
Abstract
As the first known Weyl semimetals, binary compounds including TaP, TaAs, NbAs, and NbP have received worldwide interest. This work explored the phase behaviours of NbAs and NbP under high pressure up to 200 GPa via first-principles calculations combined with intelligent particle swarm optimization. Upon compression, a new phase of NbAs with P-6m2 symmetry appeared above 23 GPa and remained stable until 38 GPa, whereupon a monoclinic structure with space group P2 1 /c became more energetically favourable. This lasted until 73 GPa, when a Pm-3m phase followed. Surprisingly, NbP underwent a single phase transition around 63.5 GPa to a new phase with Cmcm symmetry that was completely distinct from the structures shown by TaAs-class compounds such as NbAs and TaAs. All these newly pressure-stabilized structures were dynamically stable at both high and ambient pressure. Electronic band structure calculations revealed a transition from semimetal to metal under high pressure. This work is meaningful and fundamental for future studies and applications of TaAs-class Weyl semimetals under compression or extreme conditions.
Similar content being viewed by others


Introduction
Weyl semimetals are crystals hosting both Weyl fermions with definite chirality (low-energy electronic excitations) and Fermi arcs caused by topological surface states. They are a newly discovered class of topologically non-trivial materials beyond topological insulators. Many interesting physical characteristics have been reported, such as extremely large magnetoresistance1,2,3 and unusual transport mobility4 stemming from the chiral anomaly1, and thus these semimetals represent a new era in condensed matter physics. It is interesting to note that the Weyl nodes defined as the crossing points between two non-degenerate bands can be observed in the negative magnetoresistance measurements5. Furthermore, the transport evidence of the intriguing surface states can be realized through investigating Aharonov–Bohm oscillations6. First-principles simulations firstly predicted non-centrosymmetric transition-metal monophosphides to be Weyl semimetals7,8. Consequent photoemission spectroscopy of TaAs single crystals directly observed the Weyl semimetal state (Fermi arcs on the surface, together with Weyl fermion cones and Weyl nodes in the bulk)9,10. Independent angle-resolved photoemission experiments also confirmed TaAs as a Weyl semimetal11. NbAs was later identified experimentally as a Weyl semimetal by soft X-ray and ultraviolet photoemission spectroscopy12. Further works reported the Weyl state in other members of the same family such as TaP13 and NbP14. These first-known Weyl semimetals (all TaAs-class compounds) each have twelve pairs of Weyl nodes, which are entirely stoichiometric and nonmagnetic in the whole Brillouin zone. These promising discoveries suggest there are many exotic topological properties to be investigated in TaAs-class Weyl semimetals15,16,17,18,19, which might lead to potential applications in electronics, optoelectronics, and quantum computing.
Pressure is commonly used to alter the chemical environment to induce novel properties and produce new compounds. Combined theoretical simulation and synchrotron experiments have found a new Weyl semimetal phase of TaAs with P-6m2 symmetry and only six pairs of Weyl nodes that appeared above 14 GPa, followed by a P2 1 /c phase at around 34 GPa20. However, high-pressure studies of NbAs and NbP have been lacking up to date. This work reports a comprehensive and systematic study of new phases in NbAs and NbP under compression up to 200 GPa, and analyses their band structures and electron localization function in detail. A transition sequence of I4 1 md → P-6m2 → P2 1 /c → Pm-3m was observed in NbAs, while a single phase transition of I4 1 md → Cmcm took place in NbP. Our work is meaningful and fundamental to future theoretical and experimental investigations of TaAs-class topological Weyl semimetals under high pressure or extreme conditions.
Results
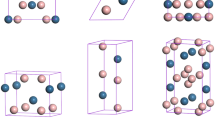
TaAs-class Weyl semimetals, comprising TaAs21, TaP22, NbAs23, and NbP24, crystallize in a body-centred tetragonal symmetry with space group I4 1 md, as shown in Fig. 1(a). The structure is distinguished by a lack of inversion symmetry, which is crucial for the Weyl semimetal state. To investigate the stable structures of NbAs and NbP under high pressure, all structures were fully geometrically optimized, allowing simultaneous variations of the unit cell and atomic positions at several pressures up to 200 GPa. The difference in enthalpy between the structures calculated at each pressure and the ground state structure is displayed in Fig. 2. The simulation identified the I4 1 md structure as the ground state in both cases, which is consistent with experimental observations. High-pressure structure searches identified the sequential emergence of three structures (with P-6m2, P2 1 /c, and Pm-3m space groups) for NbAs, but only a Cmcm symmetry structure for NbP. To fully understand the phase transitions in NbAs and NbP, we also investigated the stability of Cmcm structure of NbAs while P-6m2, P2 1 /c, and Pm-3m candidate structures of NbP since they share the same ambient structure. It is obvious to see that the NbAs with Cmcm symmetry is not energy favourable in the whole pressure from 0 GPa and 200 GPa, as illustrated in Fig. 2(a). Meanwhile, the P-6m2, P2 1 /c, and Pm-3m candidate structures of NbP have higher enthalpy as compared to the Cmcm structure from 63.5 GPa onwards, as displayed in Fig. 2(c). This further validates our structure search method adopted in this work.

Crystal structures of NbAs and NbP, in which Nb, As, and P are represented as purple, green, and yellow balls, respectively. (a) I41 md structure of NbAs at ambient pressure. (b) P-6m2 structure of NbAs at 30 GPa. (c) P21/c structure of NbAs at 50 GPa. (d) Pm-3m structure of NbAs at 80 GPa. (e) Cmcm structure of NbP at 80 GPa.

Calculated enthalpy difference per formula unit (f.u.) of different structures relative to the respective I41 md ground-state structure and calculated volume per f.u. of the lowest-enthalpy phases as a function of pressure. (a,b) NbAs up to 200 GPa. (c,d) NbP up to 120 GPa for clarity.
The lattice constants and atomic coordinates optimized for the lowest-enthalpy structures at ambient conditions, together with previous experimental data25, are listed in Table 1. The deviation of the lattice constant is within 0.67% which is in good agreement with the experimental data. The detailed structural information in Table 1 is useful to the future XRD experimental refinements. The ground state I4 1 md structure of NbAs remains stable until 23 GPa, beyond which a structure with P-6m2 symmetry is favourable up to 38 GPa. Both phases incorporate NbAs6 polyhedra. It is interesting to note that the P-6m2 phase has broken inversion symmetry and thus might be a Weyl semimetal. This possibility can be confirmed by further simulations of topological properties that are beyond the scope of our present study. At pressures beyond 38 GPa, a monoclinic P2 1 /c structure becomes the most energetically favourable phase and is predicted to be stable up to 73 GPa. Its crystal structure has inversion symmetry, and hence Weyl nodes are no longer preserved. Further compression induces a cubic phase with Pm-3m symmetry (with inversion symmetry) as most favourable from 73 to 200 GPa. These transition pressures are moderate and are accessible to the current experiment technique to further confirm our predictions. Combination of in situ Raman measurements with synchrotron XRD is commonly adopted and very effective to characterize the phase transformations behaviors under high pressure26,27,28. The coordination number of Nb increases from 6 (P-6m2) to 7 (P2 1 /c) and then to 8 (Pm-3m), as shown in Fig. 1, indicating a more dense packing under high pressure. TaAs has shown the same transition sequence under compression29. The volumetric changes are discontinuous: decreases of 0.5%, 2.9%, and 1.0% occur at 23, 40, and 73 GPa, respectively, as shown in Fig. 2(b). This is characteristic of first-order transitions. Interestingly, NbP shows only one transition, from the I41 md ground state into a new structure with Cmcm symmetry at 63.5 GPa. This is accompanied by a significant volume drop of 4.7%, as shown in Fig. 2(c,d). This transition is distinct from those in TaAs and NbAs. The Cmcm structure possesses inversion symmetry, and thus cannot be a Weyl semimetal state.
To assess dynamic stability, phonon dispersion curves are calculated for the new high-pressure structures. No imaginary phonon frequencies are found in each stable pressure range in the whole Brillouin zone (Fig. 3), indicating the phases to be dynamically stable. Calculated phonon dispersion of these structures at ambient conditions shows that the frequencies all remain positive (data not shown). These new pressure-induced phases can be inferred to remain upon decompression to ambient conditions, thereby providing the opportunity to study high-pressure exotic behaviours at ambient pressure.

Phonon dispersions of NbAs and NbP. (a) P-6m2 structure of NbAs at 30 GPa, (b) P21/c structure of NbAs at 50 GPa, (c) Pm-3m structure of NbAs at 80 GPa, and (d) Cmcm structure of NbP at 80 GPa.
It is important to explore the evolution of the electronic structures of these compounds at high pressures. Calculated electronic band structures of NbAs (I4 1 md at 0 GPa, P-6m2 at 30 GPa, P2 1 /c at 50 GPa, and Pm-3m at 80 GPa) and NbP (I4 1 md at 0 GPa and Cmcm at 80 GPa) with/without spin–orbit coupling (SOC) are shown in Fig. 4. Our results show the SOC effects give the splitting of the electronic bands degeneracies and change the crossing behaviour near the Fermi level. In the absence of SOC, NbAs shows a narrow bandgap of 0.11 eV around the N point while the SOC enhances the bandgap to 0.14 eV as illustrated in the Fig. 4(a) and its inset, indicating its semimetal character. It seems that previous crossing bands along K-Γ direction in Fig. 4(b) and crossing bands along Γ–Y direction together with D-E direction in Fig. 4(c) are fully gapped after taking SOC into account. Compression induces additional bands crossing the Fermi level and overlapping with each other, eventually leading NbAs to become a good metal with strong overlap between the conduction and valence bands in the Pm-3m phase at 80 GPa. The semimetal to metal transitions can be further confirmed through resistance measurements20. The electronic bands around Γ point change dramatically with the consideration of SOC, as indicated in Fig. 4(d). NbP is inferred to have a narrow bandgap of 0.14 eV without SOC (0.18 eV with SOC) that is also found at the N point of the Brillouin zone as shown in Fig. 4(e). With increasing pressure, the conduction band and valence band start to overlap along the Y–Γ direction, inducing a weak metallic nature. The SOC is found have a negligible influence on the band structure in the Cmcm phase of NbP, only slightly opens the gap around Y-Γ direction by 7 meV as shown in the Fig. 4(f).

Calculated electronic band structures of NbAs and NbP at different pressures without spin–orbit coupling (SOC) while the insets including the SOC effect are enlarged along prominent direction. (a) I41 md structure of NbAs at 0 GPa, (b) P-6m2 structure of NbAs at 30 GPa, (c) P21/c structure of NbAs at 50 GPa, (d) Pm-3m structure of NbAs at 80 GPa, (e) I41 md structure of NbP at 0 GPa, and (f) Cmcm structure of NbP at 80 GPa.
Electron localization function (ELF) simulation provides further understanding of high-pressure bonding behaviour. It can yield informative visual patterns of the core, binding, and lone-pair regions in chemical systems30,31,32. The results in Fig. 5 show electrons generally localized around the anions (As in NbAs and P in NbP) in all the phases. The most striking feature is the increasing localization with greater spherical symmetry around the anions in both systems as the pressure rises, indicating increasing iconicity bonding character. The charge transfer from As (P) to Nb is consistent with As (2.18) and P (2.19) being more electronegative than Nb (1.60). This pressure-induced charge transfer may also explain the phase transformations observed in NbAs and NbP.

Calculated electron localization functions of NbAs and NbP at different pressures. (a) I4 1 md structure of NbAs on the (100) surface at 0 GPa, (b) P-6m2 structure of NbAs on the \((2\bar{1}0)\) surface at 30 GPa, (c) P2 1 /c structure of NbAs on the \((30\bar{4}1)\) surface at 50 GPa, (d) Pm-3m structure of NbAs on the (110) surface at 80 GPa, (e) I4 1 md structure of NbP on the (100) surface at 0 GPa, and (f) Cmcm structure of NbP on the \((10\bar{1})\) surface at 80 GPa.
Conclusion
High-pressure structure prediction up to 200 GPa was conducted for Weyl semimetal (NbAs and NbP) using first-principles calculations combined with particle swarm optimization. NbAs showed a transition sequence of I4 1 md → P-6m2 → P2 1 /c → Pm-3m under compression with transition pressure around 23 GPa, 38 GPa, and 73 GPa, respectively. Remarkably, NbP showed a single I4 1 md → Cmcm transition around 63.5 GPa. All these newly pressure-induced structures are dynamically stable at high pressure and at ambient conditions. Electronic band structure calculations demonstrated semimetal-to-metal transitions upon compression. The influence of the SOC on the electronic band structure was examined and SOC enhances the bandgap in I4 1 md structure. Moreover, SOC effects are evidenced to give the splitting of the electronic bands degeneracies and change the crossing behaviour near the Fermi level. These results are meaningful and fundamental for future investigations of these TaAs-class Weyl semimetals under high pressure or extreme conditions.
Methods
Extensive structure searching is carried out with the swarm intelligence CALYPSO method33,34,35,36, which enables global minimization of energy surfaces realized through first-principles total-energy calculations. This method has been successfully used to investigate high-pressure structures of various materials37,38,39,40,41,42. In sampling the energy surface, chemical composition is the exclusive input parameter. Structures searching here employs a system size ranging from 2 to 4 formula units per cell, and the pressure is limited to 200 GPa. We have run 50 structure generations and 1500 random structures were generated in total. 60% of the previous generation with lower Gibbs free energy are kept while the rest 40% of current generation are generated randomly which guarantees the diversity of the structure pool. A bond characterization matrix is employed as a fingerprint technique to remove the degenerate structures which enhances the search efficiency. The underlying structural relaxations and all the electronic structure calculations are performed in the framework of density functional theory using the generalized gradient approximation (Perdew–Burke–Ernzerhof functional)43, as implemented in the Vienna ab initio simulation package (VASP)44. Projector augmented wave potentials45 with 4s24p64d45s1, 3s23p3, and 3d104s24p3 valence electrons are adopted for Nb, P, and As, respectively. To ensure the calculated enthalpy converges well to better than 1 meV/atom, plane-wave kinetic energy cutoffs of 800 eV and a Monkhorst–Pack Brillouin zone sampling grid with a resolution of 2π × 0.02 Å−1 are chosen. The electronic band structure were calculated with and without the consideration of spin–orbital coupling (SOC) duo to its essential role for the studying intriguing topological electronic states46. Previous results show that the theoretical lattice parameters, and phonon frequencies are not sensitive to SOC effects47 and thus the SOC effects have been neglected to investigate the structural optimization and lattice dynamics. The dynamic stability of the predicted structures is determined by calculating the phonon dispersion using a supercell approach48, as implemented in the PHONOPY code49.
References
Huang, X. C. et al. Observation of the Chiral-Anomaly-Induced Negative Magnetoresistance in 3D Weyl Semimetal TaAs. Physical Review X 5, 031023 (2015).
Du, J. et al. Large unsaturated positive and negative magnetoresistance in Weyl semimetal TaP. Science China Physics, Mechanics & Astronomy 59, 657406 (2016).
Zhang, C. et al. Large magnetoresistance over an extended temperature regime in monophosphides of tantalum and niobium. Physical Review B 92, 041203 (2015).
Wang, Z. et al. Helicity-protected ultrahigh mobility Weyl fermions in NbP. Physical Review B 93, 121112 (2016).
Li, C. Z. et al. Giant negative magnetoresistance induced by the chiral anomaly in individual Cd3As2 nanowires. Nature Communications 6, 10137 (2015).
Wang, L. X., Li, C. Z., Yu, D. P. & Liao, Z. M. Aharonov-Bohm oscillations in Dirac semimetal Cd3As2 nanowires. Nature Communications 7, 10769 (2016).
Weng, H. M., Fang, C., Fang, Z., Bernevig, B. A. & Dai, X. Weyl Semimetal Phase in Noncentrosymmetric Transition-Metal Monophosphides. Physical Review X 5, 011029 (2015).
Huang, S.-M. et al. A Weyl Fermion semimetal with surface Fermi arcs in the transition metal monopnictide TaAs class. Nature Communications 6, 8373 (2015).
Xu, S. Y. et al. Discovery of a Weyl fermion semimetal and topological Fermi arcs. Science 349, 613–617 (2015).
Lv, B. Q. et al. Experimental Discovery of Weyl Semimetal TaAs. Physical Review X 5, 031013 (2015).
Yang, L. X. et al. Weyl semimetal phase in the non-centrosymmetric compound TaAs. Nature Physics 11, 728−+ (2015).
Xu, S. Y. et al. Discovery of a Weyl fermion state with Fermi arcs in niobium arsenide. Nature Physics 11, 748−+ (2015).
Xu, S.-Y. et al. Experimental discovery of a topological Weyl semimetal state in TaP. Science Advances 1, e1501092 (2015).
Shekhar, C. et al. Extremely large magnetoresistance and ultrahigh mobility in the topological Weyl semimetal candidate NbP. Nature Physics 11, 645−+ (2015).
Hosur, P., Parameswaran, S. A. & Vishwanath, A. Charge Transport in Weyl Semimetals. Physical Review Letters 108, 046602 (2012).
Ojanen, T. Helical Fermi arcs and surface states in time-reversal invariant Weyl semimetals. Physical Review B 87, 245112 (2013).
Parameswaran, S. A., Grover, T., Abanin, D. A., Pesin, D. A. & Vishwanath, A. Probing the Chiral Anomaly with Nonlocal Transport in Three-Dimensional Topological Semimetals. Physical Review X 4, 031035 (2014).
Potter, A. C., Kimchi, I. & Vishwanath, A. Quantum oscillations from surface Fermi arcs in Weyl and Dirac semimetals. Nature Communications 5, 5161 (2014).
Zyuzin, A. A., Wu, S. & Burkov, A. A. Weyl semimetal with broken time reversal and inversion symmetries. Physical Review B 85, 165110 (2012).
Zhou, Y. H. et al. Pressure-Induced New Topological Weyl Semimetal Phase in TaAs. Physical Review Letters 117, 146402 (2016).
Boller, H. & Parthe, E. Transpositionstructure of Nbas and of Similar Monophosphides and Arsenides of Niobium and Tantalum. Acta Crystallographica 16, 1095−& (1963).
Schonberg, N. An X-Ray Investigation of Transition Metal Phosphides. Acta Chemica Scandinavica 8, 226–239 (1954).
Boller, H. & Parthé, E. The transposition structure of NbAs and of similar monophosphides and arsenides of niobium and tantalum. Acta Crystallographica 16, 1095–1101 (1963).
Rundqvist, S. New Metal-Rich Phosphides of Niobium Tantalum and Tungsten. Nature 211, 847−+ (1966).
Lee, C. C. et al. Fermi surface interconnectivity and topology in Weyl fermion semimetals TaAs, TaP, NbAs, and NbP. Physical Review B 92, 235104 (2015).
Kim, M. et al. Pressure and field tuning the magnetostructural phases of Mn3O4: Raman scattering and x-ray diffraction studies. Physical Review B 84, 174424 (2011).
Teredesai, P. et al. High pressure phase transition in metallic LaB6: Raman and X-ray diffraction studies. Solid State Communications 129, 791–796 (2004).
Wang, L. et al. High-pressure induced phase transitions of Y2O3 and Y2O3:Eu3+. Applied Physics Letters 94, 061921 (2009).
Lu, M. C., Guo, Y. N., Zhang, M., Liu, H. Y. & Tse, J. S. High-pressure crystal structures of TaAs from first-principles calculations. Solid State Communications 240, 37–40 (2016).
Becke, A. D. & Edgecombe, K. E. A simple measure of electron localization in atomic and molecular systems. The Journal of Chemical Physics 92, 5397–5403 (1990).
Burdett, J. K. & McCormick, T. A. Electron Localization in Molecules and Solids: The Meaning of ELF. The Journal of Physical Chemistry A 102, 6366–6372 (1998).
Savin, A. The electron localization function (ELF) and its relatives: interpretations and difficulties. Journal of Molecular Structure: THEOCHEM 727, 127–131 (2005).
Wang, H. et al. CALYPSO structure prediction method and its wide application. Computational Materials Science 112, 406–415 (2016).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Physical Review B 82, 094116 (2010).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: A method for crystal structure prediction. Computer Physics Communications 183, 2063–2070 (2012).
Li, Y., Hao, J., Liu, H., Li, Y. & Ma, Y. The metallization and superconductivity of dense hydrogen sulfide. Journal of Chemical Physics 140, 174712 (2014).
Lv, J., Wang, Y., Zhu, L. & Ma, Y. Predicted Novel High-Pressure Phases of Lithium. Physical Review Letters 106, 015503 (2011).
Wang, H., Tse, J. S., Tanaka, K., Iitaka, T. & Ma, Y. Superconductive sodalite-like clathrate calcium hydride at high pressures. Proceedings of the National Academy of Sciences of the United States of America 109, 6463–6466 (2012).
Zhu, L., Liu, H., Pickard, C. J., Zou, G. & Ma, Y. Reactions of xenon with iron and nickel are predicted in the Earth’s inner core. Nature Chemistry 6, 644–648 (2014).
Zhu, L. et al. Substitutional Alloy of Bi and Te at High Pressure. Physical Review Letters 106, 145501 (2011).
Li, Q., Zhou, D., Zheng, W., Ma, Y. & Chen, C. Global Structural Optimization of Tungsten Borides. Physical Review Letters 110, 136403 (2013).
Zhang, M. et al. Superhard BC3 in Cubic Diamond Structure. Physical Review Letters 114, 015502 (2015).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Physical Review Letters 77, 3865–3868 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Physical Review B 59, 1758–1775 (1999).
Weng, H., Dai, X. & Fang, Z. Topological semimetals predicted from first-principles calculations. Journal of Physics-Condensed Matter 28, 303001 (2016).
Zhou, D., Li, Q., Ma, Y., Cui, Q. & Chen, C. Pressure-Induced Superconductivity in SnTe: A First-Principles Study. Journal of Physical Chemistry C 117, 12266–12271 (2013).
Parlinski, K., Li, Z. Q. & Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Physical Review Letters 78, 4063–4066 (1997).
Togo, A., Oba, F. & Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Physical Review B 78, 134106 (2008).
Acknowledgements
J.Z. acknowledges funding support from the National Natural Science Foundation of China (Grant No. 11604157, 11722433, 11404148, and 11474167) and Nanjing University of Information Science & Technology (Grant No. 2016206).
Author information
Authors and Affiliations
Contributions
J.Z. performed the calculations and wrote the manuscript. Y.L. supervised the simulation and analysis. J.H., C.Y., and W.R. performed data analysis. All the authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Yang, C., Rao, W. et al. Prediction of high-pressure phases of Weyl semimetal NbAs and NbP. Sci Rep 7, 13251 (2017). https://doi.org/10.1038/s41598-017-13610-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13610-x
This article is cited by
-
High-pressure opens new windows for topological materials
Science China Physics, Mechanics & Astronomy (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
