
Abstract
Agmatine N-acetyltransferase (AgmNAT) catalyzes the formation of N-acetylagmatine from acetyl-CoA and agmatine. Herein, we provide evidence that Drosophila melanogaster AgmNAT (CG15766) catalyzes the formation of N-acetylagmatine using an ordered sequential mechanism; acetyl-CoA binds prior to agmatine to generate an AgmNAT•acetyl-CoA•agmatine ternary complex prior to catalysis. Additionally, we solved a crystal structure for the apo form of AgmNAT with an atomic resolution of 2.3 Å, which points towards specific amino acids that may function in catalysis or active site formation. Using the crystal structure, primary sequence alignment, pH-activity profiles, and site-directed mutagenesis, we evaluated a series of active site amino acids in order to assign their functional roles in AgmNAT. More specifically, pH-activity profiles identified at least one catalytically important, ionizable group with an apparent pKa of ~7.5, which corresponds to the general base in catalysis, Glu-34. Moreover, these data led to a proposed chemical mechanism, which is consistent with the structure and our biochemical analysis of AgmNAT.
Similar content being viewed by others
Introduction
The discovery and characterization of enzymes involved in fatty acid amide biosynthesis has been a longstanding focus of our research1. One possible biosynthetic route for the fatty acid amides would be the reaction between an amine and a fatty acyl-CoA: R1-NH2 + R2-CO-S-CoA → R2-CO-NH-R1 + CoA-SH. Enzymes of the GCN5-related N-acetyltransferase family (GNAT) catalyze a similar reaction using acetyl-CoA as a substrate to generate N-acetylamides2. Acetyl-CoA-dependent N-acetylation by N-acetyltransferases is known for a diversity of amines3,4,5 in a broad range of organisms2,5,6,7,8. We have long suspected that enzymes identified as N-acetyltransferases might accept longer-chain fatty acyl-CoA thioesters as substrates or that novel N-acetyltransferase-like enzymes exist that utilize fatty acyl-CoA thioesters as substrates.
Drosophila melanogaster is an excellent model organism to study fatty acid amide biosynthesis. These insects are known to produce fatty acid amides9,10, its genome has been sequenced11, these organisms can be manipulated genetically12, and are inexpensive to maintain. In addition, two N-acetyltransferases had been identified from D. melanogaster, arylalkylamine N-acetyltransferase variant A (AANATA, also called dopamine N-acetyltransferase)13 and arylalkylamine N-acetyltransferase-like 2 (AANATL2)14. Both enzymes catalyze the N-acetylation of arylalkylamines, but their respective substrate specificities, kinetic mechanisms, and chemical mechanisms were not fully defined prior to our work. A search of D. melanogaster genome using the sequences of AANATA and AANATL2 led to the identification of six other putative arylalkylamine N-acetyltransferase-like enzymes, AANATL3-814,15. A complete understanding of the structural and mechanistic features of these enzymes will provide tremendous insight into rules governing acyl-chain length specificity for GNAT enzymes. An exhaustive evaluation of these enzymes for different amine or acyl-CoA substrates may yield new chemistries and define a biosynthetic route to the fatty acid amides. To define the substrate specificities of these putative N-acyltransferases, we devised a screening strategy that involved the evaluation of a collection of amines vs. a short-chain acyl-CoA or a long-chain acyl-CoA. Our screening strategy led to the discovery that D. melanogaster AANATL2 will utilize dopamine, serotonin, and long-chain acyl-CoA thioesters as substrates16,17. These results are likely of significance to mammals because the N-fatty acyldopamines have been identified in the brain18 and the N-fatty acylserotonins in the gastro-intestinal tract19. Our application of the screening strategy to AANATL8 led to the identification of agmatine as the amine substrate with the highest (kcat/Km)app for this enzyme. Thus, we have renamed AANATL8 as agmatine N-acetyltransferase (AgmNAT). The acetylation of agmatine points to novel agmatine related metabolites and new reactions in the degradation pathways of agmatine and arginine. A thorough study of the insect AANATs contributes to our understanding of fatty acid amide biosynthesis, enables a detailed comparison between the insect AANATs to the AANATs from other organisms, and fosters the development of insecticides targeted against insect AANATs. AANATs are suggested to be a good targets for the control of insect pests20,21,22,23. Furthermore, AANAT inhibitors could also lead to drugs to treat circadian rhythm disorders because serotonin N-acetyltransferase catalyzes the rate-determining step in melatonin biosynthesis24.
Agmatine, (4-aminobutyl)guanidine, was first described in 191025 and was later identified as the product of arginine decarboxylation26. Research concerning agmatine was limited until the 1990s27, until the discovery that agmatine is produced in the mammalian brain28,29. Agmatine is distributed in many tissues, including the stomach, intestine (large and small), adrenal gland, heart, aorta, spleen, lung, vas deferens, kidney, liver, skeletal muscle, and plasma30,31,32. It is primarily located in cytoplasmic vesicles that are strongly associated with the mitochondria or endoplasmic reticulum33,34. Additionally, agmatine can translocate into the mitochondria35,36,37,38 and is likely associated with the Golgi complex, cell membrane, and nuclear membrane39. Agmatine is a neurotransmitter and neuromodulator in mammalian brain27,40, its physiological effects resulting from binding to the imidazoline (I1 and I2)28,41,42, α2-adrenergic43, nicotinic44, NMDA45, and serotonin receptors (5-HT2A and 5HT-3)46. Little is known about agmatine and its biosynthesis, degradation, elimination, and function in the fly or in insects, in general. Low levels of agmatine have been found in D. melanogaster 47 and agmatine has been reported from other insects47,48,49,50. Likewise, little is known about arginine decarboxylase from insects51. Our discovery and characterization of AgmNAT may point to unappreciated role(s) for agmatine and/or N-acetylagmatine in D. melanogaster and other insects.
Agmatine biosynthesis and degradation is shown in Supplementary Fig. S1. First, arginine decarboxylase (ADC) catalyzes the decarboxylation of arginine to generate agmatine26,27,28,29, followed by agmatine degradation via two main routes: (a) hydrolysis to urea and putrescine, as catalyzed by agmatinase (AGMAT, also known as agmatine ureohydrolase)29,52 or (b) oxidation to 4-guanidinobutanoic acid, as catalyzed by diamine oxidase (DAO) and aldehyde dehydrogenase (AlDH)53,54,55. In Thermus thermophilus, polyamine aminopropyltransferase (SpeE) catalyzes the formation of agmatine N 1-aminopropylagmatine56,57. In plants, agmatine coumaroyltransferase catalyzes the formation of p-coumaroylagmatine from p-coumaroyl-CoA and agmatine, p-coumaroylagmatine is thought to function in the defense system of the plant against infection58,59. An unexplored degradative pathway for agmatine is N-acetylation at the N1 position, catalyzed by AgmNAT to generate N-acetylagmatine – one of the subjects of this manuscript.
AgmNAT is a member of the GCN5-related N-acetyltransferase family (GNAT)2 and, in addition to the formation of N-acetylagmatine, this enzyme also catalyzes the production of N-acylpolyamines from the corresponding acyl-CoA and polyamine. We also present data showing the AgmNAT structure, substrate specificity, and kinetic and chemical mechanism for the AgmNAT-catalyzed reaction.
Results and Discussion
Crystal structure of AgmNAT
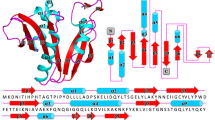
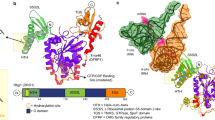
A homology model for AgmNAT was constructed using the Aedes aegypti arylalkylamine N-acetyltransferase structure21 as a template for molecular replacement. The AgmNAT (CG15766) crystal structure was determined at 2.3Å, with two monomers in the asymmetric unit of the P21 space group (Table 1). The two monomers are nearly identical with an RMSD value of 0.262 Å when aligning 862 backbone atoms. Similar to the arylalkylamine N-acetyltransferase model, the new structure is primarily composed of six α-helices and seven anti-parallel α-strands (Fig. 1A). The AgmNAT structure displays a conserved GNAT fold, similar to that observed for D. melanogaster AANATA and human spermidine/spermine N 1-acetyltransferase (SSAT) (Supplementary Fig. S2), though the sequence identity is low when compared to these N-acetyltransferase enzymes (24% with AANATA and <20% for SSAT), a known feature of GNAT enzymes2. Based on the functional and structural similarities between AgmNAT and other GNATs such as AANATA (PDB 3TE4)15,60, we predict the active site pocket to be similar, though not identical, for the binding of the acyl-CoA and amine substrates (Fig. 2). The active site is well defined in the 2Fo-Fc electron density map (Fig. 1B,C) and is located near the crystal packing interface for both monomers. Based on the structure of AANATA with acetyl-CoA bound (PDB 3TE4)60, the binding surface for the adenosine 3-phosphate 5-pyrophosphate moiety of CoA-SH is blocked by protein-protein interactions in the AgmNAT structure, but the rest of the active site is open. The splaying of β-strand four and five, a conserved structural feature in GNAT enzymes, is also displayed in AgmNAT, which is the binding site for the pantetheine arm of acetyl-CoA2. Moreover, a conserved glutamate, Glu-34, that serves as the catalytic base for other D. melanogaster N-acyltransferase enzymes, is located within an accessible pocket that can accommodate the acyl-CoA and amine substrate, similar to that observed for AANATA (Fig. 1B)15. Also observed in the active site pocket are the residues, Pro-35 and Ser-171 (Fig. 1B,C), which are conserved amino acids that regulate catalysis in other D. melanogaster N-acyltransferases15,61,62. The functional roles of Pro-35 and Ser-171 of AgmNAT are discussed in subsequent sections.

Crystal structure for AgmNAT at 2.3 Å. (A) Overall structure with labeled secondary structures. The two termini are labeled as N and C. (B) 2Fo-Fc electron density map of AgmNAT active site, contoured at 1.0 σ. Ordered water molecules are indicated by red spheres. (C) 2Fo-Fc electron density map (1.0 σ) of the Ser-171 region interacting with the 165–169 strand region.

AgmNAT active site. (A) Superimposed structures of the apo AgmNAT (green, PDB 5K9N) and the dopamine N-acetyltransferase-acetyl-CoA complex (AANATA) structure (orange, PDB 3TE4) showing active site residues and acetyl-CoA (purple) from the AANATA (3TE4) structure. All relevant residues are shown in stick. The helix bearing Tyr-170 and the loop to its left form the binding site for the adenosine 3-phosphate 5-pyrophosphate portion of coenzyme (A). (B) Highlights the relative position of Glu-34, Pro-35, Tyr-170, Ser-171, and His-206 in the active site. Additionally, the van der Waals interaction between Pro-35 and His-206 is shown.
Evaluation of acyl-CoA steady-state kinetic constants
AgmNAT showed minimal differences in the measured Km,app values for acyl-CoA substrates ranging from acetyl-CoA to decanoyl-CoA (C2–C10) (Table 2) when agmatine was used as the saturating amine substrate. However, there was an acyl chain length dependent decrease in the apparent kcat value for the acyl-CoA substrates as the chain length is increased. This apparent decrease in the turnover number of ~150-fold from acetyl-CoA to decanoyl-CoA, led to the observed acyl-chain length specific decrease in the (kcat/Km)app value. In addition, oleoyl-CoA was not a substrate at a concentration of 500 μM. These data likely result from the acyl-chain partially (decanoyl-CoA) or fully (oleoyl-CoA) occupying the amine binding site, perturbing the productive binding of agmatine; therefore, resulting in a decrease in or complete loss of catalysis. Similar results were observed for other D. melanogaster N-acyltransferases15,61,62.
Evaluation of amine substrate steady-state kinetic constants
We screened >50 amines as potential AgmNAT substrates using acetyl-CoA or oleoyl-CoA as the co-substrate because of our interests in fatty acid biosynthesis, structure function relationships of GNAT enzymes, and the development of novel insecticides targeted to this class of enzymes. Our amine substrate screen included the canonical amino acids (except for Cys because Cys reacts with DTNB), amino acid analogs, other biogenic amines, and different xenobiotic amines. Only six amines (Table 3) showed AgmNAT activity >3-fold higher than the level of background acetyl-CoA thioesterase activity, whereas none showed a greater rate for oleoyl-CoA. Also, we identified five polyamines as AgmNAT substrates: spermine, N 8-acetylspermidine, putrescine, spermidine, and cadaverine (Table 3). The (kcat/Km)app values for the polyamines were lower than that measured for agmatine, the (kcat/Km)app,agmatine/(kcat/Km)app,polyamine ratio ranging from 15 for spermine to 1900 for cadaverine. Structural evidence for the specificity for agmatine and different polyamines likely results from the acidic nature of the active site, similar to that observed for the human ortholog (human SSAT) (Fig. 3)2. A more acidic active site can accommodate an amine substrate with a basic guanidinium group better than one with a hydrophobic aromatic group, giving rise to the difference in substrate specificity when compared to an AANAT15,60. AgmNAT was originally named AANATL8 based on primary sequence similarity15; however, the substrate specificity data reported here support a new designation: agmatine N-acetyltransferase. This is the first report of agmatine serving as the best amine substrate for an N-acyltransferase. There are only a few reports of agmatine serving as a substrate within this family of enzymes17,62,63 and only two reports on the identification of N-acetylagmatine from a biological source64,65. Rats fed heavy-atom labeled agmatine yielded two major urinary products; heavy-atom labeled N-acetylagmatine and unprocessed, but labeled agmatine64, suggesting a similar conversion as that catalyzed by AgmNAT. Inactivation of agmatine neurotransmission by N-acetylation is an underappreciated reaction between arginine, agmatine, and human disease27,66,67,68, the search for a human ortholog of Drosophila AgmNAT could lead to a new target for drug development. Additionally, selective targeting of Drosophila AgmNAT could result in the development of novel insecticides for insect control20,21,22,23.

Comparison of the surface electrostatic potential of D. melanogaster AgmNAT to that of D. melanogaster AANATA and human SSAT. (A) AgmNAT (PDB code 5K9N). (B) D. melanogaster AANATA (PDB code 3TE4). (C) Human SSAT (PDB code 2JEV). (D) Close up of AgmNAT active site oriented to show the entry point for acetyl-CoA. (E) Close up of D. melanogaster AANATA active site with acetyl-CoA bound and oriented to show the entry point for acetyl-CoA. (F) Close up of the human SSAT active site with the bisubstrate inhibitor N 1-spermine-acetyl-CoA bound and oriented to show the entry point for acetyl-CoA. Blue is for positive charges and red is for negative charges. Surface electrostatic potentials reveal that the amine binding pocket for AgmNAT and SSAT are more negatively charged than the arylalkylamine binding pocket of AANATA.
We found that arginine, arginine methyl ester, N-acetylputrescine, and N 1-acetylspermidine were not AgmNAT substrates. The ~25-fold increase in kcat,app for N 8-acetylspermidine when compared to spermidine, together with our data demonstrating that N-acetylputrescine and N 1-acetylspermidine were not substrates all suggest that AgmNAT, most likely, catalyzes the mono- and N1- specific acetylation of these biogenic amines, similar to what is observed for the mammalian spermidine N-acetyltransferase69,70.
The increase in the kcat,app value, together with the small ~2-fold difference in the Km,app for N 8-acetylspermidine relative to spermidine, could result from non-productive binding of the N8-amine of spermidine in the AgmNAT active site, whereby the N1-amine is better positioned for catalysis: deprotonation and then nucleophilic attack of the –NH2 at the carbonyl of the acetyl-CoA thioester moiety. This means both of the amine moieties can bind in the active site, but only the N1-amine is acetylated.
While arginine and arginine methyl ester are not AgmNAT substrates, we further evaluated these for AgmNAT inhibition to determine if either could bind to the enzyme. Arginine methyl ester proved to weakly inhibit AgmNAT, decreasing the rate of N-acetylagmatine formation from acetyl-CoA and agmatine by ~50% at 10 mM. In contrast, we found no inhibition of N-acetylagmatine formation at both 10 mM and 25 mM arginine. These data show that a modification of the α-position of agmatine inhibits binding to AgmNAT and that the inhibition results from both electronic and steric effects. The presence of the negatively charged α-carboxylate seems to eliminate or significantly weaken AgmNAT binding, likely the result of charge-charge repulsion. Evidence for this suggestion comes from the weak inhibition by arginine methyl ester (Ki,s and Ki,i ≥ 1 mM, Supplementary Fig. S3), but no apparent inhibition by arginine at a concentration as high as 25 mM.
Kinetic mechanism
A combination of initial velocity kinetic experiments, dead-end inhibition, and product inhibition studies were used to determine the AgmNAT kinetic mechanism. Our first set of experiments was to vary the initial concentrations of one substrate at different fixed concentrations of the second substrate and fit these data to rate equations for either a sequential (Equation 3) or a ping pong (Equation 4) kinetic mechanism. Equation 3, for a sequential mechanism, provided the best fit (χ 2 is 4.6 for Equation 3 while χ 2 is 5.0 for Equation 4) which resulted in intersecting double reciprocal lines for acetyl-CoA (Fig. 4A) and agmatine (Fig. 4B). These data suggest that the AgmNAT-catalyzed formation of N-acetylagmatine occurs via a sequential mechanism; catalysis takes place only after formation of the AgmNAT•acetyl-CoA•agmatine ternary complex.

Acetyl-CoA and agmatine initial velocity double reciprocal plots. (A) Velocities measured at fixed concentrations of agmatine: 1500 μM (○), 750 μM (□), 300 μM (◇), and 60 μM (Δ). (B) Velocities measured at fixed concentrations of acetyl-CoA: 500 μM (○), 250 μM (□), 100 μM (◇), 50 μM (Δ), and 20 μM (▽). These data are best fit to the rate equation for a sequential mechanism (χ 2 is 4.6 for Equation 3) than the rate equation for a ping pong mechanism (χ 2 is 5.0 for Equation 4).
Next, we determined if the AgmNAT kinetic mechanism is an ordered or random sequential mechanism by using substrate analogs, oleoyl-CoA, arcaine, and arginine methyl ester, as dead-end inhibitors vs. acetyl-CoA and agmatine. The inhibitor data is summarized in Table 4 and we have included the double reciprocal plots for the inhibitors in the Supplementary Materials. Arcaine is structurally related to agmatine, with its primary amine moiety replaced with a guanidinium group. Arcaine serving as an AgmNAT inhibitor supports our conclusion that AgmNAT does not acetylate the guanidinium amine of agmatine. None of these inhibitors showed any rate of catalysis above the slow, background rate of acetyl-CoA or oleoyl-CoA hydrolysis. Oleoyl-CoA produced competitive and noncompetitive inhibition plots for acetyl-CoA and agmatine (Table 4 and Supplementary Fig. S4A,B). Arcaine produced uncompetitive and competitive inhibition plots for acetyl-CoA and agmatine (Table 4 and Supplementary Fig. S4C,D). As observed for arcaine, arginine methyl ester produced uncompetitive and competitive inhibition plots for acetyl-CoA and agmatine (Table 4 and Supplementary Fig. S3). These data demonstrate that AgmNAT catalyzes the formation of N-acetylagmatine through an ordered sequential mechanism: acetyl-CoA binding first followed by agmatine to generate the AgmNAT•acetyl-CoA•agmatine complex prior to catalysis. This is similar to the kinetic mechanism for other D. melanogaster GNAT enzymes, including AANATA, AANATL2, and AANATL715,16,17.
Support for ordered sequential mechanism for AgmNAT comes from a statistically better fit to Equation 3 (as shown in Fig. 4) and the noncompetitive inhibition of oleoyl-CoA vs. agmatine (Table 4 and Supplementary Fig. S4A,B). Additional support and further details for the kinetic mechanism are revealed by N-acetylagmatine product inhibition. N-Acetylagmatine produced uncompetitive and competitive inhibition plots for acetyl-CoA and agmatine (Table 4 and Supplementary Fig. S5). Uncompetitive inhibition by N-acetylagmatine vs. acetyl-CoA (Supplementary Fig. S5A) is inconsistent with a ping pong kinetic mechanism. In sum, the kinetic analyses are consistent with two kinetic mechanisms: (a) ordered sequential substrate binding with acetyl-CoA binding first followed by ordered sequential product release with N-acetylagmatine being released last or (b) ordered sequential substrate binding with acetyl-CoA binding first followed by ordered sequential product release with CoA-SH being being released last. Uncompetitive inhibition by N-acetylagmatine vs. acetyl-CoA would be explained by the formation of a non-productive AgmNAT•acetyl-CoA•N-acetylagmatine complex with no reversible connection between the AgmNAT•acetyl-CoA complex and the AgmNAT•CoA-SH complex. We favor the latter mechanism because we have demonstrated that CoA-SH will bind to other D. melanogaster AANATs15,61 and many other N-acetyltransferases exhibit ordered product release with CoA-SH being released last71,72,73,74.
Proposed AgmNAT chemical mechanism
We combined the pH-dependence of the kinetic constants, primary sequence alignment to other D. melanogaster GNAT enzymes15, determination of three-dimensional structure, and site-directed mutagenesis of a putative catalytically important residue to provide insights into the AgmNAT chemical mechanism. First, the pH-dependence of the kinetic constants was assessed for acetyl-CoA to assign apparent pKa values to ionizable groups involved in catalysis. Both the kcat,app and (kcat/Km)app pH-rate profiles produced a rising profile with a pKa,app of 7.7 ± 0.1 and 7.3 ± 0.2, respectively (Fig. 5). An apparent pKa of ~7.5 can be attributed to a general base in catalysis, likely either deprotonation of the primary amine of agmatine or the zwitterionic tetrahedral intermediate generated upon nucleophilic attack of agmatine at the carbonyl thioester of acetyl-CoA. A second, higher pKa,app, possibly resulting from the deprotonation of a catalytically important general acid, was not observed in our pH-activity data, a surprising result given that a pKa ~8.5–9.5 has been observed for many other N-acyltransferases2,75,76. Explanations for these data include: (a) AgmNAT catalysis does not require a general acid, (b) the general acid in catalysis is not rate-limiting under our assay conditions, or (c) the general acid in AgmNAT catalysis has an apparent pKa > 9.5. Because of the high rate of base-catalyzed acyl-CoA hydrolysis, we cannot perform experiments at pH > 9.5 to define a pKa > 9.5.

Wild-type pH-rate profiles. (A) kcat,app for acetyl-CoA. (B) (kcat/Km)app for acetyl-CoA.
Next, we combined information from primary sequence alignments, the AgmNAT structure, and site-directed mutagenesis to define potential amino acids that could function in catalysis. A conserved glutamate has been proposed as the catalytic base in two D. melanogaster arylalkylamine N-acetyltransferases (AANATs), which corresponds to Glu-34 in AgmNAT15,16. Additionally, the AgmNAT structure shows that Glu-34 is in the active site, a buried region with several structural waters positioned within proximity of Glu-34 (Fig. 1B), similar to D. melanogaster AANATA (PDB code: 3TE4)15. Ordered water molecules within the active site of other GNAT enzymes are thought to form a “proton wire” that assists the general base in catalysis2,15,17,63,75,76,77. Although only a number of water molecules (36 in total) were sufficiently ordered to be modeled in the current structure, the majority of them are in the active sites of the two monomers. The closest ordered water molecules to Glu-34 is ~ 3.7 Å from the Oε1, positioned slightly too far for a hydrogen bond; however, we anticipate that the conformational changes upon substrate binding could promote hydrogen bond interactions between ordered water molecules and the functional groups in AgmNAT and substrate. Such hydrogen bonds could facilitate proton transfer from the amine substrate to initiate catalysis. In addition, unlike Glu-33, which is exposed to the bulk solvent, Glu-34 is relatively sheltered and placed close to the hydrophobic core of the protein and next to residues such as Leu-36. This microenvironment could be responsible for a pKa shift of Glu-34, as that identified in the pH-rate profiles. Therefore, we sought to interrogate the catalytic role of Glu-34 by evaluating the kinetic constants of the E34A mutant. The E34A mutation produced a catalytically deficient enzyme, exhibiting only 0.05–0.07% of the wildtype kcat,app value indicating that Glu-34 does function in the catalytic cycle. Furthermore, Glu-34 seems to have a role in substrate binding because the Km,app values for both agmatine and acetyl-CoA for the E34A mutant differ from wildtype values, the Km,app for agmatine increases 20-fold and the Km,app for acetyl-CoA decreases 6-fold (Table 5). The data generated for the E34A mutant is consistent, but does not prove, that Glu-34 serves as the general base in AgmNAT catalysis. To further investigate the role of Glu-34 in catalysis, we generated pH-activity profiles for the E34A mutant (Fig. 6). The kcat,app profile produced a pH-dependent linear increase with slope of 0.7 and (kcat/Km)app profile with no slope. Attempts to titrate the pH < 8.0 were unsuccessful, by which a rate of CoA-SH release was not observed above the background hydrolysis rate. The linear profile in both the kcat,app and (kcat/Km)app pH profiles, combined with the deficiency in catalytic rate suggest that Glu-34 serves as the general base in catalysis.

E34A pH-rate profiles. (A) kcat,app for acetyl-CoA. (B) (kcat/Km)app for acetyl-CoA.
Our steady-state kinetic data identified an ordered sequential mechanism with acetyl-CoA binding first, followed by agmatine to generate the AgmNAT•acetyl-CoA•agmatine ternary complex prior to catalysis. After the ternary complex formation, Glu-34 functions as the general base to deprotonate the positively charged amine moiety of agmatine, most likely involving a “proton wire” of ordered water molecules, followed by nucleophilic attack of the carbonyl of the acetyl-CoA thioester to generate a zwitterionic tetrahedral intermediate. Breakdown of the intermediate ensues by the departure of coenzyme A, which is, most likely, protonated by the positively charged amine of the intermediate (Fig. 7). This mechanism is consistent with other proposed chemical mechanisms for the N-acyltransferases of D. melanogaster and other organisms15,16,24,78.

Proposed chemical mechanism for D. melanogaster AgmNAT.
Other amino acids in AgmNAT that function in substrate binding and modulating catalysis
In addition to Glu-34, three other amino acids were individually mutated to alanine to define their function. These residues, Pro-35, Ser-171, and His-206, are conserved between D. melanogaster GNAT enzymes15 and are proposed to function in active site formation, substrate binding, and/or regulation of catalysis16,17. The P35A mutant is catalytically deficient, with a kcat,app value that is ~2% of wildtype, while exhibiting only minimal Km,app differences when compared to wildtype for both acetyl-CoA and agmatine (Table 5). Similar results were observed for the corresponding proline in other GNAT enzymes, except most exhibited a significant Km increase for the corresponding amine, suggesting a role in substrate binding. Furthermore, the structure of sheep serotonin N-acetyltransferase (PDB code: 1CJW), co-crystalized with the tryptamine-acetyl-CoA bisubstrate inhibitor, shows that the corresponding Pro-64 interacts with this inhibitor via a CH-π interaction with the negatively charged face of the aromatic tryptamine moiety77,79. Agmatine lacks an aromatic moiety; thus, the Pro-35 of AgmNAT cannot form a CH-π interaction with agmatine, which we propose is the reason for no Km effect for the P35A mutant. The decrease in kcat,app for the P35A mutant indicates that Pro-35 is important for catalysis, but it seems unlikely that Pro-35 directly functions in catalysis. More likely, Pro-35 regulates active site dynamics contributing to the transition of AgmNAT from a low activity → high activity state, a conformational feature observed for other GNAT enzymes15,16,17,79. In the current AgmNAT structure, Pro-35 is stacked on top of the imidazole ring of His-206 side chain (Fig. 2). The extensive van der Waals interaction may make significant contributions to particular active site configurations.
Another active site residue evaluated for its role in substrate binding and catalysis is Ser-171. The S171A mutant only retained ~9% of the wildtype kcat,app and also showed a 3- to 4-fold change in the Km,app values for the substrates (a decrease in the Km,app for acetyl-CoA and an increase in the Km,app for agmatine) (Table 5). The decrease in the kcat,app could be interpreted that Ser-171 functions as a general acid in catalysis to protonate CoA-S- as it leaves the AgmNAT active site. For Ser-171 to function as a general acid during catalysis, the pKa of the serine hydroxyl would have to decrease by ~3-5 pH units to protonate the thiolate anion of the CoA product. We did not observe an apparent pKa in the pH-rate profiles that would correspond to a general acid, arguing against Ser-171 serving in this role. Alternatively, Ser-171 could have an important role in organizing the active site architecture to accommodate both substrates to enable efficient catalysis. Ser-171 is located in the active site, where its Oγ side chain atom forms hydrogen bonds with the backbone oxygen and nitrogen atoms of Ser-168, and a water-mediated interaction with the Thr-167 backbone nitrogen atom, suggesting that the 165–169 strand region in addition to Ser-171 is important in stabilizing the active site pocket to accommodate both substrates and allow for efficient catalysis to occur (Fig. 1C).
The H206A mutant resulted in a kcat,app value that is ~18-fold lower than the wild-type value, whereas the Km,app-acetyl-CoA and Km,app-agmatine increased 2.3-fold and 1.4-fold, respectively. The corresponding residue (His-220) in D. melanogaster AANATA15 was shown to interact with Tyr-185 and Pro-48 to form part of the active site, an interaction potentially resulting from a conformational change driven by acetyl-CoA binding. We assign a similar function for His-206 in AgmNAT since its general location in the active site is similar to His-220 in D. melanogaster AANATA, and the van der Waals interaction with Pro-35, as described above, is conserved (Fig. 2B). In addition, the His-206 side chain is in van der Waals contact with Ser-168 Cα and Tyr-188 Cε2, as well as several local prolines, Pro-203 and Pro-205. This means that His-206 is contributing to the formation of the active site by interacting with multiple residues. The apo-AgmNAT structure shows Tyr-170 in a position that is not optimal for a direct interaction with His-206 (Fig. 2A), unlike that shown for the corresponding residues in the AANATA structure co-crystalized with acetyl-CoA15,60. Tyr-170 occupies space near the entry point for acetyl-CoA into its binding pocket; therefore, we predict that a conformational change will occur that will move Tyr-170 into position for optimal acetyl-CoA binding, possibly by interacting with His-206.
The findings presented in this manuscript highlight mechanistic and structural insights for D. melanogaster AgmNAT, an enzyme that catalyzes the formation of N-acetylagmatine from acetyl-CoA and agmatine. We provide evidence for an underappreciated reaction in arginine metabolism; however, it still remains unclear if N-acetylation of agmatine by an N-acetyltransferase enzyme is biologically relevant. A combination of data provided herein and reported from other labs speaks to its relevancy, warranting further investigation into this chemical transformation as a part of arginine metabolism. Furthermore, we outline a chemical mechanism for the AgmNAT-catalyzed formation of N-acetylagmatine (and, by extension, other N-acylamides), which is consistent with the data presented herein. We also provide evidence for important active site residues involved in substrate binding and maintaining the structural integrity of the active site for efficient catalysis, though further work is necessary to provide more evidence for the dynamic nature of the AgmNAT active site.
Methods
Materials
The AgmNAT gene was codon optimized and synthesized by Genscript. Ambion RETROscript® Kit, ProBond™ nickel-chelating resin, and MicroPoly(A) PuristTM was purchased from Invitrogen. Oligonucleotides were purchased from Eurofins MWG Operon. PfuUltra High-Fidelity DNA polymerase was purchased from Agilent. BL21 (DE3) E.coli cells and pET-28a(+) vector were purchased from Novagen. NdeI, XhoI, Antarctic Phosphatase, and T4 DNA ligase were purchased from New England Biolabs. Kanamycin monosulfate and IPTG were purchased from Gold Biotechnology. Acyl-CoAs were purchased from Sigma-Aldrich. Cayman Chemical commercially synthesized N 1-acetylspermidine. All other reagents were of the highest quality and purchased from either Sigma-Aldrich or Fisher Scientific.
AgmNAT: sub-cloning, expression, and purification
AgmNAT was inserted into a pET-28a vector using NdeI and XhoI restriction sites, yielding the final expression vector: AgmNAT-pET-28a, that after transformation into E.coli BL21 (DE3) cells expressed a protein with an N-terminal His6-tag followed by a thrombin cleavage site. The E. coli BL21 (DE3) cells containing the AgmNAT-pET-28a vector was cultured using LB media supplemented with 40 μg/mL kanamycin at 37 °C. The culture was induced with 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at an OD600 ~ 0.6, followed by an additional four hours at 37 °C. The final culture was harvested by centrifugation at 5,000 × g for 10 min at 4 °C and the pellet was collected. The pellet was resuspended in 20 mM Tris, pH 7.9, 500 mM NaCl, 5 mM imidazole, lysed by sonication, and then centrifuged at 10,000 × g for 15 min at 4 °C. The supernatant was collected and loaded onto 6 mL of ProBond™ nickel-chelating resin, followed by two wash steps: wash one – 10 column volumes of 20 mM Tris, pH 7.9, 500 mM NaCl, 5 mM imidazole followed by wash two – 10 column volumes of 20 mM Tris, pH 7.9, 500 mM NaCl, 60 mM imidazole. AgmNAT was eluted in 1 mL fractions using 20 mM Tris, pH 7.9, 500 mM NaCl, 500 mM imidazole, the protein pooled, and extensively dialyzed at 4 °C against 20 mM Tris pH 7.4, 200 mM NaCl. The concentration of AgmNAT was determined using the Bradford assay indexed against BSA as a standard, and purity was assessed by a SDS-PAGE gel (proteins visualized using by Coomassie stain). Purification of recombinant AgmNAT by nickel affinity chromatography yielded pure protein (≥95%) as visualized by SDS-PAGE (Supplementary Fig. S6).
AgmNAT crystallography
After nickel-affinity purification, 30 mg of AgmNAT was subjected to dialysis against 50 mM HEPES pH 8.2, 200 mM NaCl, followed by removal of the His6 affinity-tag using 60 U of biotinylated thrombin for 18 h in a fresh batch of 50 mM HEPES pH 8.2, 200 mM NaCl leaving an unnatural Gly-Ser-His at the N-terminus. The protein mixture was again subjected to nickel-affinity chromatography to remove undigested AgmNAT. AgmNAT was eluted in the 20 mM Tris, pH 7.9, 500 mM NaCl, 60 mM imidazole fraction, whereas the His6-AgmNAT was retained on the column until eluted with 20 mM Tris, pH 7.9, 500 mM NaCl, 500 mM imidazole. The biotinylated thrombin was removed by using 3 mL of Pierce monomeric avidin agarose resin at 4 °C for 30 min, followed by centrifugation to recover AgmNAT, and AgmNAT concentrated to ~10 mg/mL by ultrafiltration. Further purification was performed using a HiTrap Q FF column with a linear gradient from 50 mM HEPES pH 8.2 to 50 mM HEPES pH 8.2, 0.5 M NaCl with AgmNAT eluting in fractions containing ~150 mM NaCl. A final SEC purification step was used after the ion exchange step and purified AgmNAT was concentrated to ~8 mg/ml in 50 mM HEPES pH 8.2, 100 mM NaCl for crystallization screening. The Phoenix crystallization robot and Qiagen screening kits were used to evaluate different crystallization conditions for AgmNAT. AgmNAT was crystallized using the hanging-drop vapor diffusion method in 100 mM Tris pH 8.0, 200 mM sodium acetate, 30% PEG 4000. The drop contained a 1:1 ratio of 1 μL of 8 mg/mL AgmNAT with 1 μL of well solution and incubated at 20 °C. Crystals were of elongated rod-shape. Diffraction was measured at the 22-ID-D SER-CAT beamline at the Advanced Photon Source (APS), Argonne, IL. Data were indexed, scaled, and merged with iMosflm using the CCP4 suite80. A homology model was constructed based on the AgmNAT sequence using the program SWISS-MODEL81 with mosquito arylalkylamine N-acetyltransferase (PDB ID 4FD4)21 as a template for molecular replacement. The molecular replacement program Phaser-MR was used in PHENIX. The models of refinement were first obtained using a rigid-body refinement using phenix.refine in PHENIX. PHENIX82 and Coot83 were used to complete the model rebuilding and refinement. For refinement, data was cut at 2.3 A due to relatively poor data quality at higher resolutions. The crystal structure has been deposited into the Protein Data Bank with accession code 5K9N.
Construction of AgmNAT site-directed mutants
Site-directed mutants of AgmNAT were constructed by the overlap extension method. Using the primers shown in Table S1, each mutant was amplified using pfuUltra High-Fidelity DNA polymerase with the following PCR conditions: initial denaturing step of 95 °C for 2 min, then 30 cycles of 95 °C for 30 s; 60 °C annealing temperature for 30 s; 72 °C extension step for 1 min; then a final extension step of 72 °C for 10 min. Following the amplification of the AgmNAT site-directed mutant, the sub-cloning, expression, and purification procedures are the same as discussed for the wild-type enzyme.
Measurement of enzyme activity
Steady-state kinetic constants for AgmNAT were determined by measuring the rate of coenzyme A release using Ellman’s reagent (DTNB) at 412 nm (molar absorptivity = 13,600 M−1 cm−1)15,16,17. The assay consisted of 300 mM Tris pH 8.5, 150 μM DTNB, and the desired concentration of acyl-CoA and amine substrates. Initial velocities were measured using a Cary 300 Bio UV-Visible spectrophotometer at 22 °C. Acyl-CoA kinetic constants were evaluated by holding the concentration of agmatine at a constant saturating concentration (5 mM). Amine kinetic constants were evaluated by holding the concentration of acetyl-CoA at a constant saturating concentration (500 μM). The apparent kinetic constants were determined by fitting the resulting data to equation 1 using SigmaPlot 12.0, where vo is the initial velocity, Vmax,app is the apparent maximal velocity, [S] is the substrate concentration, and Km,app is the apparent Michaelis constant. Each assay was performed in triplicate and the uncertainty for the kcat,app and (kcat/Km)app values were calculated using equation 2, where σ is the standard error.
Kinetic mechanism and inhibitor analysis
Defining the kinetic mechanism of AgmNAT was accomplished by evaluating double reciprocal plots of initial velocity data for acetyl-CoA and agmatine, followed by determining the type of inhibition for substrate analogs used as dead-end inhibitors or N-acetylagmatine for product inhibition. Initial velocities were determined by varying the concentration of one substrate, while holding the other substrate at a fixed concentration. Acetyl-CoA was evaluated at 20, 50, 100, 250 and 500 μM, whereas agmatine was evaluated at 60, 300, 750 and 1500 μM. The resulting initial velocity data was fit to equation 3 for an ordered Bi-Bi mechanism and equation 4 for a ping pong mechanism using IGOR Pro 6.34 A, where vo is the initial velocity, Vmax is the maximal velocity, Kia is the dissociation constant for substrate A, Kb is the Michaelis constant for substrate B, Ka is the Michaelis constant for substrate A, [A] is the concentration of substrate A, and [B] is the concentration of substrate B.
Inhibition experiments by either substrate analogs or N-acetylagamatine were used to discriminate between an ordered, random sequential, or ping pong kinetic mechanism. Oleoyl-CoA, arcaine, and L-arginine methyl ester were used as dead-end inhibitors for AgmNAT while N-acetylagmatine was used for product inhibition. Initial velocity patterns were generated by varying the concentration of one substrate, holding the other substrate concentration at its apparent Km, and changing the concentration of inhibitor for each data set in triplicate. The resulting data was fit to equations 5–7, for competitive, noncompetitive, and uncompetitive inhibition respectively using SigmaPlot 12.0. For equations 4–6, vo is the initial velocity, Vmax,app is the apparent maximal velocity, Km,app is the apparent Michaelis constant, [S] is the substrate concentration, [I] is the inhibitor concentration, and Ki is the inhibition constant.
Rate versus pH
The pH-dependence on the kinetic constants for acetyl-CoA was determined using intervals of 0.5 pH units, ranging from 6.5–9.5. Buffers used to measure the pH-dependence were MES (pH 6.5 and 7.0), Tris (pH 7.0–9.0), AmeP (pH 9.0 and 9.5). The resulting data were fit to equations 8 (log (kcat/Km)app – acetyl-CoA and equation 9 (log kcat,app – acetyl-CoA) to determine the apparent pKa values using IGOR Pro 6.34 A, where c is the pH-independent plateau. The wild-type enzyme is reported in triplicate, whereas the E34A mutant was evaluated in duplicate.
Synthesis of N-acetylagmatine
We synthesized N-acetylagmatine from putrescine, first converting putrescine to agmatine.
Agmatine
To a solution of putrescine (2.0 g, 22.7 mmol) in water (20 mL) was added 2-methylisouronium sulfate (2.7 g, 11 mmol). The mixture was heated to 50 °C for 6 hours, then cooled in an ice bath for 30 minutes. During this time, a white precipitate was formed, which was collected by filtration, and then washed with ice water to give agmatine (1.3 g, 44%) as a white solid that was used without further purification.
1H NMR (500 MHz, D2O) δ 3.08 (t, J = 6.0 Hz, 2 H), 2.81 (t, J = 6.8 Hz, 2 H), 1.53 (br. s., 4 H) ppm.13C NMR (126 MHz) δ 156.7, 40.5, 39.1, 25.0, 24.7 ppm. LRMS (ESI) m/z: [M + H]+ 131.1. HRMS m/z: [M + H]+ Calculated for C5H15N4 131.1297; Found 131.1290.
N-Acetylagmatine
To a mixture of agmatine (1.0 g, 7.62 mmol) in pyridine (10 mL) was added acetyl chloride (542 μL, 7.62 mmol) dropwise. The mixture was allowed to stir at room temperature for 4 hours, then was concentrated on a rotary evaporator. The crude residue was adsorbed onto silica gel and purified by flash column chromatography (methylene chloride/methanol 19:1) to give N-acetylagmatine (400 mg, 30%) as a viscous, colorless oil.
1H NMR (500 MHz, CD3OD) δ 3.15–3.19 (t, J = 6.70 Hz, 2 H), 3.16–3.20 (t, J = 6.78 Hz, 2 H) 1.94 (s, 3 H), 1.52–1.64 (m, 4 H) ppm.13C NMR (126 MHz) δ 172.0, 157.7, 40.7, 38.3, 26.2, 25.7, 21.1 ppm. LRMS (ESI) m/z: [M + H]+ 172.9. HRMS m/z: [M + H]+ Calculated for C5H15N4 173.1402; Found 173.1402.
References
Merkler, D. J., Merkler, K. A., Stern, W. & Fleming, F. F. Fatty acid amide biosynthesis: a possible new role for peptidylglycine α-amidating enzyme and acyl-CoA:glycine N-acyltransferase. Arch. Biochem. Biophys. 330, 430–434 (1995).
Vetting, M. W. et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226 (2005).
Buda, M. & Klein, D. C. Structure and Function of Monoamine Enzymes in Modern Pharmacology-Toxicology, Vol. 10 (eds Usdin, E., Weiner, N. & Youdim, M.B.H.) 527–544 (Marcel Dekker, 1977).
King, C. M. & Glowinski, I. B. Acetylation, deacetylkation and acyltransfer. Environ. Health Perspect. 49, 43–50 (1983).
Sloley, B. D. & Juorio, A. V. Monoamine neurotransmitters in invertebrates and verterbrates: an examination of the diverse enzymatic pathways utilized to synthesize and inactivate biogenic amines. Int. Rev. Neurobiol. 38, 253–303 (1995).
Hearse, D. J. & Weber, W. W. Multiple N-acetyltransferases and drug metabolism. Biochem. J. 132, 519–526.
Sim, E., Payton, M., Noble, M. & Minchin, R. An update on genetic, structural and functional studies of arylamine N-acetyltransferases in eucaryotes and procaryotes. Hum. Mol. Genet. 9, 2435–2441 (2000).
Glenn, A. E., Karagianni, E. P., Ulndreaj, A. & Boukouvala, S. Comparative genomic and phylogenetic investigation of the xenobiotic metabolizing arylamine N-acetyltransferase enzyme family. FEBS Lett. 584, 3158–3164 (2010).
Tortoriello, G. et al. Targeted lipidomics in Drosophila melanogaster identifies novel 2-monoacylglycerolsa and N-acyl amides. PLoS One 8, e67865, https://doi.org/10.1371/journal.pone.0067865 (2013).
Jeffries, K. A. et al. Drosophila melanogaster as a model system to study long-chain fatty acid amide metabolism. FEBS Lett. 588, 1596–1602 (2014).
Adams, M. D. et al. The genome sequence of Drosophila melanogaster. Science 287, 2185–2195 (2000).
Phelps, C. B. & Brand, A. H. Ectopic gene expression in Drosophila using GAL4 system. Methods 14, 367–379 (1998).
Brodbeck, D. et al. Molecular and biochemical characterization of the aaNAT1 (Dat) Molecular and biochemical characterization of the aaNAT1 (Dat) locus in Drosophila melanogaster: differential expression of two gene products. DNA Cell. Biol. 17, 621–633 (1998).
Amherd, R. et al. Purification, cloning, and characterization of a second arylalkylamine N-acetyltransferase from Drosophila melanogaster. DNA Cell Biol. 19, 697–705 (2000).
Dempsey, D. R. et al. Mechanistic and structural analysis of Drosophila melanogaster arylalkylamine N-acetyltransferases. Biochemistry 53, 7777–7793 (2014).
Jeffries, K. A. et al. Identification of an arylalkylamine N-acyltransferase from Drosophila melanogaster that catalyzes the formation of long-chain N-acylserotonins. FEBS Lett. 588, 594–599 (2014).
Dempsey, D. R., Carpenter, A.-M., Rodriguez Ospina, S. & Merkler, D. J. Probing the chemical mechanism and critical regulatory amino acid residues of Drosophila melanogaster arylalkylamine N-acyltransferase like 2. Insect Biochem. Mol. Biol. 66, 1–12 (2015).
Chu, C. J. et al. N-Oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J. Biol. Chem. 278, 13633–13639 (2003).
Verhoeckx, K. C. M. et al. Presence, formation and putative biological activities of N-acyl serotonins, a novel class of fatty-acid derived mediators, in the intestinal tract. Biochim. Biophys. Acta 1811, 578–586 (2011).
Moreteau, B. & Chaminade, N. The effects of lindane poisoning on N-acetyldopamine and N-acetyl 5-hydroxytryptamine concentrations in the brain of Locusta migratoria L. Ecotoxicol. Environ. Saf. 20, 115–120 (1990).
Han, Q., Robinson, H., Ding, H., Christensen, B. M. & Li, J. Evolution of insect arylalkylamine N-acetyltransferases: structural evidence from the yellow fever mosquito, Aedes aegypti. Proc. Natl. Acad. Sci. USA 29, 11669–11674 (2012).
Tsugehara, T., Imai, T. & Takeda, M. Characterization of arylalkylamine N-acetyltransferase from silkmoth (Antheraea pernyi) and pesticidal drug edesign base on the baculovirus-expressed enzyme. Comp. Biochem.Physiol. C Toxicol. Pharmacol. 157, 93–102 (2013).
Lourenço, B. L. A. et al. Virtual screening and molecular docking for arylalkylamine-N-acetyltransferase (aaNAT) inhibitors, a key enzyme of Aedes (Stegomyia) aegypti (L.) metabolism. Comput. Mol. Biosci. 5, 35–44 (2015).
Zheng, W. & Cole, P. A. Serotonin N-acetyltransferase: mechanism and inhibition. Curr. Med. Chem. 9, 1187–1199 (2002).
Kossel, A. Über das agmatin. Z. Physiol. Chem. 66, 257–261 (1910).
Gale, E. F. Estimation of 1 (+)-arginine in protein hydrolysates by the use of 1 (+)-arginine decarboxylase. Nature 157, 265 (1946).
Piletz, J. E. et al. Agmatine: clinical applications after 100 years in translation. Drug Discov. Today 18, 880–893 (2013).
Li, G. et al. Agmatine: an endogenous clonidine-displacing substance in the brain. Science 263, 966–969 (1994).
Halaris, A. & Plietz, J. Agmatine: metabolic pathway and spectrum of activity in brain. CNS Drugs 21, 885–900 (2007).
Raasch, W., Regunathan, S., Li, G. & Reis, D. J. Agmatine is widely and unequally distributed in rat organs. Ann. N. Y. Acad. Sci. 763, 330–334 (1995).
Molderings, G. J. et al. Gastrointestinal uptake of agmatine: distribution in tissues and organs and pathophysiologic relevance. Ann. N. Y. Acad. Sci. 1009, 44–51 (2003).
Meeley, M. P., Hensley, M. L., Ernsberger, P., Felsen, D. & Reis, D. J. Evidence for a bioactive clonidine-displacing substance in peripheral tissues and serum. Biochem.Pharmacol. 44, 733–740 (1992).
Otake, K. et al. Regional localization of agmatine in the rat brain: an immunocytochemical study. Brain Res. 787, 1–14 (1998).
Reis, D. J., Yang, X. C. & Milner, T. A. Agmatine containing axon terminals in rat hippocampus form synapses on pyramidal cells. Neurosci. Lett. 250, 185–188 (1998).
Salvi, M. et al. Agmatine is transported into liver mitochondria by a specific electrophoretic mechanism. Biochem. J. 396, 337–345 (2006).
Grillo, M. A. et al. Inhibition of agmatine transport in liver mitochondria by new charge-deficient agmatine analogouses. Biochem. Soc. Trans. 35, 401–404 (2007).
Battaglia, V. et al. Agmatine transport in brain mitochondria: a different mechanism from that in liver mitochondria. Amino Acids 38, 423–430 (2010).
Horyn, O. et al. Biosynthesis of agmatine in isolated mitochondria and perfused rat liver: studies with 15N-labelled arginine. Biochem. J. 388, 419–425 (2005).
Gorbatyuk, O. S., Milner, T. A., Wang, G., Regunathan, S. & Reis, D. J. Localization of agmatine in vasopressin and oxytocin neurons of the rat hypothalamic paraventricular and supraoptic nuclei. Exp. Neurol. 171, 235–245 (2001).
Molderings, G. J. & Haenisch, B. Agmatine (decarboxylated L-arginine): physiological role and therapeutic potential. Pharmacol. Ther. 133, 351–365 (2012).
Raasch, W., Schafer, U., Chun, J. & Dominiak, P. Biological significance of agmatine, an endogenous ligand at imidazoline binding sites. Br. J. Pharmacol. 133, 755–780 (2001).
Li, F. et al. Imidazoline receptor antisera-selected/Nischarin regulates the effect of agmatine on the development of morphine dependence. Addict. Biol. 17, 392–408 (2012).
Pinthing, D. et al. Agmatine recognizes alpha 2-adrenoceptor binding sites but neither activates nor inhibits alpha 2-adrenoceptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 351, 10–16 (1995).
Loring, R. H. Agmatine acts as an antagonist of neuronal nicotinic receptors. Br. J. Pharmacol. 99, 207–211 (1990).
Yang, X. C. & Reis, D. J. Agmatine selectively blocks the N-methyl-D-aspartate subclass of glutamate receptor channels in rat hippocampal neurons. J. Pharmacol. Exp. Ther. 288, 544–594 (1999).
Taksande, B. G., Kotagale, N. R., Tripathi, S. J., Ugale, R. R. & Chopde, C. T. Antidepressant like effect of selective serotonin reuptake inhibitors involve modulation of imidazoline receptors by agmatine. Neuropharmacology 57, 415–424 (2009).
Hamana, K., Suzuki, M., Wakabayashi, T. & Matsuzaki, S. Polyamine levels in the gonads, sperm and salivary gland of cricket, cockroach, fly and midge. Comp. Biochem. Physiol. B 92, 691–695 (1989).
Hamana, K., Niitsu, M., Samejima, K. & Matsuzaki, S. Novel polyamines in insects and spiders. Comp. Biochem. Physiol. B 100, 399–402.
Hamana, K., Uemiya, H. & Niitsu, M. Polyamines of primitive apterygotan insects: springtails, silverfish and a bristletail. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 137, 75–79 (2004).
Shen, W. et al. Development of chemical isotope labeling liquid chromatography mass spectrometry for silkworm hemolymoph metabolomics. Anal. Chim. Acta 942, 1–11 (2016).
Jeong, S. E., Lee, Y., Hwang, J. H. & Knipple, D. C. Effects of the sap of the common oleander Nerium indicum (Apocyanaceae) on male fertility and spermatogenesis in the oriental tobacco budworm Helicoverpa assulta (Lepidoptera, Noctuidae). J. Exp. Biol. 204, 3935–3942 (2001).
Morris, S. M. Jr. Recent advances in arginine metabolism. Curr. Opin. Clin. Nutr. Metab. Care 7, 45–51 (2004).
Holt, A. & Baker, G. B. Metabolism of agmatine (clonidine-displacing substance) by diamine oxidase and the possible implications for studies of imidazoline receptors. Prog. Brain Res. 106, 187–197 (1995).
Cabella, C. et al. Transport and metabolism of agmatine in rat hepatocyte cultures. Eur. J. Biochem. 268, 940–947 (2001).
Satriano, J. et al. Suppression of inducible nitric oxide generation by agmatine aldehyde: beneficial effects in sepsis. J. Cell. Physiol. 188, 313–320 (2001).
Ohnuma, M. et al. N 1-Aminopropylagmatine, a new polyamine produced as a key intermediate in polyamine biosynthesis of an extreme thermophile, Thermus thermophilus. J. Biol. Chem. 280, 30073–30082 (2005).
Ikeguchi, Y., Bewley, M. C. & Pegg, A. E. Aminopropyltransferases: function, structure and genetics. J. Biochem. 139, 1–9 (2006).
von Röpenack, E., Parr, A. & Schulze-Lefert, P. Structural analyses and dynamics of soluble and cell wall-bound phenolics in a broad spectrum resistance to the powdery mildew fungus in barley. J. Biol. Chem. 273, 9013–9022 (1998).
Muroi, A. et al. Accumulation of hydroxycinnamic acid amides induced by pathogen infection and identification of agmatine coumaroyltransferase in Arabidopsis thaliana. Planta 230, 517–527 (2009).
Cheng, K.-C., Liao, J.-N. & Lyu, P.-C. Crystal structure of the dopamine N-acetyltransferase-acetyl-CoA complex provides insights into the catalytic mechanism. Biochem. J. 446, 395–404 (2012).
Dempsey, D. R. et al. Mechanistic and structural analysis of a Drosophila melanogaster enzyme, arylalkylamine N-acetyltransferase like 7, an enzyme that catalyzes the formation of N-acetylarylalkylamides and N-acetylhistamine. Biochemistry 54, 2644–2658 (2015).
Dempsey, D. R. et al. Identification of an arylalkylamine N-acyltransferase from Drosophila melanogaster that catalyzes the formation of long-chain N-acylserotonins. FEBS Lett. 588, 594–599 (2014).
Forouhar, F. et al. Structural and functional evidence for Bacillus subtilis PaiA as a novel N 1-spermidine/spermine acetyltransferase. J. Biol. Chem. 280, 40328–40336 (2005).
Wang, H., Li, J., Meng, G., Wei, K. & Ruan, J. Identification of agmatine and its acetylation metabolities in rat urine by stable isotope labeling coupled ion trap mass spectrometry. Yaowu Fenxi Zazhi 25, 131–136 (2005).
Guillou, Y. & Robin, Y. Présence ď α-N-acétylagmatine chez des Cnidaires, Actinia equina et Actinia fragacea. C. R. Seances Soc. Biol Fil. 173, 576–579 (1979).
Uzbay, T. I. The pharmacological importance of agmatine in the brain. Neurosci. Biobehav. Rev. 36, 502–519 (2012).
Moncada, S. & Higgs, A. Mechanisms of disease - the L-arginine-nitric oxide pathway. N. Engl. J. Med. 329, 2002–2012 (1993).
Moinard, C., Cynober, L. & de Bandt, J.-P. Polyamines: metabolism and implications in human diseases. Clin. Nutr. 24, 184–197 (2005).
Matsui, I., Wiegand, L. & Pegg, A. E. Properties of spermidine N-acetyltransferase from livers of rats treated with carbon tetrachlorida and its role in the conversion of spermidine into putrescine. J. Biol. Chem. 256, 2454–2459 (1981).
Hegde, S. S., Chandler, J., Vetting, M. W., Yu, M. & Blanchard, J. S. Mechanistic and structural analysis of human spermidine/spermine N 1-acetyltransferase. Biochemistry 46, 7187–7195 (2007).
De Angelis, J., Gastel, J., Klein, D. C. & Cole, P. A. Kinetic analysis of the catalytic mechanism of serotonin N-acetyltransferase (EC 2.3.1.87). J. Biol. Chem. 273, 3045–3050 (1998).
Tanner, K. G., Langer, M. R., Kim, Y. & Denu, J. M. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J. Biol. Chem. 275, 22048–22055 (2000).
Draker, K.-a, Northrup, D. B. & Wright, G. D. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6’)-Ii from Enterococcus faecium: evidence of dimer subunit cooperativity. Biochemistry 42, 6565–6574 (2003).
Johnson, C. M., Huang, B., Roderick, S. L. & Cook, P. F. Kinetic mechanism of the serine acetyltransferase from Haemophilus influenzae. Arch. Biochem. Biophys. 429, 115–122 (2004).
Favrot, L., Blanchard, J. S. & Vergnolle, O. Bacterial GCN5-related N-acetyltransferases: from resistance to regulation. Biochemistry 55, 989–1002 (2016).
Scheibner, K. A., De Angelis, J., Burley, S. K. & Cole, P. A. Investigation of the roles of catalytic residues in serotonin N-acetyltransferase. J. Biol. Chem. 277, 18118–18126 (2002).
Hickman, A. B., Namboodiri, M. A., Klein, D. C. & Dyda, F. The structural basis of ordered substrate binding by serotonin N-acetyltransferase: enzyme complex at 1.8 Å resolution with a bisubstrate analog. Cell 97, 361–369 (1999).
Berndsen, C. E. & Denu, J. M. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 18, 682–689 (2008).
Pavlicek, J. et al. Evidence that proline focuses movement of the floppy loop of arylalkylamine N-acetyltransferase (EC 2.3.1.87). J. Biol. Chem. 283, 14552–14558 (2008).
Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763 (1994).
Schwede, T., Kopp, J., Guex, N. & Peitsch, M. C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 (2003).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132 (2004).
Acknowledgements
This work has been supported, in part, by grants from the National Institutes of Health (R03-DA034323 and R15-GM107864) to D.J.M. and a Dissertation Completion Fellowship from the Office of Graduate Studies at the University of South Florida to D.R.D. In addition, this work has also been supported by the University of South Florida (a Research Scholarship grant from the College of Arts and Sciences) and the Shirley W. and William L. Griffin Foundation. We thank Amanda Pierce for help in creating the figures.
Author information
Authors and Affiliations
Contributions
D.R.D., D.A.N., M.R.B., Y.C., and D.J.M. designed the experiments and drafted the manuscript. D.R.D., M.R.B., S.R.O., B.G.O., and A.C. carried out the biochemical experiments, A.K. and J.W.L. synthesized the N-acetylagmatine, and D.A.N., M.R.B., O.P., E.M.L., and X.Z. determined the crystal structure. All authors contributed to analyzing results and in writing/editing the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dempsey, D.R., Nichols, D.A., Battistini, M.R. et al. Structural and Mechanistic Analysis of Drosophila melanogaster Agmatine N-Acetyltransferase, an Enzyme that Catalyzes the Formation of N-Acetylagmatine. Sci Rep 7, 13432 (2017). https://doi.org/10.1038/s41598-017-13669-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13669-6
This article is cited by
-
A polyamine acetyltransferase regulates the motility and biofilm formation of Acinetobacter baumannii
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
