG129, and triple knockout (Irf3, Irf5, Irf7) TKO mice16, succumb to ZIKV within a few days of infection making it difficult to investigate vertical transmission of ZIKV in such an aggressive disease model. Therefore, we explored the SJL mouse model, which we have previously used to study foetal transmission with high doses of ZIKV4.
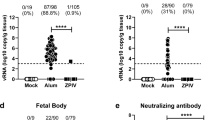
SJL males and females at 3 months of age were infected with ZIKVBR (1 × 108 PFU retro-orbitally), and circulating ZIKV RNA levels were analysed by qRT-PCR over the following 50 days. Our qRT-PCR assay is only 10-fold less sensitive than a laborious and time consuming plaque forming unit assay. Using qRT-PCR we could detect the levels of ZIKV as low as 10 plaque forming units per sample. In testing our samples, we did not record any ZIKV in the samples obtained from uninfected control mice. We found that the viral titres fluctuated over time in both males and females, ranging from 5 × 103 to 4 × 105 genome copies/µg total RNA. However, the mean titres were maintained between 104 and 105 genome copies/µg total RNA (Fig. 3a and b). Previous work has shown that ZIKV inoculation of wild-type C57BL/6 mice treated with a single dose of IFNAR1-blocking monoclonal antibody leads to infection of and damage to the testes18. We therefore investigated ZIKV titres in the testes of chronically infected SJL mice (3 months post-infection) and found readily detectable levels (103–104 ZIKV genome copies/1 µg testis RNA) (Fig. 3c). Collectively, these data indicate that SJL males and females support sustained ZIKV infection and display no signs of morbidity at 3 months post-infection. The mice therefore represent a physiologically relevant model for studying paternal and maternal vertical transmission of ZIKVBR.

Mouse model of chronic ZIKV infection for testing therapeutic interventions. (a) Female (n = 5) and (b) male (n = 8) SJL mice were infected with 108 PFU ZIKVBR retro-orbitally. Blood samples were taken every 3 days for 50 days and analysed for viral RNA by qRT-PCR. (c) qRT-PCR analysis of viral RNA in the testes of male mice 3 months after ZIKVBR infection. Mean ± SEM of 8 mice.
Vertical and horizontal transmission of ZIKV in SJL mice
We examined horizontal transmission by infecting 3-month-old SJL males and females with ZIKVBR (108 PFU retro-orbitally) and allowing them to mate with uninfected mice of the opposite sex. After 14 days, the uninfected males were separated and bled and circulating viral titres were measured by qRT-PCR. To avoid stress during pregnancy dams were allowed to deliver and then bled on the next day and circulating viral titres were measured by qRT-PCR. Interestingly, we observed efficient transmission of ZIKV from infected males to females but not vice versa (Fig. 4a). This is strikingly similar to the mode of horizontal transmission in humans, where female-to-male transmission is relatively rare19,20,21. Because we could not detect any ZIKV transmission from infected female mice to uninfected males, we concluded other routes of transmission such as via saliva or ocular secretions are insignificant.

Horizontal and vertical sexual transmission of ZIKV in SJL mice. (a) Efficient sexual transmission of ZIKVBR was observed from infected males to uninfected females, but not from infected females to uninfected males. SJL male and female mice (n = 5) were infected with ZIKVBR (108 PFU retro-orbitally) and co-housed for 14 days with uninfected females and males, respectively. Viral RNA was measured by qRT-PCR of serum samples obtained from the sires and dams on day 14. Each point represents one animal. Data are the mean ± SD. n.d.: not detected (b) Vertical transmission of ZIKVBR to the offspring of infected sires and dams. RNA from the newborn heads was prepared on postnatal day 1 for qRT-PCR analysis of viral RNA. Each point represents one day 1 pup. Data are the mean ± SEM of the offspring of 4 infected dams/uninfected sires or 4 infected sires/uninfected dams as indicated.
Our previous study investigated foetal development in SJL females directly infected with a 4 × 1010 PFU/ml of ZIKVBR on E12.54. Here, we investigated whether the virus could be transmitted vertically from ZIKV-infected dams and sires to their offspring through the natural breeding process. To this end, 3-month-old female and male SJL mice were infected with ZIKVBR (108 PFU retro-orbitally) and immediately allowed to breed with uninfected mice of the opposite sex. After regular delivery, the 1-day-old pups were euthanized and tissue samples were analysed for viral RNA by qRT-PCR. We found that all dams efficiently transmitted ZIKV to their pups (Fig. 4b). Notably, transmission from the infected sires to their pups occurred in fewer animals was less efficient (Fig. 4b), possibly reflecting variable ZIKV titres in the semen and variations in the levels of sexual transmission of ZIKV from infected sires to dams. The molecular and cellular mechanisms of ZIKV infection during pregnancy are poorly understood22, and such knowledge is critical for the development of treatment to limit ZIKV infection during pregnancy. Our results thus demonstrate that SJL males and females can transmit ZIKV vertically through the natural mating process and thus represent a unique physiological mouse model for testing of drugs that could suppress vertical viral transmission.
CQ suppresses vertical transmission of ZIKV
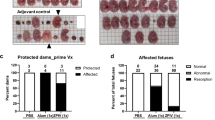
Next, we examined the effect of CQ treatment on vertical transmission of ZIKV in SJL mice using our previously published protocol4. Pregnant SJL mice (2–3 months of age) were infected with ZIKVBR (2 × 105 PFU retro-orbitally) on day E12.5. This dose is sufficient to cause a robust ZIKV infection in SJL mice. Infected dams were provided with CQ (30 mg/kg/day in drinking water) starting on day E13.5 and were euthanized on E18.5, at which point maternal blood and foetal brain samples were collected and analysed by qRT-PCR (Fig. 5a). This lower, 30 mg/kg, dosage of CQ was specifically use to protect pregnant mice from potential negative effects of the drug. We found that treatment with CQ reduced by ~20-fold the ZIKV titre in both maternal blood (Fig. 5b) and foetal brain (Fig. 5c). To confirm these results, whole embryos were immunostained with an anti-ZIKV envelope protein antibody. Consistent with the qRT-PCR data, this analysis revealed a significant reduction in the ZIKV immunostaining intensity in the foetuses of CQ-treated pregnant mice compared with the untreated mice (Fig. 5d and e).

CQ represses ZIKV infection in SJL mice and reduces vertical transmission. (a) Schematic of the experimental design. SJL dams were infected with ZIKV (2 × 105 PFU) on E12.5. On E13.5, they were randomly assigned to receive vehicle or CQ (30 mg/kg/day) in the drinking water. On E18.5, mice were euthanized for collection of blood and foetuses. (b) qRT-PCR of viral RNA. Each point represents one animal. Data are the mean ± SD of 3 (vehicle-treated) or 5 (CQ-treated) mice. *p < 0.05 by Student’s t-test. (c) qRT-PCR of viral RNA in foetal head extracts. Each point represents one foetus. Data are the mean ± SD of 8 foetuses pooled from 3 independent litters (control) or 5 foetuses pooled from 2 independent litters (CQ). *p < 0.05 by Student’s t-test. (d) Representative images of foetal brain sections from control, ZIKV-infected, and ZIKV-infected/CQ-treated mice on E18.5. Sections were stained with a primary antibody against Flavivirus Group Antigen (brown) and counterstained with Mayer’s hematoxylin (blue). Scale bar, 4 mm. (e) Quantification of ZIKV-infected cells in foetal brain sections from control, ZIKV-infected, and ZIKV-infected/CQ-treated mice. Data are the mean ± SEM of 6 sections per condition (3 embryos, 2 sections per embryo). *p < 0.05 compared with untreated ZIKV-infected mice by one-way ANOVA with Dunnett’s multiple comparisons test.
Discussion
CQ has been used worldwide for more than half a century for anti-malaria prophylaxis and therapy without evidence of foetal harm23,24,25,26. CQ can cross the placental barrier and would be expected to reach similar concentrations in maternal and foetal plasma27. The side effects of CQ have been thoroughly evaluated in a malaria prophylaxis study (400 mg/week), which found no increase in the incidence of birth defects11. High CQ concentrations (up to 500 mg/day) were administered to pregnant women with severe lupus or rheumatoid arthritis. Although a few instances of spontaneous abortion were observed (likely a consequence of the disease itself), all term deliveries resulted in normal healthy newborns28, suggesting that high doses of CQ do not interfere with foetal development in humans. The dosages of CQ we employed in our study were either comparable or significantly lower relative to the acceptable and widely used dosages in humans. Studies in rodent models have found that brain concentrations of hydroxychloroquine (CQ analogue) are 4–30 times higher than plasma concentrations29, suggesting that it has a favourable pharmacokinetic profile for inhibition of ZIKV infection in NPCs. In arthritis patients, plasma CQ concentrations reached 10 µM after daily administration of 5 mg/kg/day for a week30. Given that the half-life of CQ in humans is approximately 40 days31, people treated with 5 mg/kg/day CQ for 7 days will build up over 30 mg/kg of CQ, which is comparable to the regimen used in our animal studies. CQ treatment can be associated with retinopathy, but the reported threshold dose in humans, 5.1 mg/kg/day30, thus allowing sufficient accumulation of CQ (see above). Moreover, eye disease was not detected in a study of more than 900 rheumatoid arthritis patients treated with up to 4.0 mg/kg/day CQ for an average of 7 years30. Therefore, a level of CQ sufficient to protect SJL mice from ZIKV could be safely build up in a human body in relatively short, 7 days, time period and then maintained for many weeks or months with a minimal intake of CQ. The pharmacokinetics of CQ thus make it an excellent candidate for prophylaxis in individuals at high risk of ZIKV infection (e.g., residents or visitors in ZIKV endemic areas). Our results demonstrate that CQ effectively reduces ZIKV infection in primary human foetal NPCs and in two mouse models, and that CQ at doses comparable to or less than those broadly used in humans can markedly reduce maternal and foetal infection.
ZIKV infects cells through receptor-mediated endocytosis and membrane fusion within acidic endosomes32. CQ is thought to affect acidification of the endosomes and thus obstructs fusion of the flaviviral envelope protein with the endosomal membrane32. Cellular proteases, including furin, are essential for cleavage of the flaviviral prM during viral egress33. This transition is pH-dependent, and alterations in the intracellular pH may result in the release of less infectious virions34. Clearly, additional studies are required to determine the precise pharmacological mechanism by which CQ counters ZIKV activity.
Several approved and experimental drugs have been identified as potential anti-ZIKV agents, including daptomycin, 25-hydroxycholesterol, emricasan, niclosamide, azithromycin and sofosbuvir, and 7-deaza-2′-C-methyladenosine (inhibitors of the viral RNA polymerase)35,36,37,38,39,40,41. Thus, similar with CQ, 7-deaza-2′-C-methyladenosine (50 mg/kg/day) delayed ZIKV-induced morbidity and mortality in AG129 mice41. Further research and clinical trials will be needed to determine the optimal individual or combination drugs, for example, such as CQ and sofosbuvir or 7-deaza-2′-C-methyladenosine, for ZIKV therapy and prophylaxis. However, the long-term safety of CQ, even in pregnant women, as well as its low cost and worldwide availability, support its use alone or in combination (e.g., with sofosbuvir37,42) for the treatment and prophylaxis of ZIKV.
Methods
3-Dimensional human neurosphere assay
Control NPCs were derived from human iPSCs (WT83 c9 and c6), as previously described4. All iPSC lines were negative for mycoplasma contamination. Briefly, high-passage iPSC/hESC colonies on feeder-free plates were grown to 80% confluency. The medium was changed to N2 (DMEM/F12 medium containing 1 × N2 supplement [Invitrogen], 1 μM dorsomorphin [Tocris], and 1 μM SB431542 [Stemgent]) for 48 h. Colonies were then detached from the plate and cultured in suspension as embryoid bodies for 5 days at 90 rpm in N2 medium. Embryoid bodies were plated on Matrigel (Corning) in NGF medium (DMEM/F12 medium supplemented with 0.5 × N2, 0.5 × B27 supplement [Gibco], 20 ng/ml fibroblast growth factor, and 1% penicillin/streptomycin) for 1 week. Rosettes were manually picked, dissociated, and added to plates double-coated with polyornithine (10 μg/mL, Sigma-Aldrich) and laminin (2.5 μg/mL, Gibco) in NGF medium. To generate neurospheres, NPCs were scraped from the plates and continuously shaken for 2–5 days at 90 rpm in NGF medium. Neurospheres were then treated with NG medium supplemented with 10 μM ROCK inhibitor (Y-27632, Calbiochem) for 48 h. Neurospheres were maintained in NG medium for 4 weeks to allow neuronal maturation.
Neurosphere infection and treatment
Neurospheres were dissociated with Accutase (Thermo Fisher) and counted. For each assay, ~20 neurospheres/condition were treated as follows: uninfected (MOCK on figures), uninfected and treated with DMSO, infected with ZIKVBR at a multiplicity of infection of 1, or ZIKV infected and treated with CQ at 5, 20, or 40 µM. CQ was added during viral adsorption (1 h at 37 °C). Medium containing the appropriate concentration of CQ was changed after 2 days. At 96 h post-infection, neurospheres were transferred to polyornithine/laminin double-coated plates and maintained for 1 week to initiate neuronal maturation. Medium supplemented with CQ was changed every 2–3 days.
Immunofluorescence staining
Neurospheres were stained with anti-MAP2 (ab5392, Abcam), anti-Flavivirus Group Antigen (MAB10215, Millipore), anti-cleaved caspase 3 (9661, Cell Signaling), and DAPI. At least 3–6 images were acquired for each condition. DAPI was used to normalise cell numbers. Quantification of ZIKVBR-positive cells was based on colocalisation of Flavivirus, MAP2, and DAPI staining.
Histology and immunohistochemistry
Mouse embryos obtained on E18.5 were fixed for 48 h in 4% formaldehyde in phosphate-buffered saline (PBS), transferred to sucrose, and embedded in paraffin. Serial sections (5 μm) were cut along the sagittal axis of the embryo. Slides were deparaffinised and rehydrated using xylene and graded ethanol. Antigen retrieval was performed in a pressure cooker at 7.5 psi in 0.1 M Tris-HCl buffer (pH 9.0) for 15 min. Slides were rinsed with water 6 times at room temperature and washed for 5 min in PBS. Endogenous peroxidase activity was quenched by incubation in 3% hydrogen peroxide in PBS for 30 min at room temperature. Slides were incubated for 16–18 h at 4 °C with primary anti-Flavivirus Group Antigen (Millipore, #MAB10216) diluted 1:250 in Dako Antibody Diluent with Background Reducing Components (Agilent, #S3022). After rinsing in PBS 3 times for 5 min each, the slides were incubated with a horseradish peroxidase-conjugated goat anti-mouse secondary antibody (Abcam, #ab2891) for 30 min at room temperature. Slides were washed again in PBS, incubated for 3 min with DAB complex (ImmPACT DAB Peroxidase Substrate, Vector Laboratories, #SK-4105), and washed 3 times each in PBS and water. Finally, slides were counterstained in Mayer’s hematoxylin, mounted, visualised, and recorded using an Aperio automated system (Leica). The images were analysed using ImageScope (Leica).
Animal studies
All animal procedures were performed in accordance with the PHS Policy on Humane Care and Use of Laboratory Animals and with the approval of the Sanford Burnham Prebys Medical Discovery Institute IACUC Committee, protocol AUF#16-049.
ZIKV infection in AG129 mice
Mice received CQ (50 mg/kg in drinking water) or drinking water alone for 2 days prior to infection. ZIKVBR (2 × 103 PFU) was injected retro-orbitally, and CQ treatment was continued at 50 mg/kg in drinking water for 5 days and then at 5 mg/kg/day for the remainder of the experiment. A 6-point visual health scoring system was modified from the 7-point scale previously published by Tang et al.15. According to institutional IACUC requirements, moribund animals were euthanized and not scored. Animals were considered moribund if they could not turn themselves upright when placed on their sides.
Model of chronic ZIKV infection in SJL mice
Two groups of 3-month-old SJL mice (8 males and 5 females) were infected with ZIKVBR (1 × 108 PFU retro-orbitally), and blood samples were collected every third day for 50 days. Males were euthanized at 3 months post-infection to obtain testis samples. Viral RNA titres were measured by qRT-PCR analysis of serum and testis RNA.
ZIKV infection in SJL mice
Two- to three-month-old pregnant SJL mice (Jackson Laboratories, Bar Harbor, ME, USA) were infected on E12.5 with ZIKVBR (2 × 105 PFU retro-orbitally). CQ (30 mg/kg in drinking water) was provided for 5 days between E13.5 and E18.5. We regularly monitored and tracked water consumption in our experimental and control groups. We did not observe any difference in water consumption between the groups. On E18.5, maternal blood samples were collected and the animals were euthanized to obtain the foetal tissues. In order to prevent contamination of foetal samples from maternal blood the pregnant mice were transcardially perfused (day 18.5) with 10 ml PBS to fully remove the blood from animals.
Horizontal and vertical transmission of ZIKV in SJL mice
Three-month-old SJL male and female mice (Jackson Laboratories, Bar Harbor, ME, USA) were infected with ZIKVBR (108 PFU retro-orbitally) and then allowed to breed with uninfected females or males, respectively. After 14 days, blood samples were collected from males. On day 1 post-delivery, blood samples were collected from the mothers and the pups were euthanized to obtain brain tissues. Viral RNA levels in the blood and brain tissues were measured by qRT-PCR.
Real-time qPCR quantification of ZIKV
Total RNA was extracted from samples of blood (0.1 ml), foetal br–in, or pups using a NucleoSpin RNA Kit (Macherey-Nagel). RNA concentrations were measured using a NanoDrop spectrophotometer (NanoDrop Technologies) and the samples were kept at −80 °C until use. ZIKV-specific primers were ZIKV-835 5′-TTGGTCATGATACTGCTGATTGC-3′ and ZIKV-911c 5′-CCTTCCACAAAGTCCCTATTGC-3′, as described previously4,43. Real-time PCR was performed using QuantiTect Reverse Transcription Kit (QIAGEN), SYBR Green I Master Mix, and a LightCycler 480 II instrument (Roche) using the following conditions: initiation at 95 °C for 10 min followed by 50 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. Data were analysed using LightCycler 480 Software 1.5.0 (Roche). Assay sensitivity was determined using samples with known ZIKV concentrations. GraphPad Prism was used as the fitting software.
References
Campos, G. S., Bandeira, A. C. & Sardi, S. I. Zika Virus Outbreak, Bahia, Brazil. Emerg Infect Dis 21, 1885–1886, https://doi.org/10.3201/eid2110.150847 (2015).
Castro, L. A. et al. Assessing real-time Zika risk in the United States. BMC Infect Dis 17, 284, https://doi.org/10.1186/s12879-017-2394-9 (2017).
Azevedo, R. S. et al. Zika virus epidemic in Brazil. I. Fatal disease in adults: Clinical and laboratorial aspects. J Clin Virol 85, 56–64, https://doi.org/10.1016/j.jcv.2016.10.024 (2016).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271, https://doi.org/10.1038/nature18296 (2016).
Li, C. et al. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell, https://doi.org/10.1016/j.stem.2016.04.017 (2016).
Mlakar, J. et al. Zika Virus Associated with Microcephaly. N Engl J Med 374, 951–958, https://doi.org/10.1056/NEJMoa1600651 (2016).
Melo, A. S. et al. Congenital Zika Virus Infection: Beyond Neonatal Microcephaly. JAMA Neurol 73, 1407–1416, https://doi.org/10.1001/jamaneurol.2016.3720 (2016).
Cragan, J. D. et al. Baseline Prevalence of Birth Defects Associated with Congenital Zika Virus Infection - Massachusetts, North Carolina, and Atlanta, Georgia, 2013-2014. MMWR Morb Mortal Wkly Rep 66, 219–222, https://doi.org/10.15585/mmwr.mm6608a4 (2017).
Beckham, J. D., Pastula, D. M., Massey, A. & Tyler, K. L. Zika Virus as an Emerging Global Pathogen: Neurological Complications of Zika Virus. JAMA Neurol, https://doi.org/10.1001/jamaneurol.2016.0800 (2016).
Dirlikov, E. et al. Guillain-Barré Syndrome During Ongoing Zika Virus Transmission - Puerto Rico, January 1-July 31, 2016. MMWR Morb Mortal Wkly Rep 65, 910–914, https://doi.org/10.15585/mmwr.mm6534e1 (2016).
Wolfe, M. S. & Cordero, J. F. Safety of chloroquine in chemosuppression of malaria during pregnancy. Br Med J (Clin ResEd) 290, (1466–1467 (1985).
Delvecchio, R. et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 8, https://doi.org/10.3390/v8120322 (2016).
Cao, B., Parnell, L. A., Diamond, M. S. & Mysorekar, I. U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J Exp Med, https://doi.org/10.1084/jem.20170957 (2017).
Shiryaev, S. A. et al. Characterization of the Zika virus two-component NS2B-NS3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antiviral Res 143, 218–229, https://doi.org/10.1016/j.antiviral.2017.04.015 (2017).
Tang, H. et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 18, 587–590, https://doi.org/10.1016/j.stem.2016.02.016 (2016).
Morrison, T. E. & Diamond, M. S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J Virol 91, https://doi.org/10.1128/JVI.00009-17 (2017).
Rossi, S. L. et al. Characterization of a Novel Murine Model to Study Zika Virus. Am J Trop Med Hyg 94, 1362–1369, https://doi.org/10.4269/ajtmh.16-0111 (2016).
Govero, J. et al. Zika virus infection damages the testes in mice. Nature 540, 438–442, https://doi.org/10.1038/nature20556 (2016).
Paz-Bailey, G. et al. Persistence of Zika Virus in Body Fluids - Preliminary Report. N Engl J Med, https://doi.org/10.1056/NEJMoa1613108 (2017).
Frank, C. et al. Sexual transmission of Zika virus in Germany, April 2016. Euro surveillance: bulletin Européen sur les maladies transmissibles = European communicable disease bulletin 21, https://doi.org/10.2807/1560-7917.ES.2016.21.23.30252 (2016).
Moreira, J., Peixoto, T. M., Siqueira, A. M. & Lamas, C. C. Sexually acquired Zika virus: a systematic review. Clin Microbiol Infect 23, 296–305, https://doi.org/10.1016/j.cmi.2016.12.027 (2017).
Cao, B., Diamond, M. S. & Mysorekar, I. U. Maternal-Fetal Transmission of Zika Virus: Routes and Signals for Infection. J Interferon Cytokine Res, https://doi.org/10.1089/jir.2017.0011 (2017).
Fakeye, T. O., Fehintola, F. A., Ademowo, O. G. & Walker, O. Therapeutic monitoring of chloroquine in pregnant women with malaria. West Afr J Med 21, 286–287 (2002).
Lee, S. J. et al. Chloroquine pharmacokinetics in pregnant and nonpregnant women with vivax malaria. Eur J Clin Pharmacol 64, 987–992, https://doi.org/10.1007/s00228-008-0500-z (2008).
Chandra, R. S. et al. Creative solutions to extraordinary challenges in clinical trials: methodology of a phase III trial of azithromycin and chloroquine fixed-dose combination in pregnant women in Africa. Malar J 12, 122, https://doi.org/10.1186/1475-2875-12-122 (2013).
Radeva-Petrova, D., Kayentao, K., ter Kuile, F. O., Sinclair, D. & Garner, P. Drugs for preventing malaria in pregnant women in endemic areas: any drug regimen versus placebo or no treatment. Cochrane Database Syst Rev, CD000169, https://doi.org/10.1002/14651858.CD000169.pub3 (2014).
Law, I. et al. Transfer of chloroquine and desethylchloroquine across the placenta and into milk in Melanesian mothers. Br J Clin Pharmacol 65, 674–679, https://doi.org/10.1111/j.1365-2125.2008.03111.x (2008).
Levy, M., Buskila, D., Gladman, D. D., Urowitz, M. B. & Koren, G. Pregnancy outcome following first trimester exposure to chloroquine. Am J Perinatol 8, 174–178, https://doi.org/10.1055/s-2007-999371 (1991).
Titus, E. O. Recent developments in the understanding of the pharmacokinetics and mechanism of action of chloroquine. Ther Drug Monit 11, 369–379 (1989).
Mackenzie, A. H. Dose refinements in long-term therapy of rheumatoid arthritis with antimalarials. Am J Med 75, 40–45 (1983).
Browning, D. J. In Hydroxychloroquine and Chloroquine Retinopathy 35–63 (Springer, 2014).
Smit, J. M., Moesker, B., Rodenhuis-Zybert, I. & Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 3, 160–171, https://doi.org/10.3390/v3020160 (2011).
Maramorosch, K., Shatkin, A. J., Murphy, F. A., Chambers, T. J. & Monath, T. P. The flaviviruses: structure, replication and evolution. Vol. 59 (Academic Press, 2003).
Hsieh, S. C. et al. Highly conserved residues in the helical domain of dengue virus type 1 precursor membrane protein are involved in assembly, precursor membrane (prM) protein cleavage, and entry. J Biol Chem 289, 33149–33160, https://doi.org/10.1074/jbc.M114.610428 (2014).
Barrows, N. J. et al. A Screen of FDA-Approved Drugs for Inhibitors of Zika Virus Infection. Cell Host Microbe 20, 259–270, https://doi.org/10.1016/j.chom.2016.07.004 (2016).
Bullard-Feibelman, K. M. et al. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res 137, 134–140, https://doi.org/10.1016/j.antiviral.2016.11.023 (2017).
Sacramento, C. Q. et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci Rep 7, 40920, https://doi.org/10.1038/srep40920 (2017).
Li, C. et al. 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 46, 446–456, https://doi.org/10.1016/j.immuni.2017.02.012 (2017).
Xu, M. et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med 22, 1101–1107, https://doi.org/10.1038/nm.4184 (2016).
Retallack, H. et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc Natl Acad Sci USA 113, 14408–14413, https://doi.org/10.1073/pnas.1618029113 (2016).
Zmurko, J. et al. The Viral Polymerase Inhibitor 7-Deaza-2′-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl Trop Dis 10, e0004695, https://doi.org/10.1371/journal.pntd.0004695 (2016).
Reznik, S. E. & Ashby, C. R. Jr. Sofosbuvir: an antiviral drug with potential efficacy against Zika infection. Int J Infect Dis 55, 29–30, https://doi.org/10.1016/j.ijid.2016.12.011 (2017).
Lanciotti, R. S. et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis 14, 1232–1239, https://doi.org/10.3201/eid1408.080287 (2008).
Acknowledgements
We thank all members of the Terskikh laboratory for helpful discussions and assistance. We gratefully acknowledge Buddy Charbono, Hilda Clarke, Francisco Beltranand Mary O’Rourke-Braxtan at the Animal Facility of the Sanford Burnham Prebys Medical Discovery Institute for their invaluable assistance with the animal studies. We thank Drs. P. Mesci and A. Macia for drawing the schematic in Fig. 2. A.R.M. is supported by grants from the California Institute for Regenerative Medicine (DISC2-09649), the National Institutes of Health through the U19MH107367, and an NARSAD Independent Investigator Grant. P.M. is supported by an International Rett Syndrome Foundation fellowship. This work was supported by National Institutes of Health through R21NS100477 grant to A.V.T., A.Y.S., and S.S.
Author information
Authors and Affiliations
Contributions
S.A.S., A.Y.S., and A.V.T. conceived the research; S.A.S., P.M., A.P., I.F., N.S., S.S., C.F., C.-T.H., and A.M. designed and performed the experiments; S.A.S., A.Y.S., and A.V.T. interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shiryaev, S.A., Mesci, P., Pinto, A. et al. Repurposing of the anti-malaria drug chloroquine for Zika Virus treatment and prophylaxis. Sci Rep 7, 15771 (2017). https://doi.org/10.1038/s41598-017-15467-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15467-6
This article is cited by
-
Simultaneous LC–MS/MS method for the quantitation of Azithromycin, Hydroxychloroquine and its metabolites in SARS-CoV-2(−/ +) populations using dried blood spots
Scientific Reports (2023)
-
Intramolecular Cyclization During Bromination of the Quinoline Alkaloid Haplophyllidine
Chemistry of Natural Compounds (2022)
-
A computational approach to aid clinicians in selecting anti-viral drugs for COVID-19 trials
Scientific Reports (2021)
-
Repurposing clinical drugs is a promising strategy to discover drugs against Zika virus infection
Frontiers of Medicine (2021)
-
The risk of QTc-interval prolongation in COVID-19 patients treated with chloroquine
Netherlands Heart Journal (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
