
Abstract
(±)-Macleayins F–H (1–3), three pairs of new enantiomeric alkaloid dimers, along with four known alkaloids (4–7) as their plausible biogenetic precursors, were isolated from the aerial parts of Macleaya cordata. Compounds 1–3 were obtained under the guidance of LC-MS investigation, and their structures were elucidated by analysis of the 1D and 2D NMR spectroscopic data. The racemic mixtures were successfully separated by chiral HPLC, and the absolute configurations of enantiomers were determined by electronic circular dichroism (ECD) spectroscopy. Compounds 1–7 showed antiproliferative activity against HL-60 with IC50 values of 1.34–41.30 μM, especially compounds 1–2 exhibited the best inhibitory activity against HL-60 cell lines. In addition, the preliminary mechanism investigation for compound 2 using Annexin V/7-AAD double-staining assay, DAPI staining assay and JC-1 staining method, indicated that 2 inhibited cancer cell proliferation potentially through inducing apoptosis via the mitochondria-related pathway and arrested cell cycle of HL-60 cells at S phase.
Similar content being viewed by others

Introduction
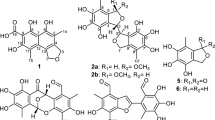
Benzophenanthridine and protopine alkaloids occur in Macleaya cordata (Willd) R. Br. and have been investigated for their intriguing bioactivity, such as anticancer, anti-bacterial, insecticidal, and anti-inflammatory effects1,2. Macleaya cordata is a perennial plant in the family of Papaveraceae, which has been used in folk medicine for the treatment of cervical cancer, thyroid cancer, inflamed wounds, ringworm infection, arthritis, and trichomonas vaginalis3,4. In our previous investigation, two pairs of enantiomeric alkaloid dimers (macleayins A and B) with cytotoxicity were isolated from M. cordata 5. In our present study, LC-MS- guided fractionation (Fig. 1) led to the isolation of three pairs of analogous dimers, macleayins F–H (1–3) biogenetically derived from a benzophenanthridine and protopine alkaloid through C6–C13′ linkage. Four possible biogenetic precursors, sanguinarine, chelerythrine, protopine, and allocryptopine (4–7) were obtained (Fig. 2). Herein, we reported the isolation, and structural elucidation of new compounds. In addition, the antiproliferative properties and action mechanism were also investigated.

LC-MS analysis chromatogram of the crude ethanol extract from M. cordata.

Structures of compounds 1–7 and macleayins A, B.
Results and Discussion
Structural Elucidation of Compounds 1a/1b–3a/3b
Macleayin F (1) was isolated as white amorphous powder, with the molecular formula of C41H36N2O9, deduced from HRESIMS [M + H]+ (m/z 701.2491, calcd for 701.2494), appropriate for 25 degrees of unsaturation. The IR spectrum indicated the presence of ketone carbonyl (1656 cm−1), methylenedioxyl group (2792, 941 cm−1), and aromatic ring (1616, 1486, 1462 cm−1). In the UV spectrum, the absorption maxima at 227 and 287 nm were detected. The HRESIMS/MS spectrum exhibited the fragment peak at m/z 348.1281 assigned to chelerythrine6. The 1H NMR (Table 1) spectrum revealed the presence of three AB spin systems of aromatic protons in ortho-position ((δ H 6.68 (br d, J = 7.3 Hz, H-9) and 7.23 (br d, J = 7.3 Hz, H-10), 7.46 (d, J = 8.5 Hz, H-12) and 7.61 (d, J = 8.5 Hz, H-11), 6.97 (br d, J = 8.0 Hz, H-11′) and 7.50 (br s, H-12′)), four aromatic protons in singlet (δ H 7.05, 6.85, 6.83, and 6.21), two sp3 methine protons (δ H 5.13 (d, J = 9.0 Hz) and 4.59 (d, J = 9.0 Hz)), as well as three methylenedioxyl groups ((δ H 5.99 (d, J = 1.1 Hz), 5.96 (d, J = 1.1 Hz), 5.91 (d, J = 1.1 Hz), 5.83 (d, J = 1.1 Hz), 5.87 (d, J = 1.4 Hz), and 5.82 (d, J = 1.4 Hz)), two methoxyl groups (δ H 3.94 and 3.83), and two N-methyl groups (δ H 2.51 and 1.63). The 13C NMR (Table 1) combined with the HSQC spectroscopic data identified twenty-eight aromatic, three methylenedioxyl, two methoxyl, two N-methyl, three methylene, and two sp3 methine carbons (Table 1). Considered its molecular formula, one carbonyl carbon signal was absent in its 13C NMR spectrum. The above data suggested that 1 was a dimeric alkaloid consisting of a chelerythrine and a protopine moiety5.
The HMBC correlations (Fig. 3) of H-1/C-3, C-4a, C-12; H-10/C-6a, C-8; H-11/C-4b, C-10a, C-12a; H-12/C-1, C-4a, C-10b; 7-OCH3/C-7; 8-OCH3/C-8; -NCH3/C-4b, and from the protons of OCH2O (δ H 5.99 and 5.96) to C-2, and C-3, as well as the1H−1H COSY correlations (Fig. 3) of H-9/H-10 and H-11/H-12, evidenced the presence of chelerythrine moiety. This was furthermore confirmed by the fragment peak at m/z 348.1281 in the HRESIMS/MS (Supplementary Figure S13). In the 1H NMR spectra of 1, the signals in the high field region were substantially overlapped. Hence, the presence of protopine structure could not be completely assigned with the observed HMBC cross-peaks of H-1′/C-3′, C-4′a; H-4′/C-2′, C-5′, C-14′a; H-11′/C-9′, C-10′, C-12′a; as well as cross-peaks of two methylenedioxys at δ H 5.91, 5.83/C-2′, C-3′ and δ H 5.87, 5.82/C-9′, C-10′. Comparison of the NMR and HRESIMS data of 1 with those of macleayins A and B5 which were previously obtained as a single crystal molecule from M. cordata indicated that 1 and macleayin B possessed protopine moiety. In addition, the1H-1H COSY of H-6/H-13′ proved the direct linkage of C6–C13′. Similarly, one carbonyl and several crucial HMBC correlations were not observed in 1. Accordingly, the planar structure of compound 1 was defined as shown.

HMBC,1H-1H COSY, and NOESY correlations of macleayin F (1).
The relative stereochemistry of 1 was established by the NOESY experiment (Fig. 3). The coupling constant (J = 9.0 Hz) and no NOE correlation between H-6 and H-13′, suggested that H-6 and H-13′ may be on the opposite side. The proposed relative stereochemistry is a reasonable hypothesis, as well as the fact that this is due to the lacunar nature of the available data set. Compound 1 was a racemic mixture like macleayins A and B due to the lack of optical rotation and Cotton effect (CE)7. Subsequently, the enantioseparation of 1 was performed on a chiral column (Daicel Chiralpak IB) to yield the enantiomers 1a and 1b in an approximate 1:1 ratio (Supplementary Figure S4)8, which were opposite in terms of optical rotation (1a:\({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) + 274 (c 0.07, MeOH), 1b: \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) −248 (c 0.07, MeOH)). The assignments of their absolute configuration at the stereogenic centers were determined by comparing the ECD spectra (Fig. 4) of both enantiomers with those of two similar compounds (+)-macleayin A and (−)-macleayin A. The measured ECD curves of 1a and 1b matched with that of (6 R,13′R)-macleayin A and (6 S,13′S)-macleayin A, respectively. Hence, the absolute configuration for 1a (6 R,13′R) and 1b (6 S,13′S) were unambiguously determined as shown in Fig. 1 and named (+)-macleayin F and (−)-macleayin F, respectively.

CD spectra for compounds 1a, 1b, 2a, 2b, 3a, 3b, and (+)-/(−)-macleayin A, (+)-/(−)-macleayin B.
Macleayin G (2) was obtained as white amorphous powder. The molecular formula of 2 was established as C42H40N2O9 on the basis of positive HRESIMS at m/z 717.2799 [M + H]+ (calcd 717.2807), indicating 24 indices of hydrogen deficiency. The IR spectrum showed absorption bands due to ketone carbonyl (1652 cm−1), methylenedioxyl group (2794, 938 cm−1), and aromatic ring (1619, 1491, 1458 cm−1) functionalities. The UV spectrum exhibited maximum absorption at 229 and 286 nm. Its1H and 13C NMR data (Table 1) revealed structural similarity to 1, expected that one methylenedioxy in 1 was replaced by two methoxyl groups (δ H 3.95, 3.50; δ C 56.0, 60.9). It was confirmed by the HMBC correlations of 8-OCH3 with C-8, 7-OCH3/H-6 with C-7; 9′-OCH3/H-8′/H-11′ with C-9′, and of 10′-OCH3/H-11′ with C-10′ (Supplementary Figure S2). Furthermore, the HRESIMS/MS showed the fragment ion peak at m/z 348.1306 which was recognized as chelerythrine, suggested that 2 was made up of chelerythrine and allocryptopine, which was supported by comparison of NMR data of 2 with those of macleayin B5. 1H-1H COSY correlations (Supplementary Figure S2) of H-6/H-13′ confirmed that two parts were connected by C-6 with C-13′. Similarly, compound 2 was proposed to be a racemic mixture on account of the lack of optical rotation and CE. Subsequently, 2a and 2b were obtained by chiral-phase HPLC separation in a ratio of 1:1 (Supplementary Figure S5), which exhibited the mirror image-like ECD curves and owned the opposite specific optical rotations (2a: \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) + 238 (c 0.10 MeOH)), 2b: \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) −210 (c 0.10 MeOH)). Thus, the absolute configuration of 2a and 2b were assigned as (6 R,13′R) and (6 S,13′S) by comparing their experimental ECD spectra with those of (+)-macleayin A and (−)-macleayin A (Fig. 4), and given the names (+)-macleayin G and (−)-macleayin G, respectively.
Macleayin H (3) was isolated as white amorphous powder and yielded a quasi-molecular ion peak at m/z 701.2485 [M + H]+ (calcd for 701.2494) in the HRESIMS, indicative of molecular formula C41H36N2O9, the same as that of 1. The identical fragment peak at m/z 348.1323 in HRESIMS/MS as 1, together with 1D and 2D NMR data (Table 1) deduced that 3 was a similar dimer comprising chelerythrine and protopine as 1. But the chemical shifts of H-4 and H-4′ in compound 3 were significantly shifted upfield (ca. −1.6 and −0.3 ppm), and H-13′ was shifted downfield (ca. +0.4 ppm). Compound 3 showed more similar NMR data (Supplementary Tables S1 and S2) to macleayin B than 1, deducing that the relative configuration of 3 was the same as macleayin B. Subsequent chiral HPLC resolution (Supplementary Figure S6) of 3 afforded 3a (\({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) + 71 (c 0.11 MeOH)) and 3b (\({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) −94 (c 0.11 MeOH)). The absolute configuration at the stereocenters of 3a (6 S,13′R) and 3b (6 R,13′S) were established by comparison of their experimental ECD spectra with those of (+)-macleayin B and (−)-macleayin A (Fig. 4), and named (+)-macleayin H, and (−)-macleayin H, respectively.
Antiproliferative activity
Our previous study revealed that alkaloids from M. cordata exhibited promising antiproliferative effects, so all the isolated compounds were tested for the growth inhibitory activities against HL-60, A-549, MCF-7 human cancer cell lines by the trypan blue method and MTT method9,10. The results were summarized in Table 2. All compounds inhibited the growth against all the tested cancer cells with IC50 values ranged from 1.34 to 41.30 μM. In particular, 1a, 1b, 2a, and 2b showed more potent antiproliferative property against HL-60 cell lines than their biogenetic precursors 5–7, and the enantiomers displayed similar inhibitory effects. Therefore, compound 2 was selected for further investigation on the antiproliferative mechanism in HL-60 cells.
Influence of compound 2 on the HL-60 cell cycle
Cell cycle arrest was an important sign for inhibition of proliferation and the series of events that took place in a cell leading to its division and duplication11,12. In order to explore whether the growth inhibition induced by 2 was caused by the regulation of the HL-60 cell cycle, the cell cycle distribution in the presence of 2 was detected by flow cytometry (Fig. 5). HL-60 cells were treated with compound 2 at concentrations of 3.75, 7.5, and 15.0 μM for 72 h, which resulted in a remarkable increase of 31.49%, 46.11%, and 48.36% of cells at S phase compared with that of the control (28.00%), while there was a concomitant decline in the number of cells in G1 and G2 phases. This information indicated that compound 2 influenced cell cycle of HL-60 arrested at S phase in a dose-dependent manner.

The influence of HL-60 cell cycle by compound 2. Left red part: cell in G1 phase; Right red part: cell in G2 phase; Oblique line part: cell in S phase; White part: total cells.
Induction of apoptosis by compound 2
Apoptosis is an ordered and orchestrated cellular process that takes play in physiological and pathological conditions13,14. Therefore, drug-induced apoptosis in tumor cells is important for cancer treatment. The effects of 2 on the apoptosis of HL-60 cells by staining them with Annexin-V APC/7-AAD and analysis by flow cytometry were examined. As shown in Fig. 6, the treatment with 3.75, 7.50, and 15.0 μM of 2 for 72 h in HL-60 cells, the total percentage of early apoptotic cells (right low quadrant) and late apoptotic and necrotic cells (right upper quadrant) were increased from 5.41% to 61.34%. The data indicated that compound 2 remarkable induced apoptotic cells death in HL-60 cells in a concentration-dependent manner.

Induction of apoptosis by 2 in HL-60 cells.
Effect of mitochondria depolarization by compound 2
The destruction of mitochondrial membrane potential is widely considered to be a crucial event in the process of cell apoptosis15,16. In order to further research the apoptosis induced effect of compound 2, the fluorescent probe JC-1 was carried out to detect the changes of mitochondrial membrane potential. JC-1 (a kind of lipophilic cationic dye) can pass the plasma membrane into cells and accumulate in mitochondria. Meanwhile, membrane potential and degree of accumulation of JC-1 in mitochondria exist close contact. Normal cells, normal membrane potential, at the higher concentration of dye JC-1, with the dye aggregation, fluorescence emission gradually changes red. While apoptotic cells, the mitochondrial transmembrane potential depolarization, the monomer of JC-1 is formed, and fluorescence emission changes from red to green. Therefore, it can easily be used to test the changes of mitochondrial membrane potential by detecting the changes of fluorescence color. HL-60 cells dealt with compound 2 at 3.75, 7.50, and 15 μM for 72 h respectively were stained with JC-1, meanwhile untreated cells was used as control. The percentage of green fluorescence increased (0.12%, 25.11%, 36.70%, and 64.80%) in a concentration-dependent manner was observed in Fig. 7. The results demonstrated that 2 could induce apoptosis in HL-60 cells through mitochondrial-related pathway.

Effect of 2 on the mitochondrial membrane potentials in HL-60 cells.
Methods
General experimental procedures
Optical rotations were measured on Rudolph Autopol-V digital polarimeter and Jasco P-2000 polarimeter. UV spectra were recorded using a Shimadzu UV-2201 spectrometer. IR spectra were recorded on a Bruker IFS-55 spectrometer (using a KBr disk method). ECD spectra were measured on Bio-logic MOS 450 spectropolarimeter. 1D and 2D NMR spectra were acquired with Bruker ARX-300 and AV-600 NMR spectrometers using solvent signals (CDCl3: δ H 7.26/δ C 77.16; CD3OD: δ H 3.31/δ C 49.00), with tetramethylsilane (TMS) as an internal standard. HRESIMS data were obtained using Bruker micro-TOFQ-Q mass spectrometer. LC-MS analysis was performed with Thermo Fisher LCQ Fleet Ion Trap LC/MSn, and ESI-IT-MS spectra were performed on a Thermo LCQ Advantage MAX Fleet mass spectrometer. A Shimadzu LC-6 AD equipped with a SPD-20A (UV/DAD) detector was used for HPLC. The chiral HPLC isolation was accomplished on Daicel Chiralpak IB column (4.6 × 250 mm, 5 μm; Daicel Chemical Ltd, Tokyo, Japan). Column chromatography (CC) were performed with silica (100–200 and 200–300 mesh, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), neutral alumina (100–200 mesh, Sinopharm Chemical Reagent Co. Ltd., Shanghai, China), ODS (50 µm, YMC Co. Ltd., Kyoto, Japan), and Sephadex LH-20 (GE Healthcare, Sweden). TLC analyses were carried out with glass plate precoated silica gel (GF254, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China). Spots were visualized under UV light or by spraying with 10% H2SO4 in 95% EtOH followed by heating or with bismuth potassium iodide solution.
Plant materials
The plant material was purchased from Anguo Medicines Ltd (Hebei), China, in November 2013, and was identified as the aerial parts of Macleaya cordata (Willd.) R. Br. by Prof. Jincai Lu (School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang, China). The voucher sample (BLH-20131108) was deposited in the Department of Natural Products Chemistry, Shenyang Pharmaceutical University, Shenyang, China.
Extraction and isolation
The air-dried aerial parts of M. cordata (40.0 kg) were extracted with 95% ethanol (400 L) under reflux 2 times, and 75% ethanol (1 × 400 L), each time for 2 h, respectively. After the solvent was removed under reduced pressure, the crude extract (14.6 kg) was suspended in H2O, successively partitioned with CH2Cl2 and n-BuOH, to afford CH2Cl2, n-BuOH and aqueous extracts. Part of the CH2Cl2 extract (500 g) was subjected to silica gel column chromatography (CC) and eluted with petroleum ether (60–90 °C) –acetone (100:5, 100:10, 100:20, 100:50, 1:1 and 0:100, v/v) to yield six crude fractions (Fr. A– Fr. F). Fr. A was subsequently loaded onto a silica gel column using petroleum ether–EtOAc (100:0, 100:3, 100:5, 100:8, 100:20, v/v) as the eluent to give five subfractions (Fr. A1 –Fr. A5). Fr. A2 was applied to neutral alumina CC eluting with petroleum ether–EtOAc, and further purified by preparative TLC (PTLC) to afford 4 (4.0 mg). Fr. D was further subjected to ODS CC with MeOH–H2O (50:50, 60:40, 65:35, 70:30, 80:20, 90:10, v/v) as the mobile phase to give subfractions (Fr. D1-Fr. D6). The residue of Fr. D1 and Fr. D4 after recrystallization was separated by Sephadex LH-20 CC eluting with CH2Cl2/MeOH (1:1, v/v) to yield total alkaloids (23.9 g) based on TLC analysis. The total alkaloids fraction was further subjected to ODS CC (MeOH–H2O, 30:70 to 90:10, v/v). The fraction of 40% methanol elution was recrystallized to give 5 (4.5 mg). The fraction of 75% methanol elution was separated by performing repeated neutral alumina, silica gel, Sephadex LH-20 CC, and PTLC to yield 1 (5.0 mg), and 3 (7.5 mg). Fr. E was subjected to silica gel CC eluted with CH2Cl2–MeOH (100:0, 200:1, 100:1, 100:2, v/v) to afford four subfractions Fr. E1–Fr. E4. Fr. E1 was subjected to ODS CC eluting with MeOH–H2O gradient system, and further recrystallization to yield 6 (30 mg), and then performed on Sephadex LH-20 CC eluting with CH2Cl2/MeOH (1:1, v/v) to afford 2 (6.0 mg). The n-BuOH extract (185 g) was separated on macroporous resin (D101) using EtOH–H2O (30:70, 50:50, 70:30, 95:5, v/v) as the eluent to give four subfractions Fr. G–Fr. J. Fr. H was subjected to ODS CC with MeOH–H2O gradient eluting, and the part of 20% MeOH eluting was further purified by reversed-phase preparative HPLC (ODS; 5 μm, 250 × 20 mm; MeOH/H2O/DEA, 63:37:0.05, v/v/v; flow rate, 5.0 mL/min) to afford 7 (35 mg, t R = 45 min).
Chiral separation of 1, 2, and 3 was performed on Daicel chiralpak IB column (250 × 4.6 mm), eluted with n-hexane–EtOH–DEA (40: 60: 0.1, v/v/v), flow rate 1.0 mL/min to yield 1a (1.1 mg, tR 5.930 min), 1b (1.1 mg, t R 8.022 min), 2a (1.5 mg, t R 5.813 min), 2b (1.5 mg, t R 12.305 min), 3a (1.7 mg, t R 12.087 min), and 3b (1.7 mg, t R 8.074 min), respectively.
Macleayin F (1). White amorphous powder. UV (CH2Cl2) λ max (log ε): 227 (4.6), 287 (4.6) nm. IR (KBr) ν max: 2790, 1656, 1616, 1486, 1462, 941 cm−1. (+)-HRESIMS m/z 701.2491 [M + H]+ (calcd for C41H37N2O9, 701.2494).
(+)-Macleayin F (1a). \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) + 274 (c 0.07, MeOH); ECD (MeOH) λmax (∆ɛ) 224 (+9.4), 243 (−22.6), 268 (−3.9), 300 (−7.9) nm.
(−)-Macleayin F (1b). \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) −248 (c 0.07, MeOH); ECD (MeOH) λmax (∆ɛ) 221 (−13.5), 243 (+24.8), 269 (+3.4), 300 (+7.5) nm.
Macleayin G (2). White amorphous powder. UV (CH2Cl2) λ max (log ε): 229 (4.6), 286 (4.6) nm. IR (KBr) ν max: 2794, 1652, 1619, 1491, 1458, 938 cm−1. (+)-HRESIMS m/z 717.2799 [M + H]+ (calcd for C42H41N2O9, 717.2807).
(+)-Macleayin G (2a). \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) + 238 (c 0.10, MeOH); ECD (MeOH) λ max (∆ɛ) 222 (+15.9), 241 (−30.3), 264 (−6.8), 283 (−14.3) nm.
(−)-Macleayin G (2b). \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) −210 (c 0.10, MeOH); ECD (MeOH) λ max (∆ɛ) 223 (−19.4), 241 (+36.0), 265 (+7.3), 283 (+15.1) nm.
Macleayin H (3). White amorphous powder. UV (MeOH) λ max (log ε): 232 (4.6), 287 (4.6) nm. IR (KBr) ν max: 2798, 1658, 1617, 1485, 1463, 939 cm−1. (+)-HRESIMS m/z 701.2485 [M + H]+ (calcd for C41H37N2O9, 701.2494).
(+)-Macleayin H (3a). \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) + 71 (c 0.11, MeOH); ECD (MeOH) λ max (∆ɛ) 204 (+19.3), 233 (−24.0), 271 (−0.2), 291 (−5.7) nm.
(−)-macleayin H (3b). \({[\alpha ]}_{{\rm{D}}}^{{\rm{20}}}\) −94 (c 0.11, MeOH); ECD (MeOH) λ max (∆ɛ) 205 (−17.2), 233 (+19.3), 276 (+0.4), 291 (+6.0) nm.
Sanguinarine (4). Red amorphous powder. 1H NMR (400 MHz, CD3OD) δ H: 9.97 (1 H, s, H-6), 8.67 (1 H, d, J = 8.8 Hz, H-10), 8.57 (1 H, d, J = 8.9 Hz, H-11), 8.25 (1 H, d, J = 8.9 Hz, H-12), 8.18 (1 H, s, H-4), 7.98 (1 H, d, J = 8.8 Hz, H-9), 7.60 (1 H, s, H-1), 6.54 (2 H, s, -OCH2O-2,3), 6.28 (2 H, s, -OCH2O-7,8), 4.97 (3 H, s, N-CH3). 13C NMR (150 MHz, CD3OD) δ C: 107.0 (C-1), 150.8 (C-2), 150.9 (C-3), 105.0 (C-4), 122.0 (C-4a), 132.9(C-4b), 52.8 (N-CH3), 151.0 (C-6), 111.4 (C-6a), 148.3 (C-7), 149.6 (C-8), 121.3 (C-9), 118.3 (C-10), 129.2 (C-10a), 127.7 (C-10b), 119.7 (C-11), 133.3 (C-12), 134.3 (C-12a), 104.3 (-OCH2O-2,3), 106.5 (-OCH2O-7,8).
Chelerythrine (5). Yellow amorphous powder. 1H NMR (400 MHz, CD3OD) δ H: 9.99 (1 H, s, H-6), 8.71 (1 H, d, J = 9.0 Hz, H-10), 8.68 (1 H, d, J = 9.2 Hz, H-11), 8.24 (1 H, d, J = 9.0 Hz, H-9), 8.23 (1 H, d, J = 9.2 Hz, H-12), 8.21 (1 H, s, H-4), 7.59 (1 H, s, H-1), 6.28 (2 H, s, -OCH2O-2,3), 4.30 (3 H, s, 7-OCH3), 4.15 (3 H, s, 8-OCH3), 5.01 (3 H, s, N-CH3). 13C NMR (100 MHz, CD3OD) δ C: 107.1 (C-1), 151.8 (C-2), 150.8 (C-3), 105.1 (C-4), 121.9 (C-4a), 132.6 (C-4b), 52.9 (N-CH3), 152.1 (C-6), 119.9 (C-6a), 147.6 (C-7), 151.8 (C-8), 127.5 (C-9), 121.0 (C-10), 130.2 (C-10a), 127.2 (C-10b), 119.5 (C-11), 132.7 (C-12), 134.4 (C-12a), 104.3 (-OCH2O-2,3), 62.8 (7-OCH3), 57.6 (8-OCH3).
Protopine (6). Colourless tetragonal crystal (CH2Cl2: MeOH = 1:1). 1H NMR (400 MHz, CDCl3) δ H: 6.90 (1 H, s, H-1), 6.69 (1 H, d, J = 7.8 Hz, H-12), 6.66 (1 H, d, J = 7.8 Hz, H-11), 6.64 (1 H, s, H-4), 5.95 (2 H, s, -OCH2O-2,3), 5.92 (2 H, s, -OCH2O-9,10), 3.78 (2 H, br s, H-13), 3.58 (2 H, br s, H-8), 2.2–3.2 (4 H, br s, H-5, 6), 1.91 (3 H, s, N-CH3). 13C NMR (100 MHz, CDCl3) δ C:108.3 (C-1), 146.5 (C-2), 148.1 (C-3), 110.6 (C-4), 132.9 (C-4a), 31.9 (C-5), 57.9 (C-6), 50.9 (C-8), 118.0 (C-8a),146.0 (C-9), 146.1 (C-10), 106.9 (C-11), 125.2 (C-12), 129.1 (C-12a), 46.6 (C-13), 195.1 (C-14), 136.3 (C-14a), 101.3 (-OCH2O-2,3), 101.0 (-OCH2O-9,10), 41.6 (N-CH3).
Allocryptopine (7). White amorphous powder. 1H NMR (400 MHz, CDCl3) δ H: 6.95 (1 H, s, H-1), 6.91 (1 H, d, J = 8.2 Hz, H-12), 6.80 (1 H, d, J = 8.2 Hz, H-11), 6.63 (1 H, s, H-4), 5.94 (2 H, s, -OCH2O-2,3), 3.86 (3 H, s, 10-OCH3), 3.78 (3 H, s, 9-OCH3), 3.73 (2 H, br s, H-13), 3.0−3.4 (2 H, br s, H-8), 2.2−3.0 (4 H, br s, H-5, 6), 1.86 (3 H, s, N-CH3). 13C NMR (100 MHz, CDCl3) δ C: 110.6 (C-1), 146.2 (C-2), 148.2 (C-3), 109.4 (C-4), 136.2 (C-4a), 32.5 (C-5), 57.7 (C-6), 50.3 (C-8), 128.7 (C-8a), 151.7 (C-9), 147.6 (C-10), 110.8 (C-11), 127.9 (C-12), 129.8 (C-12a), 46.5 (C-13), 193.5 (C-14), 133.0 (C-14a), 101.3 (-OCH2O-2,3), 60.9 (9-OCH3), 55.8 (10-OCH3), 41.4 (N-CH3).
Antiproliferative activity
The HL-60 (human leukaemia cell lines), MCF-7 (human breast cancer cell lines), A-549 (human lung adenocarcinoma cell lines) used in this study were purchased from America Type Culture Collection, ATCC (Rockville, MD, USA). All were cultured in RPMI-1640 medium (Gibco, New York, NY, USA) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM glutamine and 10% heat-inactivated fetal bovine serum (Gibco) at 37 °C in humidified atmosphere with 5% CO2. Cytotoxic activity was evaluated by the trypan blue method against HL-60, and MTT assay against MCF-7 and A-549.
Cell cycle study
HL-60 cells in logarithmic growth were plated in 6-well plates and incubated for 24 h, then incubated with different concentrations (0, 3.75, 7.50, 15.0 μM) of 2 (DMSO only as control) at 37 °C for 72 h. The cells were washed with ice-cold PBS buffer, and then collected, fixed with 70% EtOH at −4 °C for 24 h. The fixed cells were washed with ice-cold PBS and then were treated with 100 μL RNase A at 37 °C for 30 min, and finally stained with 400 μL propidium iodide (PI) in the dark at 4 °C for 30 min. The cycle distribution analysis was performed using a flow cytometer (FACS Calibur, Becton-Dickinson, America).
Cell apoptosis analysis
HL-60 cells in logarithmic growth were plated in 6-well plates and incubated for 24 h, then incubated with different concentrations (0, 3.75, 7.50, 15.0 μM) of 2 (DMSO only as control) at 37 °C for 72 h. The cells were washed twice with ice-cold PBS buffer, and then collected. 500 μL binding buffer suspension cells were added, and finally double stained Annexin V–APC/7-AAD at room temperature for 15 min in the dark. The apoptotic cells were detected to analyze apoptosis by flow cytometry.
Mitochondrial membrane potential assay
HL-60 cells in logarithmic growth were plates in 6-well and incubated for 24 h, then incubated with different concentrations (0, 3.75, 7.50, 15.0 μM) of 2 (DMSO only as control) at 37 °C for 48 h. The cells were washed with ice-cold PBS buffer, subsequently collected and adjusted the cells concentration to 1 × 106/ml, and finally stained according to the manufacture’s instruction (Keygen, KGA601, Nanjing, China) with JC-1. The percentage of green fluorescence was detected by the flow cytometry to analyze the cells apoptosis and collapsed mitochondrial membrane potentials.
References
Qing, Z. X. et al. Structural speculation and identification of alkaloids in Macleaya cordata fruits by high-performance liquid chromatography/quadrupole-time-of-flight mass spectrometry combined with a screening procedure. Rapid Commun. Mass Spectrum 28, 1033–1044 (2014).
Zeng, J. G. et al. Integration of transcriptome, proteome and metabolism data reveals the alkaloids biosynthesis in Macleaya cordata and Macleaya microcarpa. PloS One 8, 1–18 (2013).
Liu, M. et al. In vitro assessment of Macleaya cordata crude extract bioactivity and anticancer properties in normal and cancerous human lung cells. Exp. Toxicol. Pathol. 65, 775–787 (2013).
Yu, X. L. et al. Alkaloids from the tribe Bocconieae (Papaveraceae): A chemical and biological review. Molecules 19, 13042–13060 (2014).
Sai, C. M. et al. Two pairs of enantiomeric alkaloid dimers from Macleaya cordata. Org. Lett. 17, 4102–4105 (2015).
Qing, Z. X. et al. Research progress on mass spectra fragmentation behaviour of alkaloids in Macleaya cordata. Tradit. Herb. Drugs 44, 2929–2939 (2013).
Wu, Z. D. et al. Enantiomeric lignans and neolignans from Phyllanthus glaucus: Enantioseparation and their absolute configurations. Sci. Rep. 6, https://doi.org/10.1038/srep 24089 (2016).
Luo, Q. et al. (±)-Sinensilactam A, a pair of rare hybrid metabolites with Smad3 phosphorylation inhibition from Ganoderma sinensis. Org. Lett 17, 1565–1568 (2015).
Wang, K. B. et al. A series of β-carboline alkaloids from the seeds of Peganum harmala show G-quadruplex interactions. Org. Lett. 18, 3398–3401 (2016).
Sai, C. M. et al. Racemic alkaloids from Macleaya cordata: structural elucidation, chiral resolution, and cytotoxic, antibacterial activities. RSC Adv 6, 41173–41180 (2016).
Li, D. H. et al. Synthesis, biological activity, and apoptotic properties of NO-donor/enmein-type ent-kauranoid hybrids. Int. J. Mol. Sci. 17, https://doi.org/10.3390/ijms17060747 (2016).
Tran, T. D. et al. Lignans from the Australian endemic plant Austrobaileya scandens. J. Nat. Prod. 79, 1514–1523 (2016).
Luo, R. et al. Synthesis and biological evaluation of baicalein derivatives as potent antitumor agents. Bioorg. Med. Chem. Lett. 24, 1334–1338 (2014).
Yu, X. L. et al. Usnic acid derivatives with cytotoxic and antifungal activities from the lichen Usnea longissimi. J. Nat. Prod. 79, 1373–1380 (2016).
Kang, W. J. et al. New chalcone and pterocarpoid derivatives from the roots of Flemingia philippinensis with antiproliferative activity and apoptosis-inducing property. Fitoterapia 112, 222–228 (2016).
Zhao, N. et al. Antiproliferative activity and apoptosis inducing effects of nitric oxide donating derivatives of evodiamine. Bioorg. Med. Chem. 24, 2971–2978 (2016).
Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (Grant No. 81172958), the Shandong Natural Science Foundation (No. ZR2017LC003), and the Foster Foundation of Jining Medical University (No. JYP 201716). We thank Prof. Jincai Lu of School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University for identification of the plant. We gratefully acknowledge Mr. Yi Sha and Mrs. Wen Li, Department of Analytical Testing Center, Shenyang Pharmaceutical University, for measurements of the NMR data.
Author information
Authors and Affiliations
Contributions
S.C. and H.H. conceived the project, and designed the experiments. S.C. performed the main experiments, data analyzes, and wrote the manuscript; L.S. assisted the chiral HPLC resolutions; S.C., L.Z. and H.H. analyzed the spectroscopic data; L.D., H.T., and G.Y. performed the antiproliferative activity and mechanism experiments. S.C. and H.H. revised and polished this manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sai, C., Li, D., Li, S. et al. LC-MS guided isolation of three pairs of enantiomeric alkaloids from Macleaya cordata and their enantioseparations, antiproliferative activity, apoptosis-inducing property. Sci Rep 7, 15410 (2017). https://doi.org/10.1038/s41598-017-15423-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15423-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
