
Abstract
Herein, we report the synthesis of chromium oxide nanoparticles, α -Cr2O3 NPs, followed by full characterization via XRD, SEM, XPS, and N2 sorptiometry. The synthesized nanoparticles were tested as catalysts toward the oxidation of CO. The impact of calcination temperature on the catalytic activity was also investigated. CO conversion (%), light-off temperature, T50, data were determined. The results revealed that chromia obtained at low calcination temperature (400 °C) is more active than those obtained at high calcination temperatures (600° or 800 °C) and this is ascribed to the smaller particle size and higher surface area of this sample. The results revealed a superior catalytic activity of Cr2O3 NPs at lower temperature as we reached a complete conversion at 200 °C which is high value in the forefront of the published results of other non-noble catalysts. The high activity of Cr2O3 nanoparticles (T50 as low as 98 °C) where found to be dependent on a careful selection of the calcination temperature. These results may provide effective and economic solutions to overcome one of the major environmental threats.
Similar content being viewed by others
Introduction
The primary pollutants from vehicles comprised of carbon monoxide (CO), hydrocarbons (HCs) and nitrogen oxides (NOx)1. These three harmful pollutants are major source of air pollution and it affects humans, vegetation, and atmosphere in number of ways. Among all types of exhaust gases carbon monoxide is most harmful2. Carbon monoxide is an odourless, colourless and toxic gas. It is also called the silent killer and its known to contributes indirectly to global warming and ozone depletion3. Thus, CO levels in the ambient air play a role in determining the air quality of a region. Noble metals are known for their high oxidation power and terms as paramount in automobile industry since the seventeenth century. So far, most of effective catalysts for this system have been reported to use the supported noble metals4,5,6,7,8,9. These catalysts exhibited high activities for CO oxidation; however, they have some disadvantages with a high cost, a limited availability and low selectivity at high temperatures. Although not as efficient as noble catalysts, some non-noble metal oxides (e.g. CeO2-based catalyst) show high activity for CO oxidation, and hence, still advantageous due to their lower cost. For example, the light-off temperature, T50, for a CoOx/CeO2 catalyst was reported to be 135 °C (i.e. 50% of CO converted to CO2 at that temperature)10. Reported also CuO-CeOx hybrid ceria catalyst for CO oxidation and showed T50 around 94 °C11. Also, Fe-Cu/CeO2 composite catalysts were tested for CO oxidation and showed T50 around 158 °C12. Also, Co3O4@CeO2 core shell cubes with optimized CeO2 shell thickness exhibited 100% conversion of CO at 190 °C in CO oxidation13.
Recently, CO oxidation at low temperature with nonprecious metal based catalysts was an important research goal14,15,16,17,18. With a special focus to Cr2O3, very rare reports disclosed its usage for CO oxidation. Ghandhi et al.19 reported the CO oxidation using Cr2O3 and they obtained T50 values of 265 °C. Ren et al.20 reported the T50 values of 200 °C-pretreated mesoporous Cr2O3 at 151 °C and they found it to be higher than those of the corresponding 400 °C-pretreated Cr2O3 which was 147 °C.
The present work aimed to produce a non-noble metal catalyst (other than the extensively studied cerium oxide catalyst) with a high activity for the CO catalytic oxidation at lower temperature. Based on a literature survey, it is the first time to utilize nano chromia as a catalyst, without doping it with other metal oxide or using a support, for CO oxidation with such low-temperature efficiency. In this context, we prepared chromia nanoparticles via simple method at different calcination temperatures. Full characterization to the synthesized nanoparticles was investigated to stand on the most promising characteristics leading to efficient catalytic activity.
Experimental part
Preparation of chromia nanoparticles
25% aqueous ammonia solution was added dropwise to a continuously stirred 0.1 M aqueous solution of the nitrate salt of Cr. The resulting mixture was stirred for another hour, then was left overnight. The precipitate thus obtained was filtrated through a Wattman filter paper (No. 42), washed with double distilled water, and dried overnight at 100 °C. The dried precipitate was ground, sealed in vials, and stored over silica gel in a desiccator till further use. The dried precipitates were treated thermally by calcination on heating in a still atmosphere of air at various temperatures (400, 600, or 800 °C) for 2 h. The calcination products thus obtained are indicated below by corresponding oxide formula and an added Arabic numeral to symbolize the temperature applied.
Characterization of Nanoparticles
The X-ray diffraction (XRD) measurements were conducted by using a Bruker AXS D8 Advance X-Ray Powder Diffractometer with a copper target and a nickel filter with Cu Kα radiation (λ = 0.154 nm). Measurements were performed in the range 20–80° (2θ). The morphology of the particles, as well as electron diffraction patterns were obtained by scanning electron microscopy (SEM) using a JEOL JSM-7001F operating at 120 kV.
X-ray photoelectron spectroscopy (XPS) surface elemental analysis was conducted using a model Thermo ESCA Lab 250xi equipped with Mg Kα radiation (1253 eV) and operated at 23 kV and 13 mA. The binding energy was referenced to the C 1s line at 284.76 eV for calibration. N2 adsorption-desorption isotherms were measured on test samples at liquid nitrogen temperature (−195 °C) using a model ASAP 2010 automatic Micromeritics sorptiometer (USA) equipped with a degassing platform.
Catalytic activity measurements
A weighed portion (250–300 mg) of the test catalyst, was placed at ambient temperature on a G1-porous quartz disc mounted in the middle of a tubular reactor (i.d. = 2 cm; length = 15 cm) equipped with a tubular, sealed sheath for a Pt/Rh thermocouple compatible with a Type-J Cole-Parmer (USA) digital thermometer for reaction temperature reading accurate to within ±2 °C. The catalyst was pre-activated by in situ heating (at 20 °C min−1) in a 200 Torr portion of O2 gas at 200 °C for 15 min. The heating was enabled by a temperature-controlled OMEGA (USA) tubular furnace mounted around the reactor. Subsequently, the gas was pumped off to 10−2 Torr, and a fresh 200 Torr portion of O2 was admitted into the reactor and maintained for 15 min, prior to further outgassing. This process of gassing and outgassing of O2 at 200 °C was repeated for two more times, before a final outgassing at 200 °C and cooling to room temperature (RT). A 300 Torr portion of the reactants mixture (CO + O2 with a 1:3 mass ratio) was expanded into the reactor at RT, then temperature was increased at 20 °C min−1 to certain higher temperatures in the range 50–400 °C keeping an almost constant interval of ca. 50 °C. Each temperature was maintained for a 15-min period through which the gas samples were withdrawn from the reaction mixture (reactants plus products). It is worth noting, that the reactor hot zone was counter-parted by a cold zone (a refrigerated water jacket) maintained at 10 °C, adopting a design similar to that described previously by Schwab et al.21.
50-μl gas samples were withdrawn from the reaction atmosphere of catalytic CO oxidation by means of a Hamilton gas syringe at different reaction temperatures (three samples at each temperature) and analyzed by a model CP-9001 Chrompack gas chromatograph (The Netherlands). The GC was equipped with a TCD detector (maintained at 120 °C) and a packed column of PORAPACK Q (maintained at 110 °C). A 99.99% pure He (KOAC) was employed as the carrier gas (20 ml min−1), and an installed MOSAIC software facilitated a computer processing of the experimental results versus pre-constructed calibration curves. For accurate quantitative results, calibration procedures are required. Calibration involves the correlation between a known concentration of a component and the resultant detector signal generated when that component is detected. Electronic integrators are used to convert the detector signal to either peak area or height. Calibration algorithms are incorporated into the integrator or computer software used for data acquisition and analysis. Thus, one creates calibration tables in the data system through the use of prompted dialogs or menu items. The simplest procedure involves creating a single-level calibration and is achieved by the following process. First one optimizes the chromatographic parameters for the desired separation and identification of components (such as choice of column, detector, injection process, and oven temperature).
Results and Discussions
Characterization of chromia nanoparticles
IR spectra obtained for the as-obtained chromia, Cr2O3-RT, and its calcination products at 400–800 °C are shown in Fig. 1. IR spectrum of the Cr2O3 sample displays a strong absorption at 523 cm−1, a broad shoulder at 839 cm−1, a sharp band at 1384 cm−1, a weak shoulder at 1468 cm−1 and a weak band 1628 cm−1. The spectrum also exhibits a strong and broad band centered around 3416 cm−1. The broad band shown at 3416 cm−1 is due to υO-H of H-bonded hydroxyl groups. The peak at 1628 cm−1 can be assigned to δH-O-H bending vibration of physically adsorbed water molecule. The band at 1467 cm−1 is attributed to υCOO of carbonate impurities species. The absorption at 1384 cm−1 is due to υNO3 − vibrations, which originate from the metal precursor. The bands below 1000 cm−1 is related to the υCr-O bands. IR spectra of the calcination products (at 400–800 °C), are significantly different from that of the as-prepared Cr2O3. The IR spectrum of Cr2O3-4 only exhibits two sharp, strong bands at 640 and 581 cm−1, in addition to a much weaker absorption at 3453 cm−1. Upon further increase of the calcinations temperature up to 800 °C two additional low-frequency bands emerge at 445 and 414 cm−1. The four absorption at 661, 574, 445 and 414 cm−1 are assignable to υCr−O lattice vibration of α-Cr2O3 crystallites22.

FT-IR spectra of the as-obtained Cr2O3 and its calcined products at 400°, 600° and 800 °C.
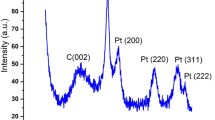
XRD diffractogram recorded for the as-obtained Cr2O3 and its calcination products (400–800 °C) are exhibited in Fig. 2. It is obvious from the results that the as-obtained Cr2O3 is largely non-crystallite, which is reflected in its gelatinous nature. The calcination products are shown to be crystalline. The crystallinity is shown to increase with calcination temperature. In spite of that, the calcination products are shown to exhibit the same diffraction pattern, which is similar to that filed in JCPDS card no. 01-129423 for α-Cr2O3. The crystal size can be calculated according to Debye-Scherrer formula:
where K, a shape factor, k = 0.89, λ is the wavelength of the Cu-Kα radiations, ß is the full width at half maximum and θ is the angle obtained from 2θ values corresponding to maximum intensity peak in XRD pattern. The mean crystallite size values were found to be an increase function of temperature and equal 36 nm, 40 nm and 56 nm for Cr2O3-4, Cr2O3-6 and Cr2O3-8 respectively.

X-ray powder diffractograms for the as-obtained Cr2O3 and its calcination products (400°–800 °C). *-labelled peaks are due to the Pt/Rh sample holder.
XPS results obtained for a chromia, Cr2O3-4, are shown in Fig. 3. The binding energy of all the deconvoluted peaks of the studied samples are tabulated in Table 1. Cr(2p) electron binding energy values were found to be located at 586 and 587ev for Cr 2p1/2, and 576 and 579 eV for Cr 2p3/2 respectively. The lower binding energy peak located at 576 eV is assigned to the Cr3+ state24. While the peak located at 579 eV is attributed to higher oxidation state Cr6+ 24. The results indicate that despite the fact that the calcination products are essentially α-Cr2O3, the surface is partially oxidized to expose Cr≥3+ ions. Similar results have been reported by Fahim, et al.25. The Cr≥3+/Cr3+ surface atomic ratios, as were calculated from 2p XPS signal of Cr, are 0.18, 0.32, 0.42, 0.31 for Cr2O3-RT, Cr2O3-4, Cr2O3-6, and Cr2O3-8, respectively. The slight amount of surface OH-1 is due to the unavoidable physical water adsorption at the surface26. The O1s XPS spectrum showed three components with O1s binding energies at 529.7, and 531.1 eV, assigned to lattice oxygen and the peak at 533.2 eV is assigned to surface defects and/or adsorbed surface hydroxyls27. The O/Cr surface atomic ratio were found to be 4.6, 2.8, 2.1, and 2.1 for Cr2O3-RT, Cr2O3-4, Cr2O3-6, and Cr2O3-8, respectively. The higher ratio at low calcination temperature is expected and it is due to the formation of Cr2O3.nH2O structure (oxide-hydroxide structure). This is also evident from the presence of two type of O2− signals shows in Table 1, the lower binding energy one’s is believed to be for surface lattice oxygen from the nonhydrated Cr2O3 formula while the higher bending energy is from water associated with the chromia formula (i.e. hydrated Cr2O3). The third peak at around 533 eV are corresponding to the OH−/H2O species physisorbed at the surface. In other words, beside the XPS signal of the lattice oxygen, there is two other signals resulted from two type of water interacting with the catalyst surfaces. One resulted from chemisorbed water encouraged by the presence of lattice defects/vacancies and the other is due to physisorbed water molecules experiencing dipole-dipole interactions with the lattice oxygen.

Deconvoluted XPS peaks of (A) O (1s) and (B) Cr (2p) and for Cr2O3-4.
We believe that, the chemical composition of Cr2O3-4 sample play a major role in this, lowest reported, light-off temperature (98 °C) for undoped and unsupported chromia (i.e. the occurrence of the metal oxide surfaces of sample Cr2O3-4 at a transition intermediate between Cr2O3.nH2O ⇔ Cr2O3 play a major role in such low light-off value).
N2 adsorption-desorption isotherms determined at −195 °C on the as-obtained Cr2O3 and its calcination products (400°−800 °C) are shown in Fig. 4. It is obvious from the classification reviewed elsewhere28, the isotherms obtained are of type-IV. Type-IV isotherms imply largely mesoporous surfaces. Except for the as-obtained α-Cr2O3, i.e., non-crystalline chromia, the hysteresis loop displayed has closure points at p/p◦ < 0.45 which may account for narrow mesopores. Hysteresis loops exhibited by as-obtained α-Cr2O3 and its calcination products (400°–600 °C) are of type-H2. Whereas α-Cr2O3-8 displays type-H3 loop. Type-H2 loop is believed to be associated with ink-bottle-like pores of varying radius, often generated by agglomerates or compacts of spheroidal particles of non-uniform size and arrangement. A type-H3 loop is attributed to adsorbate condensation in capillary spaces between parallel plates or open slit-shaped capillaries.

N2 adsorption-desorption isotherms for the as-obtained Cr2O3 and its calcination products.
The SB values observed for the as-obtained Cr2O3 is 127 m2/g. Upon calcination at 400°−800 °C, the SB value decreased to 29 and further to 17 m2/g (see Table 2). A SB ≈ SC indicates presence of cylindrical mesopores while the result (SC > SB) exhibited by Cr2O3-8 implies the presence of non-cylinderical mesopores. The Cr2O3 showed lowest pore volume value (0.0385 cm3/g) at 800 °C (Table 2), which is a direct result of the high temperature calcination and consequent sintering of particles. The as-obtained Cr2O3 itself shows the highest pore volume (0.235 cm3/g). Upon calcination (400°–800 °C) α-Cr2O3 particles are produced, which exhibit a higher V c p compared to V p thus implying the presence of non-cylindrical mesopores (see Table 1). The as-obtained α-Cr2O3 shows narrower pore size than the calcined ones. As the calcination temperature increases from 400° up to 800 °C, a pore widening is observed (r p /nm = 11 → 23 nm). Concomitantly, a drop in pore volume (0.0937 → 0.0385 cm3/g) is observed. This supports a pore widening mechanism (Table 2).
SEM images obtained for as-obtained Cr2O3 and its calcination products at 400°−800 °C are shown in Fig. 5. SEM indicates that the as-obtained Cr2O3 and Cr2O3-4 products exhibit irregular clumps of particles of ill-defined contours, whereas the calcined samples at ≥600°C produce a different morphology characterized by uniform particle size showing quazi-spherical particles. It is also seen that the size of these particles increases with calcination emperature from 600° to 800 °C.

SEM micrographs for the as-obtained Cr2O3 and its calcined products at 400°, 600° and 800 °C.
CO oxidation activity
On chromia surfaces, redox couples of Cr3+/Cr>+3are important catalytic oxidation sites29. Figure 6A compares plots of CO conversion versus reaction temperature for chromia catalysts derived from Cr2O3 at various calcination temperatures.

(A) Plots of CO conversion vs. reaction temperature for Cr2O3 nanoparticles at different calcination temperatures. (B) The Turn Over Number of the three-studied catalysts at 100 °C.
The results brought about reveal that CO oxidation on the chromia catalysts occurs actively at low temperatures as shown in Fig. 6A. T100 is the temperature at which 100% of CO was oxidized and T50 is the light-off temperature and their corresponding values are represented in Table 3 in which the present results were compared with their corresponding non-noble catalysts reported in the literature. Our results also reveal that chromia obtained at low calcination temperature (400°C) is more active than those obtained at high calcination temperatures (600° or 800 °C). From the XPS data, we found that, the higher the calcination temperature, the lower the surface content of Cr>+3 sites. Since it is known that on chromia surfaces intimately coupled Cr3+-Cr>+3 sites facilitate the electron mobility required for CO oxidation, and, thus, considered the active sites29. Hence, the drop in activity upon increasing calcination temperature may be linked to the concomitant loss or excess changes on chromia surfaces. It is worth noting, that the three test samples of chromia are of comparable surface area, thus the surface area may not be a key parameter in shaping up the CO oxidation activity on the present test samples. Moreover, chromia is an acidic oxide, and, hence, surface basic sites are also of less importance to the surface catalytic activity in the present case.
The results presented in Fig. 6B, are for the intrinsic (i.e. exclude the specific surface area from consideration) activity of the calcined catalyst. In other words, the graphs show the turn over number, TON, per CO molecules per min per m2. Accordingly, TON-based ranking of the test oxides shows that at 100 °C, the Cr2O3-4 samples exhibit a better intrinsic activity than Cr2O3-6 and Cr2O3-8. This means that, among the studied catalysts, Cr2O3-4 exhibits the best activity and 100 °C intrinsic activity. It is worth mentioning that, from the XPS results, the Cr≥3+/Cr3+ surface atomic ratios of Cr2O3-4 is 0.32 which is equal to that found for Cr2O3-8 (0.31) but less than that of Cr2O3-6 (0.42). This means that, for the studied samples, the Cr≥3+/Cr3+ surface ratio is not the only factor that control the intrinsic activity and other factors are playing roles. This also support our conclusion that the occurrence of Cr2O3-4 at the transition intermediate between Cr2O3.nH2O ⇔ Cr2O3 is the determining factor here. The different morphological structure of the Cr2O3-4 catalyst (see Fig. 5) may also play role in such superiority30.
Kinetic catalysis studies have shown the pathway adopted by CO oxidation on metal oxides to be dependent on the reaction temperature regime applied31: (i) at ≤150 °C, Eley-Rideal mechanism (CO(g) + O2(ads) or O2(g) + CO(ads), ∆E ≤ 10 kCal/mol; (ii) at 150–250 °C, Langmuir-Hinshelwood mechanism (CO(ads) + O2(ads)), ∆E ca. 10–20 kCal/mol; and (iii) at ≥250 °C, Mars-van-Krevelen mechanism (CO(g) + O(lattice)), ∆E ≥ 20 kCal/mol. Based on the studied temperature, the Langmuir–Hinshelwood mechanism is expected (both the reactants (CO and O2) are adsorbed on the catalyst surface and O2 get activeated and reacted with the CO. The involvement of lattice oxygen is rolled out since 100% CO conversion is reach at relatively low temperature (200–300 °C). The four elementary steps of the Langmuir-Hinshelwood mechanism of CO oxidation. with the following elementary reaction Eqs (2–5):
where “X” denotes the active site.
Conclusion
In this study, we reported a new CO oxidation catalyst of Cr2O3 nanoparticles prepared via hydrothermal technique. The effect of calcination temperature on the crystal structure and composition was investigated via XRD and XPS measurements. The catalytic activities of Cr2O3 calcined at different temperature were determined and revealed superior activity (T50 as low as 98 °C) was achieved for Cr2O3-4 samples that occur at the intermediate composition transition between Cr2O3.nH2O ⇔ Cr2O3 (i.e. hydrated to nonhydrated chromite transition). We attributed this superior activity to this chemical composition nature in addition to its other surface characteristics. The results of this study will make its applicability to be economically possible in automotive exhausts and factory chimneys.
References
Heck, R. M. & Farrauto, R. J. Automobile exhaust catalysts. Applied Catalysis A: General 221, 443–457, https://doi.org/10.1016/S0926-860X(01)00818-3 (2001).
Brugge, D., Durant, J. L. & Rioux, C. Near-highway pollutants in motor vehicle exhaust: A review of epidemiologic evidence of cardiac and pulmonary health risks. Environmental Health 6, 23–23, https://doi.org/10.1186/1476-069x-6-23 (2007).
Mohan Kumar, S., Sampath, S., Jeena, V. S. & Anjali, R. Carbonmonoxide pollution levels at Environmentally Different Sites. J. Ind. Geophys. union 12, 31–40 (2008).
Mariño, F., Descorme, C. & Duprez, D. Noble metal catalysts for the preferential oxidation of carbon monoxide in the presence of hydrogen (PROX). Applied Catalysis B: Environmental 54, 59–66, https://doi.org/10.1016/j.apcatb.2004.06.008 (2004).
Kim, Y. H., Park, E. D., Lee, H. C., Lee, D. & Lee, K. H. Preferential CO oxidation over supported noble metal catalysts. Catalysis Today 146, 253–259, https://doi.org/10.1016/j.cattod.2009.01.045 (2009).
Soni, K. C., Krishna, R., Chandra Shekar, S. & Singh, B. Catalytic oxidation of carbon monoxide over supported palladium nanoparticles. Applied Nanoscience 6, 7–17, https://doi.org/10.1007/s13204-015-0419-5 (2016).
Schubert, M. M. et al. CO Oxidation over Supported Gold Catalysts—“Inert” and “Active” Support Materials and Their Role for the Oxygen Supply during Reaction. Journal of Catalysis 197, 113–122, https://doi.org/10.1006/jcat.2000.3069 (2001).
Wang, F. Effect of support carbon materials on Ag catalysts used for CO oxidation in the presence and absence of H2. Journal of Environmental Chemical Engineering 4, 4258–4262, https://doi.org/10.1016/j.jece.2016.08.012 (2016).
Allian, A. D. et al. Chemisorption of CO and Mechanism of CO Oxidation on Supported Platinum Nanoclusters. Journal of the American Chemical Society 133, 4498–4517, https://doi.org/10.1021/ja110073u (2011).
Royer, S. & Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. Chem Cat Chem, 3, 24–65, https://doi.org/10.1002/cctc.201000378 D. (2011)
Chen, S. X., Zhao, S. F., Xu, Z., Liu, Z. G. & Zhu, R. L. Influence of pH on the catalytic performance of CuO-CoOx-CeO2 for CO oxidation. RSC Adv. 5, 61735–61741 (2015).
Hinokuma, S., Yamashita, N., Katsuhara, Y., Kogami, H. & Machida, M. CO oxidation activity of thermally stable Fe-Cu/CeO2 catalysts prepared by dual-mode arc-plasma process. Catal. Sci. Technol. 5, 3945–3952 (2015).
Zhen, J. et al. Co3O4@CeO2 core@shell cubes: Designed synthesis and optimization of catalytic properties. Chemistry A European Journal 20, 4469–4473 (2014).
Hinokuma, S., Yamashita, N., Katsuhara, Y., Kogami, H. & Machida, M. C. O. oxidation activity of thermally stable Fe-Cu/CeO2 catalysts prepared by dual-mode arc-plasma process. Catalysis Science & Technology 5, 3945–3952, https://doi.org/10.1039/c5cy00370a (2015).
Xie, X., Li, Y., Liu, Z.-Q., Haruta, M. & Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 458, 746–749, http://www.nature.com/nature/journal/v458/n7239/suppinfo/nature07877_S1.html (2009).
Li, Y., Peng, H., Xu, X., Peng, Y. & Wang, X. Facile preparation of mesoporous Cu-Sn solid solutions as active catalysts for CO oxidation. RSC Advances 5, 25755–25764, https://doi.org/10.1039/c5ra00635j (2015).
Gonçalves, R. V. et al. Easy Access to Metallic Copper Nanoparticles with High Activity and Stability for CO Oxidation. ACS Applied Materials & Interfaces 7, 7987–7994, https://doi.org/10.1021/acsami.5b00129 (2015).
Shinde, V. M. & Madras, G. Kinetics of carbon monoxide oxidation with Sn0.95M0.05O2-[small delta] (M = Cu, Fe, Mn, Co) catalysts. Catalysis Science & Technology 2, 437–446, https://doi.org/10.1039/c1cy00421b (2012).
Shelef, M., Otto, K. & Gandhi, H. The oxidation of CO by O2 and by NO on supported chromium oxide and other metal oxide catalysts. Journal of Catalysis 12(4), 361–375, https://doi.org/10.1016/0021-9517(68)90121-8 (1968).
Ren, Y. et al. Ordered Crystalline Mesoporous Oxides as Catalysts for CO Oxidation. Catalysis Letters 131(1), 146–154, https://doi.org/10.1007/s10562-009-9931-0. (2009).
Schwab, G.-M. & Karatzas, A. Katalytische Wirkung Intermetallischer Phasen und Ihrer Mischungen. Zeitschrift für Elektrochemie und angewandte physikalische Chemie 50(11–12), 242–249 (1944).
Marchall, R., Mitra, S. S., Gielisse, P. J., Plendi, J. N. & Mansur, L. C. Infrared lattice spectra of a-Al2O3 and Cr2O3. J. Chem. Phys. 43, 2893–2894 (1965).
Zhao, G. et al. Two-dimensional Cr2O3 and interconnected graphene-Cr2O3 nanosheets: synthesis and their application in lithium storage. Journal of Materials Chemistry A 2, 944–948, https://doi.org/10.1039/c3ta13535g (2014).
Gupta, P., Bhargava, R., Das, R. & Poddar, P. Static and dynamic magnetic properties and effect of surface chemistry on the morphology and crystallinity of DyCrO3 nanoplatelets. RSC Advances 3, 26427–26432, https://doi.org/10.1039/c3ra43088j (2013).
Fahim, R. B., Gabr, R. M., Zaki, M. I. & Mansour, S. A. A. Nonstoichiometry and surface characterization of chromia gel. Journal of Colloid and Interface Science 81, 468–476, https://doi.org/10.1016/0021-9797(81)90428-8 (1981).
Moulder, T. F., Stickle,W. F., Sobol, P. E. and Bomben, K. D. Handbook of X-ray photoelectron spectroscopy. Perkin Elmer, Eden Prairie, Minnesota,1992.
Yao, H. B., Li, Y. & Wee, A. T. S. An XPS investigation of the oxidation/corrosion of melt-spun Mg. Applied Surface Science 158, 112–119, https://doi.org/10.1016/S0169-4332(99)00593-0 (2000).
Lecloux, A. J. In Catalysis-Science and Engineering, Anderson, Vol. 2, J. R. & Boudart, M. (Eds), Berlin: Springer-Verlag, pp. 171–229 (1981).
Zaki, M. I., Hasan, M. A. & Fouad, N. E. Stability of surface chromate – A physicochemical investigation in relevance to environmental reservations about calcined chromia catalysts. Applied Catalysis A: General 171, 315–324, https://doi.org/10.1016/S0926-860X(98)00088-X (1998).
Yu Yao, Y.-F. The Oxidation of Hydrocarbons and CO Over Metal Oxides II. alpha-Cr2O3. J. Catal. 28, 139–149 (1973).
Zaki, M. I., Hasan, M. A. & Pasupulety, L. Influence of CuOx additives on CO oxidation activity and related surface and bulk behaviours of Mn2O3, Cr2O3 and WO3 catalysts. Applied Catalysis A: General 198, 247–259 (2000).
Pantaleoa, G. et al. Support effect on the structure and CO oxidation activity of Cu-Cr mixed oxides over Al2O3 and SiO2. Materials Chemistry and Physics 114, 604–611 (2009).
Yao, Y. et al. Enhanced low temperature CO oxidation by pretreatment: specialty of the Au-Co3O4 oxide interfacial structures. Catalysis Science & Technology. 6(7), 2349–60 (2016).
Zhou, L. et al. Transition-Metal Doped Ceria Microspheres with Nanoporous Structures for CO Oxidation. Scientific Reports 6, 23900, https://doi.org/10.1038/srep23900 (2016).
Cai, L., Hu, Z., Branton, P. & Li, W. The effect of doping transition metal oxides on copper manganese oxides for the catalytic oxidation of CO. Chinese Journal of Catalysis 35, 159–167, https://doi.org/10.1016/S1872-2067(12)60699-8 (2014).
Avgouropoulos, G. et al. A comparative study of Pt/γ-Al2O3, Au/α-Fe2O3 and CuO–CeO2 catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catalysis Today 75, 157–167, https://doi.org/10.1016/S0920-5861(02)00058-5 (2002).
Mountapmbeme Kouotou, P. et al. Structure-activity relation of spinel-type Co-Fe oxides for low-temperature CO oxidation. Catalysis Science & Technology 4, 3359–3367, https://doi.org/10.1039/c4cy00463a (2014).
Mountapmbeme Kouotou, P., Tian, Z.-Y., Vieker, H. & Kohse-Höinghaus, K. Pulsed-spray evaporation CVD synthesis of hematite thin films for catalytic conversion of CO. Surface and Coatings Technology 230, 59–65, https://doi.org/10.1016/j.surfcoat.2013.06.023 (2013).
Zhao, Z., Yung, M. M. & Ozkan, U. S. Effect of support on the preferential oxidation of CO over cobalt catalysts. Catalysis Communications 9, 1465–1471, https://doi.org/10.1016/j.catcom.2007.12.013 (2008).
Acknowledgements
The authors gratefully acknowledge the support of Kuwait University Research Administration, college of graduate studies and RSPU Facilities No. (GS 01/01, GS 03/01, GS 01/05 and GS 02/08 and GE 03/08). In addition, Nanoscopy Science Centre is highly acknowledged.
Author information
Authors and Affiliations
Contributions
S.A. performed the experimental work. A.B. and F.A. proposed the idea and drafted the manuscript. A.B. and M.M. wrote the manuscript text and M.M. prepared the figures. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bumajdad, A., Al-Ghareeb, S., Madkour, M. et al. Non-noble, efficient catalyst of unsupported α-Cr2O3 nanoparticles for low temperature CO Oxidation. Sci Rep 7, 14788 (2017). https://doi.org/10.1038/s41598-017-14779-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14779-x
This article is cited by
-
Growth of Cr2O3 on n-Silicon Substrate using AACVD and its Application as a Hole Selective Layer
Silicon (2024)
-
Flower-like Cr2O3-Cr(OH)3/poly-2-chloroaniline nanocomposite photoelectrode grown on polypyrrole film for hydrogen generation from sewage water
Optical and Quantum Electronics (2024)
-
Mist CVD-based growth of crystalline alpha chromium oxide (α-Cr2O3) on c-plane sapphire substrate with chromium acetylacetonate as a precursor
Chemical Papers (2023)
-
Synthesis and characterization of PANI-ZrWPO4 nanocomposite: adsorption-reduction efficiency and regeneration potential for Cr(VI) removal
Environmental Science and Pollution Research (2023)
-
Evolution of micro-pores in Ni–Cr alloys via molten salt dealloying
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
