
Abstract
Ammonia borane (AB) is among the most promising precursors for the large-scale synthesis of hexagonal boron nitride (h-BN) by chemical vapour deposition (CVD). Its non-toxic and non-flammable properties make AB particularly attractive for industry. AB decomposition under CVD conditions, however, is complex and hence has hindered tailored h-BN production and its exploitation. To overcome this challenge, we report in-depth decomposition studies of AB under industrially safe growth conditions. In situ mass spectrometry revealed a time and temperature-dependent release of a plethora of NxBy-containing species and, as a result, significant changes of the N:B ratio during h-BN synthesis. Such fluctuations strongly influence the formation and morphology of 2D h-BN. By means of in situ gas monitoring and regulating the precursor temperature over time we achieve uniform release of volatile chemical species over many hours for the first time, paving the way towards the controlled, industrially viable production of h-BN.
Similar content being viewed by others

Introduction
Hexagonal boron nitride (h-BN) is a layered ceramic material with an atomic structure similar to that of graphite, but with a wide band gap of 5.7 eV1. Monolayer or few-layer h-BN has received considerable attention recently as a substrate for graphene and other 2D-nanomaterials. Due to the low number of charge impurities in h-BN the mobility of supported graphene improved by more than an order of magnitude2 compared to graphene on the commonly used silicon dioxide. Highly crystalline h-BN domains serve as a better template for the epitaxial growth of monolayer graphene3 and WS2 4, showing good alignment of the domains. The possibility of forming heterostructures wholly from the 2D family of nanomaterials opens many exciting opportunities in the electronics industry. Radio frequency electronics5, nanocapacitors6, flexible and transparent transistors7 have been demonstrated using h-BN and other 2D-nanomaterials.
The synthesis of high-quality h-BN by chemical vapour deposition (CVD) has many challenges, some of which are similar to those of graphene where the right CVD parameters have to be found. But an inherent difficulty for the controlled h-BN production arises from the limited choice of suitable precursors containing both nitrogen and boron. A variety of precursors have been investigated, some of which are diborane and ammonia8, b-tricholoroborazine9, decaborane and ammonia10 or borazine11,12,13,14,15,16,17,18. For example, borazine allowed the growth of micron-sized h-BN domains on Cu at temperatures between 950–1000 °C19 and is one of the most commonly used precursors due to its high vapour pressure. Nevertheless, these compounds are known to be toxic, highly flammable, corrosive, unstable, or difficult to store, presenting obvious safety concerns, practical challenges and higher processing costs. For this reason, ammonia borane (AB) precursor, classed as a non-hazardous substance or mixture, has been gaining popularity as the precursor of choice for h-BN synthesis20,21,22,23,24,25,26,27. Often, however, AB is used in combination with high hydrogen flows, defeating the purpose of using a safe precursor.
Atmospheric pressure CVD (APCVD) is a scalable 2D method that does not require costly investment or maintenance, and is suitable for e.g. an open roll-to-roll process28. To obtain sufficient concentration of NxBy-containing species in the synthesis gas, the AB precursor must be heated to temperatures where its decomposition occurs21. Previous decomposition studies of AB focused on the suppression of NxBy-containing species for the use of AB in hydrogen storage applications29,30,31. In contrast to this, the CVD synthesis of h-BN requires efficient conversion of AB into volatile NxBy-containing species. Several studies on h-BN synthesis refer to thermoanalytical measurements31,32,33 of AB decomposition resulting in the release of H2, borazine and monomeric aminoborane, but failing to note the differences in temperatures or conditions. Additionally, such measurements are performed either in an Ar carrier gas or in vacuum. The former causes difficulties in subtracting the overlapping mass spectra background31, while the latter does not replicate the dynamic equilibrium in an APCVD system. With the increased need for controlled and scalable production of h-BN, identifying the precise nature of the precursor species in CVD is an urgent priority.
Here, we report a time dependent release of the chemical species that are generated from a commercially-available AB precursor at various temperatures (TAB) using in situ mass spectrometry (MS). We find that seven N and B-containing species are present in the temperature range typically used for h-BN growth at atmospheric pressure. Moreover, their time-dependent flow profiles do not correlate with each other, suggesting rather complicated reaction pathways. We also find that at higher temperatures (TAB >80 °C) the time-dependent profiles show a rapid peaking and decaying behaviour (within minutes), while at lower temperatures (TAB: 60–80 °C) the precursor decomposition is slow and varies on the order of hours. Experimental observations of h-BN deposits from different stages of the precursor decomposition show a variation in the growth rate and shape. As such, reproducible production of high quality h-BN can be difficult to achieve unless the release of volatile species is controlled. We also report solutions for controlling the occurrence of the volatile NxBy-containing species with good accuracy by regulating the precursor temperature with time to achieve reproducibility and larger h-BN domains (~50 µm).
Results
Identification of the chemical species released from AB decomposition
The decomposition of the AB precursor was initially measured in different gas atmospheres to determine targeted information. First, the mass spectrum was recorded in pure H2 carrier gas to avoid the background resulting from the Ar carrier gas. The system was thoroughly purged (>12 hours) and the background was subtracted before AB was heated (TAB = 100 °C). The resulting spectrum when the decomposition reaction reached a maximum is shown in Fig. 1a. AB gas (m/z 31) was not detected due to low vapour pressure at this temperature, compared to its decomposition products31. Some of the observed species from the plethora of fragments are commonly-known chemical compounds and their patterns can be identified. Currently there are no reports that detect all of these species simultaneously and assess their influence on h-BN synthesis. The readily identified chemical species were ammonia34 (NH3, Fig. 1d); borazine31,35 (N3B3H6, Fig. 1h), borane (BH3, Fig. 1b) and diborane34 (B2H6, Fig. 1c). We find negligible quantity (<0.3%) of borazine in our measurements (Fig. 1a,h), in agreement with previous reports where borazine is mainly produced at higher precursor temperatures of TAB > 130 °C35. Little identification data is available relating to the two remaining fragmentation patterns between m/z 26–30 and m/z 36–44. Monomeric aminoborane, BNH4, matches with the m/z 26–30 fragment and has been reported in previous studies31,35. The species in the m/z 36–44 range preliminary matched with convoluted patterns of hydrogen-rich triborane36 (B3Hx) and aminodiborane37,38 (NB2Hx). To confirm presence of these two species and to identify their fragmentation patterns an experiment was designed where the spectrum was recorded in a H2/NH3 atmosphere, such that NH3 was highly in excess. It is expected that excess ammonia can shift the chemical equilibrium of the reaction towards BxNyHz containing species. Notably, the B2H6 and B3Hx fragmentation patterns disappeared (Supplementary Figure 1) allowing us to deconvolute them from the overlapping BxNyHz species. Fragmentation intensities of monomeric aminoborane, triborane and aminodiborane are thus identified as shown in Fig. 1e–g respectively (Supplementary Table 1). It must be noted that no other fragments were observed at different temperatures (TAB: 50–110 °C), only their intensity varied depending on the temperature and time, as will be discussed later in the text.

Mass spectrometry analysis of AB decomposition. (a) Mass spectrum of ammonia borane decomposition when heated to 100 °C in the background-free H2 atmosphere at ambient pressure. NxBy-containing species are assigned to different fragmentation patterns. The patterns between m/z 21–30 are deconvoluted to diborane and monomeric aminoborane; and the patterns between m/z 31–45 are deconvoluted to aminodiborane and triborane as discussed in the main text. The intensity profiles between m/z 50 and 80 (borazine) are magnified ten times. (b–h) Bar plots of the deconvoluted intensity profiles assigned to various chemical species.
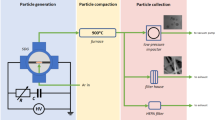
While secondary decomposition and deposition during the transfer of molecules to the MS may be possible, it is expected to be low since the majority of the detected species are stable in the timeframe of the measurement. For example, a reaction between ammonia and diborane requires heating at 200 °C for several hours39 and borazine decomposes >340 °C40. We estimate that it takes ca. 30 s for the gas molecules to travel through the precursor container and the manifold to the position of the MS sampling capillary (also furnace), however, it only takes ca. 0.3 s to travel through the sampling capillary. Such configuration ensures the best match between the measured species and the species actually entering the reaction chamber during the synthesis. Additionally, the temperatures of the precursor chamber and the transfer manifold (including the sampling capillary) were controlled separately allowing to maintain comparable molecule flight path conditions at different precursor temperatures. The detailed CVD system configuration and the procedure for the MS decomposition measurements are described in the methods section.
Temporal profiles of AB decomposition
Once the chemical species were identified in the pure H2 and ammonia-rich H2 atmospheres, follow-on measurements were performed in a non-flammable, industrially-safe 2.5 % H2 in Ar gas mixture to match exactly the CVD synthesis experiments for h-BN. To improve reproducibility and to avoid AB foaming, the precursor was mixed with an inert filler (methods). Exemplar time-dependent evolution profiles of all the significant NxBy-containing species released at a precursor temperature of TAB = 90 °C over 2 hours are shown in Fig. 2a–f. Notably, the anticipated products of AB decomposition: NH3 and B2H6 appear first after around 10 minutes (including a 5 minute 50 °C pre-heating time; methods), as shown in Fig. 2a,b. The latter can be considered a product of two borane molecules that rapidly react41. This simple reaction pathway corresponds to the symmetrical splitting of the AB molecule,

Time-dependent flow evolution profiles of individual NxBy-containing species released at TAB = 90 °C. (a–f) Molar flows of ammonia, diborane, triborane, aminodiborane, borane and monomeric aminoborane respectively; showing variation throughout the time of measurement. Aminoborane becomes the dominant species at peak decomposition, while aminodiborane, borane and borazine have low contributions to the total flow.
As time progresses, other species start to appear. After ca. 30 minutes, there is a rapid increase in their intensity, most significantly triborane and aminoborane (Fig. 2c,f). As will be shown later in the text the profiles measured at different temperatures also displayed two such stages. At lower temperatures (TAB ≤ 90 °C) these stages could be deconvoluted in time, whereas at higher temperatures the transition took place very quickly. These observations suggest that the precursor changes in the duration of the measurement. Previous studies42 showed that AB indeed undergoes a solid-state phase change to its ionic isomer, diammoniate of diborane (DADB), albeit in different experimental conditions. This phase is more mobile42, explaining the observed increase in total flow of NxBy-containing species. The following chemical reaction describes this solid-state phase change:
To further confirm the phase change of AB during heating (TAB = 90 °C) we recorded Fourier transform infrared spectra (FTIR) of the precursor at different stages of its decomposition: (i) pristine precursor, (ii) at the maximum flow rate of volatile species (partially decomposed), and (iii) after full decomposition (Fig. 3). The FTIR features can be readily assigned to different modes43. Fundamentally different bonding between B-N in AB and DADB means that these can be distinguished from the B-N stretch mode in the 600 cm−1–900 cm−1 region43. AB has three characteristic peaks at 726 cm−1, 782 cm−1 and 798 cm−1, while DADB has two wider separated main peaks at ~667 cm−1, ~853 cm−1 and two smaller peaks43. Notably, AB is no longer detected when the release of N and B-species reaches a maximum, only peaks for DADB can be observed. After complete decomposition of the precursor, the intensity of B-H and N-H modes becomes very low and a broad feature appears in the 600–1500 cm−1 region (Fig. 3). This spectrum matches well with polyiminoborane (PIB), reported by previous studies43 and it is the solid residue that remains in the precursor chamber after full AB decomposition.

Spectroscopic precursor characterization. FTIR spectra of pristine AB (room temperature), partially decomposed AB (maximum NxBy species flow; TAB = 90 °C, 90 min) and after full decomposition (low NxBy species flow; TAB = 90 °C, 150 min). Due to the difference in B-N bonding of the AB and DADB molecules, the B-N stretch region can be used to identify AB and DADB phases.
From the exemplar profiles at TAB = 90 °C (Fig. 2a–f) it is evident that the release of N-species is dominated by ammonia during the AB decomposition phase (~80 %), but after the conversion to DADB (~90 min), the majority originates from monomeric aminoborane (~73 %). Various chemical species contribute to the presence of B atoms throughout the measurement: initially diborane and then monomeric aminoborane respectively. These results are summarised in Supplementary Table 2.
Choice of the precursor temperature for h-BN synthesis
A range of heating conditions for AB have been reported (TAB: 60 °C − 130 °C), where often the choice resulted from observations of h-BN deposits after the synthesis. Using the approach proposed here, based on time-dependent measurement of chemical species, it is now possible to systematically assess the suitability of different heating regimes for the tailored production of h-BN. For clarity in further discussion, due to the chemical similarities we distinguish the contribution to the total molar flows of N and B atoms from the “asymmetric” molecules (BH3, B2H6, B3Hx, NH3, NB2Hx, B≠N) and also a “symmetric” molecule, aminoborane (the other, heavier symmetric molecules, N2B2Hx, N3B3Hx, are ignored because of their negligible contribution). The data analysis steps are discussed in the methods section. Figure 4 shows the temporal profiles of total flows of N and B atoms from the asymmetric species and aminoborane for TAB: 60 °C − 110 °C: showing the rapid profiles at higher temperatures first followed by the slower profiles. Above 60 °C the profiles exhibit three stages: (i) an induction period where the flows of N and B are nearly constant, followed by a (ii) sigmoidal increase in the concentration of the products (as predicted by the “nucleation and growth” mechanism30 for AB polymerisation), followed by (iii) a rapid decay in the concentration. These measurements match the explanation of the solid-state phase change to DADB from previous studies42. As would be expected, different time scales and different magnitudes of N, B fluxes are observed at different precursor temperatures (TAB). At higher temperatures, the reaction is faster and produces significantly more NxBy-containing species. For example, at TAB = 70 °C (Fig. 4e), the induction period can be observed up to 400 minutes with the total flow of around 4 nmol min−1 for B atoms (similar for N), while at TAB = 90 °C (Fig. 4c) the initial phase shortens to 30 minutes and produces about 50 nmol min−1 flow of B atoms. As such, the precursor is exhausted much quicker at higher temperatures (TAB). Importantly, the ratio of flows of N to B is not consistent throughout the measurement, which is discussed later in the text.

Temporal profiles of ammonia borane decomposition at various precursor temperatures (TAB). (a–f) Profiles of molar flows of N and B from asymmetric species (left) and of N (or B) from monomeric aminoborane (right) in the temperature range between TAB: 60 – 110 °C. For temperatures above 60 °C an apparent change in precursor behaviour can be observed corresponding to the solid-state phase change from AB to DADB (overlapped for 110 °C), as discussed in main text. At lower temperatures the peak is significantly delayed, while the flux of NxBy-containing species is also lower. At 60 °C simple decomposition and decay occurs. Flows are scaled per 1 mg of AB precursor. The first 5 minutes account for pre-heating the container to 50 °C, followed by rapid heating to the set temperature (methods).
Notably, the contribution of aminoborane is very significant during the peak in the flow of volatile species for TAB > 70 °C. A number of previous reports44,45,46,47 observed small circular deposits around triangular h-BN domains, especially with APCVD44. These deposits were hypothetically attributed to monomeric aminoborane and its tendency to form oligomers35,45. Akin to amorphous C contamination if, for example, ethylene and methane precursors are used simultaneously for the production of graphene. In order to verify this assertion, we performed synthesis at the peak of the flow of volatile species (TAB = 90 °C, 90 min), where the monomeric aminoborane flow accounted for about ~55 % of the total molar flow. The furnace temperature was kept at 1070 °C for this and all other synthesis experiments (methods). We observed circular, “fuzzy” deposits at these conditions (Fig. 5a,b). The deposits either surrounded triangular h-BN domains or were evenly distributed on the surface of Cu. However, they did not significantly grow in size or interfere with the formation of an h-BN film (Fig. 5c). Nevertheless, further studies are needed to examine if these amorphous deposits can cause increased number of defects in the film. When the experiment was performed at lower precursor temperatures (TAB = 60 °C) or before the maximum decomposition rate was reached, such that monomeric aminoborane flow accounted for < 30 % of the total molar flow, only crystalline triangular deposits were observed (Fig. 5d). These observations suggest that monomeric aminoborane is not desirable and thus heating regimes and times should be selected to reduce its presence.

Monomeric aminoborane influence on the morphology of h-BN deposits. (a–c) Deposits produced with high aminoborane fraction (~55 % of the total flow) at different stages of the growth: after nucleation; with partial and full coverage. Crystalline triangular domains are surrounded with a “fuzzy”, likely amorphous deposit that does not grow in size as the triangular domains. Additionally, separate circular “fuzzy” deposits are observed. The full-coverage h-BN film does not contain the amorphous regions after triangular domains coalesce. (d) h-BN domains produced with a lower aminoborane fraction of the total volatile species flow (~30 %). White particles arise from contaminations on the Cu surface. Scale bars (a–c) 2 μm; inset: 0.5 μm; (d) 10 μm.
The profile at TAB = 60 °C does not exhibit the precursor phase change and simply decays over the course of the measurement. Analogously to graphene, the flow of any h-BN precursor should be sufficiently constant or increasing to avoid reaching a self-limiting growth regime and the inability to achieve full coverage at larger scale48. From the measurements in Fig. 4 it can be noted that the AB decomposition profiles do not have a sufficiently long region of constant volatile species flows. In comparison, for graphene synthesis on Cu, experiments were run for up to 48 hours49. As such, none of the precursor decomposition profiles is optimal for the tailored h-BN synthesis; however, the profiles TAB ≤ 70 °C are preferable and were studied further.
Sequential synthesis experiments at a constant precursor temperature
From the time-dependent precursor decomposition measurements (Fig. 4a–f), it was found that not only the molar N, B flows varied, their ratio also changed with time, which has not been reported before. The sum of N, B flows (both asymmetric and symmetric) and their ratio is shown in Fig. 6a. The concentration of B was initially lower than N in the gas atmosphere, which inverted (B > N) during the decomposition maximum (and is expected to revert to B < N in the final decomposition stage in line with the shorter profiles in Fig. 4a–d). The cause of this behaviour is unclear, but could be a result the formation of structurally different NxBy-containing species during different precursor phases. Most importantly, this finding is expected to have a profound effect on the formation of h-BN. It was established in previous studies50 that the shape of h-BN domains strongly depends on the ratio of N:B atoms. For example, when N > B, nitrogen edge termination occurs resulting in triangular shape, however, when N = B, hexagonal shapes are formed. An entirely different and more difficult question is what factors control the N:B ratio in the vicinity of growing h-BN domains. Several reasons have been proposed: different N, B dissolution rates51, substrate contamination or roughness20, different stability of the h-BN edge52, imbalance due to an external chemical, e.g., N2 carrier gas50. Our in situ studies now revealed that the AB precursor itself cannot be considered a source of a constant ratio of N to B, even though its chemical formula has a 1:1 stoichiometry.

Precursor decomposition profile and selective h-BN synthesis at 70 °C. (a) A profile for the total (symmetric and asymmetric) N and B species flows and their ratio are given for the 70 °C precursor temperature (scaled per 1 mg of AB precursor). Notably, there is a variation in the ratio of N and B flows throughout the measurement. (b) Exemplar optical images of h-BN domains (white regions) on oxidised (post-synthesis) Cu corresponding to evenly divided time intervals between 0 and 1800 minutes (as marked: 1–5). Initially only triangular domains are observed, followed by domains with various shapes: triangles, truncated triangles, hexagons or mixtures of shapes. To counter the variation in the total molar flow of N and B species, experiments were performed with different synthesis times as to obtain individual h-BN domains (30 min, 20 min, 20 mins, 15 min, 15 min). The growth temperature was 1070 °C. All the scale bars in (b) are 10 μm.
To understand how different regions of the varying AB decomposition profile (Fig. 6a) affect h-BN growth we performed systematic CVD synthesis experiments from a single precursor loading at different times. The precursor was heated to TAB = 70 °C and the Cu substrate was exposed to the released volatile vapours sampling a period of 30 hours (Tsynthesis = 1070 °C, 15–30 minutes, methods). The resulting h-BN deposits are shown in Fig. 6b. Only in the first experiment triangular domains were obtained; where the ratio of N:B was sufficiently greater than 1. Notably, AB phase change started soon after this experiment, evident by an increase in the total volatile species release. At later times (experiments 2–5, Fig. 6a,b), a variety of shapes appeared: truncated triangles, hexagons and star-like shapes. Additionally, the synthesis time had to be progressively changed to obtain individual domains and account for the varying release of volatile species.
Without the possibility to control the shape of h-BN domains it is difficult to control domain stitching, leading to inferior film crystallinity. Because of the variation in shape morphology and the level of coverage, methods need to be developed that result in a constant flux of volatile NxBy-containing species.
Precursor regulation with varying temperature
We devised a strategy to achieve constant flows of N and B atoms by varying the precursor temperature with time; which was optimised empirically from a series of measurements and is based on the constant TAB = 60 °C decomposition profile (Fig. 4f). As shown in Fig. 7a, the precursor temperature is slowly increased to counter the decay in the concentration of volatile NxBy-containing species. The initial ramping rate is very low, around 0.06 °C per hour. Nevertheless, increasing the precursor temperature by only 2 °C after ~1500 minutes started AB phase change to DADB. To prevent a significant increase in the decomposition product formation, the precursor was controllably cooled down. Again, at a very low rate of approximately 0.06 °C per hour. Finally, the precursor temperature did not need significant changes between ~3600–4200 minutes to achieve sufficient consistency. The details of the precursor temperature control points are given in Supplementary Table 3. The resulting total N, B flows (symmetric and asymmetric) and their ratio are shown in Fig. 7b. The ratio of N:B > 1 persisted from the constant TAB = 60 °C profile (Fig. 4f), while the molar flows were made sufficiently constant. As before, a series of synthesis experiments were performed at different points of the developed temperature-regulated profile (Tsynthesis = 1070 °C, 30 min, methods). In this case, most domains had triangular shape and the size of domains between different experiments was similar (Fig. 7c); without the need to adjust experimental conditions. These measurements also show that temperature deviations of one or few degrees can cause considerable differences in precursor behaviour and thus lead to irreproducibility. As such, it is important to accurately control the precursor in a separate chamber as we suggest (methods), different to the “two-stage furnace” apparatus20,21,23,27, where infrared radiation causes significant overheating. Such regulation of AB heating may be a necessary requirement for the controlled growth of h-BN, akin to the need to cool borazine to control its vapour pressure.

Time-dependent AB decomposition control with temperature regulation. (a) The temperature control chart; starting at 60 °C and running for 70 hours. (b–d) The corresponding N and B total flow profiles and the N:B ratio. (c) Optical images of h-BN domains on oxidized Cu showing that the size and shape did not change significantly in the duration of the profile over 70 hours. The synthesis time was also constant between the experiments (30 min; Tsynthesis = 1070 °C). The observation matched well with the measurement. All the scale bars in (c) are 10 μm.
Interestingly, in addition to improved shape and size uniformity, these methods allowed us to identify a strategy to re-use the precursor in multiple runs. We found that if partially exhausted precursor was cooled down and then re-heated to the same temperature, the profile of NxBy-containing species returned to the last value after a short delay (Supplementary Figure 2). This means that the same precursor loading can be used for multiple experiment by simply recording the cumulative time of precursor heating, making experiments more convenient and reducing waste, cost, and time.
Synthesis optimisation and h-BN characterization
With a uniform, reproducible precursor supply through temperature-controlled decomposition of AB, all the other CVD parameters became much more relevant and strikingly comparable to the established methods for graphene synthesis. It was possible to systematically study and optimise h-BN synthesis towards larger crystal size. We investigated three strategies for lowering the nucleation density in analogy with graphene: 1) electropolishing; 2) oxidation of Cu foil followed by Ar annealing; 3) increasing the H2 to precursor flow ratio to attain slower growth rate but reduced nucleation density. Electropolishing significantly reduced the nucleation density, from around 5 × 105 mm−2 to 103 mm−2, in agreement with previous studies20,53 and was used for all samples. Recently, Cu oxidation has been established as the mainstream route towards mm-sized graphene domains with two possible mechanism at play: Cu surface passivation54 or scavenging of impurities55 that could promote nucleation. However, our experiments showed that this approach was not directly applicable to h-BN because of the formation of elongated and distorted domains as a result (Supplementary Figure 3), suggesting O2 interference in the synthesis chemistry. Such influence indicates incompatibility of utilising O2 in h-BN growth, while it seems beneficial for graphene. However, it was possible to avoid the formation of the elongated features by further prolonged annealing in a reducing Ar/H2 atmosphere for 3 hours. This treatment allowed sufficient time to negate the effects of O2 and resulted in appreciable reduction in the h-BN domain nucleation density (103 mm−2 to 400 mm−2), possibly simply due to enhanced Cu restructuring during oxidative annealing56. Finally, the nucleation density was lowered even further by using higher H2 flows and longer synthesis times57,58. For example, 50 µm domains and nucleation density of ~4 mm−2 could be achieved with 500 sccm flow of 2.5% H2 in Ar (Tsynthesis = 1070 °C, 6 hours, methods). Two comparative images of h-BN deposits on simply electropolished Cu and on electropolished Cu with the above optimised synthesis conditions are shown in Fig. 8a,b.

Synthesis optimisation and characterisation of h-BN domains. (a,b) Comparative images of the improvement in domain size and nucleation density on simply electropolished Cu and electropolished Cu with optimised annealing and synthesis parameters (methods). Domain size is increased by one order of magnitude, while the nucleation density is reduced 3 orders of magnitude as discussed in the main text. (c) Raman spectrum of the deposits; exhibiting a significant blue-shift in the E2g peak position (1370 cm−1) and a wide FWHM (23 cm−1) compared to bulk deposits (1366 cm−1, 15 cm−1). Consistent with monolayer or very thin h-BN domains27,45,58. The scale bars in (a,b) are 50 μm.
Raman characterization (Fig. 8c) showed that the deposits exhibited a large blue shift in the E2g peak (1370 cm−1) and a large full width at half maximum (FWHM) value (23 cm−1) compared to bulk or few layer deposits (1366 cm−1 and 15 cm−1 respectively), suggesting monolayer or very thin h-BN, consistent with previous studies44,59,60. Aberration-correct transmission electron microscopy (AC-TEM) was also used to confirm the result by imaging a single layer of h-BN atoms near the domain edge which exhibited hexagonal symmetry (Supplementary Figure 4).
Discussion
In summary, we show that while ammonia borane is a safe and inexpensive precursor for h-BN synthesis, its behaviour needs to be better understood in order to develop appropriate control methods. In situ MS measurements revealed that the stochastic polymeraization and a solid-state phase change of AB are accompanied by the release of multiple volatile NxBy-containing species upon heating. Moreover, their abundance and the ratio of N:B atoms are not constant with time. Different species can produce different types of deposits, while the time-dependent variation of the synthesis atmosphere leads to poor control of the nucleation density, thickness and shape. We overcome these complexities by simply regulating the precursor temperature with time and monitoring the precursor decomposition with mass spectrometry. A consistent flux of suitable volatile NxBy-containing species can be achieved and thus, the ability to tailor the type of h-BN deposits. We utilise non-flammable conditions and develop a strategy to re-use the precursor, paving the way towards industrially-applicable production.
Methods
Apparatus and controls implementation
Our APCVD system consisted of a stainless steel (SS) gas mixing manifold, precursor evaporation chamber, precursor delivery manifold connected to a fused silica tube and a high temperature furnace (Supplementary Figure 5a). An electrical heater and electrical tape were used to heat the precursor chamber and the SS manifold to 50–110 °C and 90 °C respectively. The set temperature of the heater could be reached quickly with a heating rate of approximately 10 °C min−1 and maintained with < 0.1 °C accuracy. An SS needle in the precursor chamber allowed convenient air purging and precursor vapour displacement. It was found that the commercially-available AB precursor released considerable tetrahydrofuran impurity (Supplementary Figure 6) in the early stages of heating, which is used as an intermediate step of the industrial production of AB61. For this reason, a by-pass exhaust line connected directly to the evaporation chamber was employed (Supplementary Figure 5a), allowing precursor pre-heating and purging of the system in parallel with annealing of the substrate.
AB polymerisation and foaming is well documented62 and presented an additional challenge (Supplementary Figure 5b). This process meant that experiments performed under the same conditions produced appreciably different response due to the random nature of the polymer seed formation and subsequent rapid propagation. To overcome this problem, we mixed AB with an inert filler (BN powder) with weight ratio of 1:10 AB to BN, such that the AB granules were sufficiently separated from each other. Although each granule could still start the polymerisation reaction at random when heated, on average, the time-dependent evolution of NxBy-containing species was more reproducible and the foaming was suppressed. Supplementary Figure 5c,d schematically summarises the mechanism behind controlling the polymerisation process.
MS measurements and data analysis
MS measurements were performed on a Hiden Analytical HPR20 quadrupole mass spectrometer with a quartz inert fast sampling capillary. MS pressure was kept at 1E-6 torr. Measurements were performed with a secondary electron multiplier detector at 10−11 torr acquisition range and extended dwell times (2000–5000%). Electron energy of 70 V and emission current of 100 μA were used for ionisation. The manifold and the MS sampling capillary were always heated to 90 °C as to obtain comparable flight path conditions for the decomposition products. The travel time of the analytes through the capillary is around 0.3 seconds. The probe was inserted into the fused silica tube, at a position where the furnace would be placed during a CVD experiment. For all measurements, the electrical heater was pre-heated to 50 °C and raised over the evaporation chamber for 5 minutes to avoid any variation in the heating time from ambient temperature; followed by setting the precursor temperature for the measurement63,64,65,66.
MS intensity profiles are approximately quantified by comparing the base peak of each detected species to the Ar-38 isotope which appears in the ~630 part per million (ppm) concentration67. It is understood that this approach could carry an error due to the relative apparatus sensitivity factors (RSF) between the Ar-38 isotope and the measured molecules. It was not possible to obtain correction factors for all of the uncommon species that were detected. Nevertheless, we developed a strategy to approximately correct for the RSF values by first measuring the RSF value of NH3 vs. Ar-38 in a calibrated Ar-NH3 mixture, which was found it to be only 1.03. After this correction, the absolute error in the asymmetric N-species (dominated by NH3 in the early stages of decomposition) was very low, < 3 %. The AB precursor conveniently gave us a way to also correct the scaling of the asymmetric B-species since the ratio of N:B after full AB decomposition is 1:1. Thus, the error in the quantification of the absolute values of the summed flows is estimated to be < 10 %. However, the error in the relative values of the summed flows (e.g. N:B ratio) is expected to be even lower, since RSFs divide out. Overall, such MS measurements and quantification are broader and more versatile than what is available with other techniques and is appropriate for the purposes of this study. Data analysis of the 3D MS spectra (m/z, time, intensity) were performed in MATLAB. Briefly, the intensities were scaled to the Ar-38 isotope and multiplied by the total molar gas flow and scaled to 1 mg of the precursor. The background cycle (the cycle before the precursor was heated) was subtracted from all the subsequent cycles allowing to set to zero the background levels of impurity. Any fluctuation in the water level (m/z 18) was corrected for ammonia (m/z 17, m/z 16) and the level of ammonia (m/z 16 scaled to the base peak) was recorded as the final ammonia profile. Similarly, fragmentation patterns presented in the Supplementary Table 1 were used to deconvolute and record profiles of the other NxBy-containing species. Diborane profile was calculated using the m/z 24 peak, as to avoid a significant overlap with aminoborane; this peak was scaled to the base peak. Diborane contribution was then subtracted from m/z 10–15, 21–28. Monomeric aminoborane, aminodiborane, triborane, borane and borazine profiles were calculated from m/z 27, 43, 37, 13, 80 (scaled to base peaks). For the asymmetric N-species flow, ammonia and aminodiborane flows were added; for the asymmetric B-species flow, 2x diborane, 3x triborane, 2x aminodiborane and 1x borane flows were added together; for the symmetric N-(B-) species flow only the monomeric aminodiborane flow was used due to negligible borazine concentration.
h-BN synthesis
Cu foil (Alfa Aesar, 99.8 % purity, 25 µm thick) was washed in acetone, followed by electropolishing in 85 % phosphoric acid at 1.8 V for 15 minutes and then annealed in 2.5 % H2 in Ar mixture for 30 minutes. The growth temperature was 1070 °C for all CVD experiments. Basic h-BN domains were synthesised with 350 flow of 2.5 % H2 in Ar and 110 mg of a mixture of AB/BN (1:10) powder ; at specified precursor temperatures and precursor decomposition times. For the larger, optimised h-BN domain growth, electropolished Cu was oxidized for 60 seconds at 200 °C, followed by 30 min annealing in pure Ar and 3 hour annealing in a 2.5 % H2 in Ar mixture. The synthesis was then performed with 500 sccm flow of Ar/H2 for 6 hours while the precursor was heated according to the profile in Fig. 7a. The full temperature control chart is given in Supplementary Table 3.
Characterization
For optical microscopy, the samples were heated to 200 °C for 30 seconds to oxidize the uncovered Cu surface, which changed its colour. Images were recorded on a Yenway-CX40 microscope.
Scanning electron microscope (SEM) images of the samples were recorded using a Carl Zeiss Merlin SEM operating at 5 kV accelerating voltage and 1 nA probe current.
The h-BN transfer was performed using the standard polymer-assisted technique described elsewhere50. Raman spectra were recorded on a Horiba LabRAM Aramis microscope equipped with a 532 nm laser. FTIR spectra were recorded on Bio-Rad Varian Excalibur FTS 3500 spectrometer. AFM scans were performed in AC mode on an Oxford Instruments Asylum Research Cypher AFM. An AC160TS probe was used from Olympus Probes. AC-TEM was performed on a double aberration-corrected JEOL JEM-2200MCO microscope at 80 kV.
Change history
22 November 2017
A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
References
Watanabe, K., Taniguchi, T. & Kanda, H. Direct-bandgap properties and evidence for ultraviolet lasing of hexagonal boron nitride single crystal. Nature Materials 3, 404–409 (2004).
Gannett, W. et al. Boron nitride substrates for high mobility chemical vapor deposited graphene. Appl Phys Lett 98, 242105 (2011).
Tang, S. et al. Precisely aligned graphene grown on hexagonal boron nitride by catalyst free chemical vapor deposition. Sci. Rep. 3 (2013).
Okada, M. et al. Direct Chemical Vapor Deposition Growth of WS2 Atomic Layers on Hexagonal Boron Nitride. ACS Nano 8, 8273–8277 (2014).
Wang, H. et al. BN/Graphene/BN Transistors for RF Applications. Ieee Electr Device L 32, 1209–1211 (2011).
Shi, G. et al. Boron Nitride-Graphene Nanocapacitor and the Origins of Anomalous Size-Dependent Increase of Capacitance. Nano Letters 14, 1739–1744 (2014).
Lee, G. H. et al. Flexible and Transparent MoS2 Field-Effect Transistors on Hexagonal Boron Nitride-Graphene Heterostructures. ACS Nano 7, 7931–7936 (2013).
Ismach, A. et al. Toward the Controlled Synthesis of Hexagonal Boron Nitride Films. ACS Nano 6, 6378–6385 (2012).
Auwarter, W., Suter, H. U., Sachdev, H. & Greber, T. Synthesis of one monolayer of hexagonal boron nitride on Ni(111) from B-trichloroborazine (ClBNH)(3). Chem Mater 16, 343–345 (2004).
Chatterjee, S. et al. Chemical Vapor Deposition of Boron Nitride Nanosheets on Metallic Substrates via Decaborane/Ammonia Reactions. Chem Mater 23, 4414–4416 (2011).
Shi, Y. M. et al. Synthesis of Few-Layer Hexagonal Boron Nitride Thin Film by Chemical Vapor Deposition. Nano Letters 10, 4134–4139 (2010).
Morscher, M., Corso, M., Greber, T. & Osterwalder, J. Formation of single layer h-BN on Pd(111). Surface Science 600, 3280–3284 (2006).
Corso, M., Greber, T. & Osterwalder, J. h-BN on Pd(110): a tunable system for self-assembled nanostructures? Surface Science 577, L78–L84 (2005).
Preobrajenski, A. B. et al. Influence of chemical interaction at the lattice-mismatched h-BN/Rh(111) and h-BN/Pt(111) interfaces on the overlayer morphology. Phys Rev B 75 (2007).
Sutter, P., Lahiri, J., Albrecht, P. & Sutter, E. Chemical Vapor Deposition and Etching of High-Quality Monolayer Hexagonal Boron Nitride Films. ACS Nano 5, 7303–7309 (2011).
Joshi, S. et al. Boron Nitride on Cu(111): An Electronically Corrugated Monolayer. Nano Letters 12, 5821–5828 (2012).
Gibb, A. L. et al. Atomic Resolution Imaging of Grain Boundary Defects in Monolayer Chemical Vapor Deposition-Grown Hexagonal Boron Nitride. Journal of the American Chemical Society 135, 6758–6761 (2013).
Kim, K. K. et al. Synthesis and Characterization of Hexagonal Boron Nitride Film as a Dielectric Layer for Graphene Devices. ACS Nano 6, 8583–8590 (2012).
Kidambi, P. R. et al. In Situ Observations during Chemical Vapor Deposition of Hexagonal Boron Nitride on Polycrystalline Copper. Chem Mater 26, 6380–6392 (2014).
Tay, R. Y. et al. Growth of Large Single-Crystalline Two-Dimensional Boron Nitride Hexagons on Electropolished Copper. Nano Letters 14, 839–846 (2014).
Song, L. et al. Large Scale Growth and Characterization of Atomic Hexagonal Boron Nitride Layers. Nano Letters 10, 3209–3215 (2010).
Lee, Y. H. et al. Growth selectivity of hexagonal-boron nitride layers on Ni with various crystal orientations. Rsc Adv 2, 111–115 (2012).
Guo, N. et al. Controllable growth of triangular hexagonal boron nitride domains on copper foils by an improved low-pressure chemical vapor deposition method. Nanotechnology 23 (2012).
Gao, Y. et al. Repeated and Controlled Growth of Monolayer, Bilayer and Few-Layer Hexagonal Boron Nitride on Pt Foils. ACS Nano 7, 5199–5206 (2013).
Lee, K. H. et al. Large-Scale Synthesis of High-Quality Hexagonal Boron Nitride Nanosheets for Large-Area Graphene Electronics. Nano Letters 12, 714–718 (2012).
Wang, L. F. et al. Monolayer Hexagonal Boron Nitride Films with Large Domain Size and Clean Interface for Enhancing the Mobility of Graphene-Based Field-Effect Transistors. Adv Mater 26, 1559–1564 (2014).
Tay, R. Y. et al. A systematic study of the atmospheric pressure growth of large-area hexagonal crystalline boron nitride film. Journal of Materials Chemistry C 2, 1650–1657 (2014).
Zhong, G. et al. Growth of continuous graphene by open roll-to-roll chemical vapor deposition. Appl Phys Lett 109, 193103 (2016).
Hu, M. G., Geanangel, R. A. & Wendlandt, W. W. The thermal decomposition of ammonia borane. Thermochim Acta 23, 249–255 (1978).
Bowden, M., Autrey, T., Brown, I. & Ryan, M. The thermal decomposition of ammonia borane: A potential hydrogen storage material. Current Applied Physics 8, 498–500 (2008).
Baitalow, F., Baumann, J., Wolf, G., Jaenicke-Rossler, K. & Leitner, G. Thermal decomposition of B-N-H compounds investigated by using combined thermoanalytical methods. Thermochim Acta 391, 159–168 (2002).
Wolf, G., Baumann, J., Baitalow, F. & Hoffmann, F. P. Calorimetric process monitoring of thermal decomposition of B–N–H compounds. Thermochim Acta 343, 19–25 (2000).
Baumann, J., Baitalow, F. & Wolf, G. Thermal decomposition of polymeric aminoborane (H2BNH2)x under hydrogen release. Thermochim Acta 430, 9–14 (2005).
Stein, S.E. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69. (eds P. J. Linstrom & W. G. Mallard) (March 28, 2017).
Frueh, S. et al. Pyrolytic Decomposition of Ammonia Borane to Boron Nitride. Inorg Chem 50, 783–792 (2011).
Paine, R.T., Sodeck, G. & Stafford, F. E. M-B Mass-Spectra and Pyrolyses of Fluorophosphine-Triborane(7)Complexes - Formation and Mass-Spectrum of Triborane(7). Inorg Chem 11, 2593-& (1972).
Chen, X. N. A., Zhao, J. C. & Shore, S. G. Facile Synthesis of Aminodiborane and Inorganic Butane Analogue NH3BH2NH2BH3. Journal of the American Chemical Society 132, 10658–10659 (2010).
Schlesinger, H. I., Ritter, D. M. & Burg, A. B. Hydrides of boron X The preparation and preliminary study of the new compound B2H7N. Journal of the American Chemical Society 60, 2297–2300 (1938).
Stock, A. & Pohland, E. Borwasserstoffe, IX.: B3N3H6. Berichte der deutschen chemischen Gesellschaft (A and B Series) 59, 2215–2223 (1926).
Laubengayer, A., Porter, R.F. & Moews, P.C. Condensation of Borazine to Polycyclic Boron-Nitrogen Frameworks by Pyrolytic Dehydrogenation. Journal of the American Chemical Society 83, 1337-& (1961).
Page, M., Adams, G. F., Binkley, J. S. & Melius, C. F. Dimerization energy of borane. The Journal of Physical Chemistry 91, 2675–2678 (1987).
Stowe, A. C., Shaw, W. J., Linehan, J. C., Schmid, B. & Autrey, T. In situ solid state 11B MAS-NMR studies of the thermal decomposition of ammonia borane: mechanistic studies of the hydrogen release pathways from a solid state hydrogen storage material. Phys Chem Chem Phys 9, 1831–1836 (2007).
Fang, Z. et al. Facile solid-phase synthesis of the diammoniate of diborane and its thermal decomposition behavior. Phys Chem Chem Phys 13, 7508–7513 (2011).
Koepke, J. C. et al. Role of Pressure in the Growth of Hexagonal Boron Nitride Thin Films from Ammonia-Borane. Chem Mater 28, 4169–4179 (2016).
Lu, G.Y. et al. Synthesis of large single-crystal hexagonal boron nitride grains on Cu-Ni alloy. Nat Commun 6 (2015).
Sharma, S. et al. Morphology-Controlled Synthesis of Hexagonal Boron Nitride Crystals by Chemical Vapor Deposition. Cryst Growth Des 16, 6440–6445 (2016).
Khan, M.H. et al. Synthesis of Large and Few Atomic Layers of Hexagonal Boron Nitride on Melted Copper. Sci Rep-Uk 5 (2015).
Vlassiouk, I. et al. Large scale atmospheric pressure chemical vapor deposition of graphene. Carbon 54, 58–67 (2013).
Zhou, H. et al. Chemical vapour deposition growth of large single crystals of monolayer and bilayer graphene. Nat Commun 4, 2096 (2013).
Stehle, Y. et al. Synthesis of Hexagonal Boron Nitride Monolayer: Control of Nucleation and Crystal Morphology. Chem Mater 27, 8041–8047 (2015).
Caneva, S. et al. Controlling Catalyst Bulk Reservoir Effects for Monolayer Hexagonal Boron Nitride CVD. Nano Letters 16, 1250–1261 (2016).
Liu, Y., Bhowmick, S. & Yakobson, B. I. BN White Graphene with “Colorful” Edges: The Energies and Morphology. Nano Letters 11, 3113–3116 (2011).
Murdock, A. T. et al. Targeted removal of copper foil surface impurities for improved synthesis of CVD graphene. Carbon 122, 207–216 (2017).
Hao, Y. F. et al. The Role of Surface Oxygen in the Growth of Large Single-Crystal Graphene on Copper. Science 342, 720–723 (2013).
Braeuninger-Weimer, P., Brennan, B., Pollard, A. J. & Hofmann, S. Understanding and Controlling Cu-Catalyzed Graphene Nucleation: The Role of Impurities, Roughness, and Oxygen Scavenging. Chem Mater 28, 8905–8915 (2016).
Reckinger, N. et al. Oxidation-assisted graphene heteroepitaxy on copper foil. Nanoscale 8, 18751–18759 (2016).
Vlassiouk, I. et al. Role of Hydrogen in Chemical Vapor Deposition Growth of Large Single-Crystal Graphene. ACS Nano 5, 6069–6076 (2011).
Babenko, V. et al. Rapid epitaxy-free graphene synthesis on silicidated polycrystalline platinum. Nat Commun 6, 7536 (2015).
Gorbachev, R. V. et al. Hunting for Monolayer Boron Nitride: Optical and Raman Signatures. Small 7, 465–468 (2011).
Song, X. et al. Chemical vapor deposition growth of large-scale hexagonal boron nitride with controllable orientation. Nano Res 8, 3164–3176 (2015).
Ramachandran, P. V. & Kulkarni, A. S. The role of ammonia in promoting ammonia borane synthesis. Dalton T 45, 16433–16440 (2016).
Choi, Y. J. et al. Kinetics study of solid ammonia borane hydrogen release - modeling and experimental validation for chemical hydrogen storage. Phys Chem Chem Phys 16, 7959–7968 (2014).
Meysami, S.S., Grobert, N. Device, method and apparatus for the transfer of analytes. http://patentscope.wipo.int/search/en/WO2013030544 (2013).
Meysami, S. S., Snoek, C. L., Grobert, N. Versatile in Situ Gas Analysis Apparatus for Nanomaterials Reactors. Analytical Chemistry 86(17), 8850–8856 (2014).
Meysami, S. S., Koós, A. A., Dillon, F., Grobert, N. Aerosol-assisted chemical vapour deposition synthesis of multi-wall carbon nanotubes: II. An analytical study. Carbon 58, 159–169 (2013).
Meysami, M. M., Koós, A. A., Dillon, F., Dutta, M., Grobert, N. Aerosol-assisted chemical vapour deposition synthesis of multi-wall carbon nanotubes: III. Towards upscaling. Carbon 88, 148–156 (2015).
De Laeter, J. R. et al. Atomic weights of the elements: Review 2000 - (IUPAC technical report). Pure Appl Chem 75, 683–800 (2003).
Acknowledgements
The work at Oxford University Materials Department is supported by the DTA-EPSRC (V.B.), the Commonwealth Scholarship Commission (A.T.M.), University of Oxford Clarendon Fund (A.T.M.), the Royal Society (N.G.), the European Research Council (ERC-2009-StG-240500-DEDIGROWTH, ERC-2015-POC-680559-CONDUCT, ERC-2011-POC-309786-DEVICE) (N.G.), the Engineering and Physical Sciences Research Council IAA block grants (N.G.).
Author information
Authors and Affiliations
Contributions
V.B., N.G. conceived and designed the investigation. V.B., G.L., carried out the MS measurements, optimisation and MS data analysis, V.B., G.L., A.A.K., A.T.M. performed CVD synthesis and optimisation, transfer experiments, SEM, optical, Raman characterization. K.S. carried out FTIR measurements. J.M. performed AFM characterization and data analysis. J.B. performed AC-TEM measurements. S.S.M. assisted with MS measurements and advised on MS data analysis, N.G. oversaw project direction, planning and infrastructure. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Change History: A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A correction to this article is available online at https://doi.org/10.1038/s41598-017-16514-y.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Babenko, V., Lane, G., Koos, A.A. et al. Time dependent decomposition of ammonia borane for the controlled production of 2D hexagonal boron nitride. Sci Rep 7, 14297 (2017). https://doi.org/10.1038/s41598-017-14663-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14663-8
This article is cited by
-
Controllable Synthesis of High-Quality Hexagonal Boron Nitride Films on Ni-B Alloy
Journal of Electronic Materials (2023)
-
Direct Fabrication of Vertically Stacked Double Barrier Tunnel Junctions Based on Graphene and h-BN
Electronic Materials Letters (2022)
-
Vapor–liquid–solid growth of large-area multilayer hexagonal boron nitride on dielectric substrates
Nature Communications (2020)
-
Loading of AgNPs onto the surface of boron nitride nanosheets for determination of scopoletin in Atractylodes macrocephala
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
