
Abstract
MicroRNA is responsible for the fine-tuning of fundamental cellular activities and human disease development. The altered availability of microRNAs, target mRNAs, and other types of endogenous RNAs competing for microRNA interactions reflects the dynamic and conditional property of microRNA-mediated gene regulation that remains under-investigated. Here we propose a new integrative method to study this dynamic process by considering both competing and cooperative mechanisms and identifying functional modules where different microRNAs co-regulate the same functional process. Specifically, a new pipeline was built based on a meta-Lasso regression model and the proof-of-concept study was performed using a large-scale genomic dataset from ~4,200 patients with 9 cancer types. In the analysis, 10,726 microRNA-mRNA interactions were identified to be associated with a specific stage and/or type of cancer, which demonstrated the dynamic and conditional miRNA regulation during cancer progression. On the other hands, we detected 4,134 regulatory modules that exhibit high fidelity of microRNA function through selective microRNA-mRNA binding and modulation. For example, miR-18a-3p, −320a, −193b-3p, and −92b-3p co-regulate the glycolysis/gluconeogenesis and focal adhesion in cancers of kidney, liver, lung, and uterus. Furthermore, several new insights into dynamic microRNA regulation in cancers have been discovered in this study.
Similar content being viewed by others


Introduction
Mature microRNAs (miRNAs) are typically 20–25 nucleotides long. They hybridize with complementary sequences in the 3′-untranslated regions (UTRs) in messenger RNAs (mRNAs) and silence protein-coding genes through destabilizing mRNA or preventing translation of mRNA1. In humans, it was estimated that 2,588 miRNAs regulate over 60% of human genes and participate in every aspect of cellular activities in cell growth and cell death2. During the past decade, numerous studies have disclosed the regulatory roles of miRNAs in both the fundamental cellular processes3,4,5,6,7,8,9 and the development of complex human diseases such as cancer10,11,12,13,14.
Functional study of miRNA largely depends on the reliable identification of miRNA-mRNA regulatory interactions. Most state-of-the-art computational prediction methods, such as miRanda15, RNA2216, DIANA-microT17 and Targetscan18, are focused on primary search for nucleotide sequences complementary to the miRNA seed region (2nd–8th bases on the 5′ end)18 and suffer from high false prediction because of the dramatic complexity in miRNA binding19,20. Recent sequencing-based interactome data provide different views in this issue. For examples, through the crosslinking, ligation, and sequencing of hybrids (CLASH) analysis21, 18,514 miRNA-mRNA interactions were detected and only ~22% were associated with seed region, via either contiguous or gapped complementary RNA base pairing. The same study also unveiled that ~60% of the binding sites are within coding region of mRNA, as opposed to the 3′UTR centric search by the existing algorithms. Similar findings were observed using the covalent ligation of endogenous Argonaute–bound RNAs (CLEAR)-CLIP in human hepatoma (Huh7.5) cells, where ~26% of the interactions are seed-associated and ~57% are non-3′UTR interactions22. In addition, compelling evidence also shows the stochastic nature of miRNA-mRNA interactions that 1) multiple miRNAs can bind to the same mRNA sequence or different copies of the same transcript - combinatorial interactions23,24,25,26 and 2) multiple different mRNAs, possibly along with other long non-coding RNAs and circular RNAs27,28,29, can compete for binding to the same miRNA - competitive interactions23,30,31,32, which is very similar to transcription factor (TF) regulation33,34. Other factors, including genetic mutations35,36,37,38, the competition with other RNA binding proteins39,40 and the conditional expression of miRNA and mRNA41 can also affect the status of miRNA-mRNA interactions. Each of these mechanisms stresses the dynamic miRNA regulation from a different perspective and many of them are conditional (e.g., associated with certain phenotypes or development stages). It is notable the conditional information can be captured by genomic data collected under associated conditions. However, to the best of our knowledge, currently there is no systematic assessment available on the dynamic miRNA regulation that assesses the changes across conditions. The first such attempt, reported in24, only briefly touched the combinatorial module that a set of miRNA can regulate a common set of targets in a specific condition, which hinders the practical use as a dynamic system.
We notice that the actual challenges in tackling dynamic miRNA regulation involve several major trends. First, while the reliable stratification of gene regulation networks has been lagging, the associations between miRNAs and TFs were reported in different scenarios26,42,43,44. For example, one TF gene, CTCF, controls the expression of HOXC5, which is the host gene of miR-615-3p45,46 while miR-615-3p interacts with CTCF transcript in CALSH experiment21. This type of bi-directed regulation between TF and miRNA may introduce the feedback loops to the whole gene regulation network. Likewise, in order to determine the miRNA-mediated regulation on gene expression, one should also consider other major gene regulatory factors, such as copy number variation (CNV) and DNA methylation47. Secondly, evidence also shows that multiple miRNAs can regulate a set of functionally relevant genes instead of the same gene in a cooperative manner, e.g., miR-17-5p, -18a-5p, -19a-3p, and -92a-3p co-regulate 44 functionally related genes, such as CCND2, TNRC6B, and PHF1248. Although our knowledge of cooperative miRNA regulation is extremely limited, it is envisioned that the mechanisms underlying TF non-complex co-regulation49,50 can be adopted in miRNA study.
In this study, we propose a new method to identify dynamic miRNA-gene interactions, particularly focusing on the conditional interactions that are associated with tumor progression, based on the integration of heterogeneous genomic information on multiple human cancers. Other gene expression regulators such as TF, CNV, and DNA methylation have also been considered when determining miRNA-mediated gene regulatory networks in each developmental stage of cancer. Moreover, we demonstrated the discovery of modularized miRNA regulation based on the identified miRNA-gene interactions.
Results
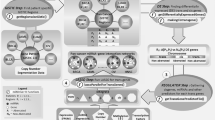
The workflow of this study is demonstrated in Fig. 1 with the detailed methodologies explained in Methods. We applied the proposed pipeline on cancers from seven tissues including lung, kidney, stomach, breast, liver, uterine, and pancreas, and have identified miRNA-mRNA interactions associated with each specific cancer. Note that the interactions were identified in the regulator detection step (Fig. 1B), which was performed for each cancer and each stage. While the complete results were archived in our online database (http://sbbi.unl.edu/miRDR), main discoveries were presented in the following sections. Particularly, we focused on the kidney and lung cancers and compare our discoveries between two well-defined subtypes, i.e., Kidney Renal Clear Cell Carcinoma (KIRC) versus Kidney Renal Papillary Cell Carcinoma (KIRP), and Lung Adenocarcinoma (LUAD) versus Lung Squamous Cell Carcinoma (LUSC).

Discovery of dynamic and modularized miRNA regulation during cancer progression. (A) The identification pipeline of conditional miRNA regulatory interactions; (B) Meta-Lasso Regression approach to the detection of microRNA regulators of each gene in each cancer stage.
Transcript availability and binding affinity are not the only factors in miRNA regulation
After reanalyzing CLASH data (details in Methods), 17,400 miRNA-mRNA interactions resulting from 17,651 high-confidence binidng sites have passed the quality-filtering step and remained for the downstream analysis. Figure 2A shows the abundance distribution of these interactions and Fig. 2B shows the distribution of corresponding corrected p-values from the Binomial test. 95.3% of the interactions have their counts significantly higher compared to random interaction (p-value ≤ 0.05 after Bonferroni multiple test adjustment). Detailed statistics were provided in Supplementary Table S2.

Overview of the interactions detected by the CLASH analysis. (A) Distribution of the miRNA-gene interactions (y-axis) with respect to their abundances (x-axis). (B) Distribution of the corrected p-values of each interaction from Binomial test for quality filtering. (C) Projection of each interaction based on the abundance of the corresponding miRNA (x-axis), mRNA (y-axis), and interaction (z-axis). (D) The relationship between the abundances of each miR-92a-3p interaction and the corresponding mRNA. The colored points indicated the three levels of binding affinities based on MFE values: High (red, [−31.2, −21.8]], Medium (green, [−21.8, 12.3]), and Low (blue, [−12.3, −2.9]).
We also observed weak correlations of miRNA regulatory interaction abundance with both mRNA and miRNA abundance (Fig. 2C). For example, 57.9% of miRNAs has Pearson correlation coefficient r ≤ 0.03 between the abundance of their target mRNAs and the number of interactions, contradicting the generally-believed assumption that miRNA binds to its different mRNA targets proportionally with respect to their sequence availability. Figure 2D shows that the most “versatile” miRNA, i.e., miR-92a-3p, regulates 956 different mRNAs through 1,065 different interaction sites. The interaction counts range from 63 to 23,870 (Mean = 296.5), significantly correlated with the corresponding mRNA abundance (r = 0.26). Similar weak correlations were also observed between binding affinities, e.g., minimum free energy (MFE)51, and interaction abundances (r = 0.01).
In summary, these findings suggest that in addition to mRNA sequence availability and binding affinity, there must be some other factors impacting the miRNA-mRNA binding, which makes miRNA regulation a highly selective process. For example, one possible factor can be mRNA structure that determines the accessibility for miRNA binding.
MiRNA-mRNA interactions can be specific to cancer type and stage
The initial analysis of genomic data has identified differentially expressed (DE) genes and miRNAs in different stages of cancer (summary statistics are in Supplementary Table S3 while more details are available online). Many changes were found to be stage-specific. For example, there are 4,273 DE-genes with fold-change (FC) over 2 (fold) in at least one stage of KIRC, but only 21.2% (908) of them have more than 2-fold changes in all four stages. Similar observations were made on miRNAs, i.e., 174 DE-miRNAs were identified in KIRC and only 43.7% (76) were consistent across all stages. This analysis provides a list of 15,901 genes and 419 miRNAs that are cancer-associated for downstream analysis. However, this simple correlation analysis each time only checks one pair of miRNA and mRNA. It fails to acknowledge the fact that in reality there can be multiple miRNA regulators for one individual gene. Considering the fact of simultaneous multiple interactions, we turn to our proposed pipeline to solve this issue.
Using our meta-Lasso regression approach (Methods), we predicted interactions between miRNA and miRNA-targetable genes under each cancer developmental stage. Overall, there were 14,554 unique miRNA-mRNA interactions identified across all conditions. Similar to DE genes and miRNAs, those interactions tend to be cancer stage- or type- specific. Figure 3A shows the statistics of interactions identified in each cancer and each stage where the blue bars indicate the portion specific to the corresponding stage within that cancer type. First, we observed in most cancers (8 out 9), miRNA interactions are more abundant in early stages (Stage I + II) compared to the advanced stages, which highlights the miRNA’s implication in the early cancer progression. In total, 73.7% of the interactions are detected in only one stage of that cancer. In different stages of a cancer, the same miRNA may regulate different genes while the same gene can be regulated by different miRNAs. For example, a kidney developing gene, Homeobox A11 (HOXA11), was regulated by three miRNAs in KIRC and suppressed expression was observed during progression (3-fold, miR-769-3p (Stage I); 2.9-fold, miR-484 (Stage II); 2.1-fold, miR-30d-5p (Stage III); 2.2-fold, none (Stage IV)) (Fig. 4A). Similar observation was made on Kinesin Family Member 1 C (KIF1C) gene: five miRNAs interacted with KIF1C in three stages of KIRC (1.8-fold, miR-484 and miR-769-3p (Stage I); 1.9-fold, miR-1260b, let-7b-5p, miR-149-5p, miR-769-3p and miR-484 (Stage II); 2.0-fold, miR-769-3p (Stage III); 2.2-fold, none (Stage IV)) (Fig. 4B). This may indicate that different miRNAs can play a similar role in cancer development by targeting the same target.

Overview of the miRNA-mRNA interactions identified in nine cancers. (A) Number of interactions detected in a specific stage (Blue) versus multiple stages (Orange), in each cancer. (B) Pie chart shows the percentages of interactions shared by different cancers.

Illustration of the dynamic miRNA-mediated gene regulation (miRNA: pink diamonds and genes: purple nodes). (A) Three miRNAs regulate HOXA11 gene in different stages of KIRC. (B) Five miRNAs repress the same KIF1C gene in KIRC while the miR-484 and miR-769-3p (pink diamonds with red border) consistently interact with KIF1C across stages.
In addition to the low consistency across stages of the same cancer, we also observed limited common miRNA regulatory interactions across cancers. Up to 92.7% (12,742) of the interactions were shared by less than five types of cancer and 22.8% (3,142) were identified specifically in one cancer type (Fig. 3B). Interestingly, even within the same tissue type, the number of common interactions across cancer subtypes was much lower than expected, e.g., 17.8% (1,264 out of 7,118) between KIRC and KIRP and 16.2% (1,051 out of 6,496) between LUAD and LUSC. This may be explained by the distinct expression profiles of genes and miRNAs in different subtypes of cancer. For example, KIRC and KIRP only share 34.5% of 6,332 DE genes and 23.5% of 136 DE miRNAs. The distinct miRNA regulatory patterns among stages and cancers demonstrate again the conditional feature of miRNA regulation. Moreover, it signifies that some miRNAs can only contribute to certain types or phases of tumor developments.
Functional implication of miRNA regulation in kidney cancers
To assess the functional impacts of miRNA regulation in cancer progression, we further examined the interactions identified in two subtypes of kidney cancer (KIRC and KIRP), particularly focusing on those associated with cancer driver genes reported by Vogelstein et al.52. Per our identification, 19 cancer genes were regulated by miRNAs in both early-stage KIRC and KIRP (purple nodes in the upper panel, Figure S1), where only 16 out of the 118 related interactions are commonly shared by both cancers (red edges) and 12 genes are regulated by totally different miRNAs. For example, Neurogenic Locus Notch Homolog Protein 2 gene (NOTCH2) was regulated by miR-744-5p, -25-3p, -92a-3p, and -27b-3p in KIRC and by miR-744-5p, -106a-5p, -130b-5p, and let-7c-5p in KIRP. Another critical cancer suppressor gene TP53 was regulated by miR-454-3p in KIRC, and by miR-324-5p and -222-3p in KIRP. On the other hand, we observed that common miRNAs regulated different targets in two cancer subtypes. Among 27 common miRNAs, 65.0% of the interactions were linked to different cancer genes in both early-stage tumors.
Given the distinct mechanisms underlying the development of these two subtypes of cancer53,54, we are also interested in exploring how miRNA regulation contributes to the general or divergent growth of kidney cancers. Pathway and gene ontology (GO) enrichment analysis was performed using The Database for Annotation, Visualization and Integrated Discovery (DAVID v6.8)55 based on the suppressed genes in the early-stage cancers. Table 1 lists the top enriched KEGG pathways that are either common or specific in KIRC and KIRP (selected according to p-values and the complete list of GO and pathways are provided in Supplementary Table S4). Among the three common pathways (PI3K-ATK signaling pathway, Cell cycle, and Thyroid hormone signaling pathway), 22 genes were miRNA-regulated in the early-stage kidney cancer and 9 of them occurred in both subtypes. Only 7 miRNA-gene interactions were observed in both subtypes, which indicates that the same functional regulation can be realized by miRNAs interacting with different gene targets.
Figure 5 demonstrates how miRNAs regulated PI3K-Akt signaling pathway (in 5 A) and cell cycle pathway (in 5B) in early-stage KIRC and KIRP through various conditional interactions. For example, the common genes (purple nodes), e.g., TSC1, MYC, and CDKN2A, were targeted by different miRNAs in two cancer subtypes while the common miRNA regulators (pink diamonds with red border), e.g., miR-320a, -17-5p, and 92a-3p, can interact with different targets, which leads to a different set of interactions that control the same functional pathway. Moreover, within the cancer-specific pathways, the distinct miRNA regulation patterns (Table 1) also provide new insights into cancer subtyping in the miRNA regulation perspective. In addition, similar observations were found by comparing LUAD and LUSC (Supplementary Table S5).

MiRNA-mediated gene regulation involved in two functional pathways in the early stage of KIRC and KIRP (miRNAs: pink diamonds; genes: ellipses). (A) PI3K-Akt Signaling Pathway; (B) Cell Cycle Pathway. Purple nodes denote the common genes between KIRC and KIRP; pink diamonds with red border indicate the common miRNA regulators; green ellipses are the condition-specific cancer genes.
Prediction of miRNA regulatory modules
Based on consistent observations in all nine cancers, we hypothesized that miRNAs can exert a regulatory role in certain biological functions through interacting gene targets that may be different across conditions. Moreover, multiple miRNAs can effectively cooperate in the same functional pathway by repressing the same or associated targets. Therefore we conducted a module detection based on the miRNA-gene interactions derived from the previous steps (Methods). As a result, 4,134 miRNA modules were identified and each included a set of 4–5 miRNAs consistently co-regulating the same set of pathways across more than two cancer conditions. Table 2 lists the summary statistics on the derived modules and details are provided in Supplementary Table S6.
We observed that most miRNA modules play their regulatory roles in different stages of cancer, and that only a small proportion is common across cancers. As expected, we observed in each single module that miRNAs coregulate one or multiple module, miRNAs co-regulate one or more biological processes through co-binding to the same target or different targets in the same pathway across conditions. Table 3 provides an example of a 4-miRNA module that regulates two pathways (glycolysis and focal adhesion) in three cancers (KIRC, LIHC, and UCEC). Under different conditions, miRNAs tend to target different genes but miR-320a always interacts with the same gene (PKM2) when regulating the glycolysis pathway. Figure 6 illustrates how the same miRNA module regulates critical signaling transduction genes in the focal adhesion pathway. For example, MET and LAMB1/COL4A1/LAMA5 were regulated in different stages. Since those genes are the initial activators and triggers in focal adhesion, the regulatory model is capable of controlling the whole process more efficiently by each miRNA member targeting different genes under different stages.

Illustration of the cooperative regulation of a miRNA module that contains four miRNAs: miR-18a-3p, -320a, -193b-3p, and -92b-3p (pink diamonds). This miRNA module consistently regulates the Focal Adhesion pathway in KIRC, LUSC, and UCEC. The target genes are color-coded (each color represents one cancer condition and the bi-colored nodes indicate the corresponding genes have been regulated by miRNAs in two conditions). The detailed miRNA-gene interactions are listed in Table 3. The network is visualized using Cytoscape 3.2.0 72 with KEGGscape73 plugin.
Validation of identified regulatory interaction in cancers
Through comparison, 2,014 miRNA-gene interactions of our prediction have been annotated as experimentally validated entries in miRTarBase56, which are reported by various assays, such as Luciferase reporter assay, qRT-PCR or Western blot analysis with miRNA transfections (Supplementary Table S7).
To further validate the interations in cancer, we also performed an extensive literature search on all miRNA/cancer-related publications during 2001–2017 in PubMed database by utilizing the NCBI API tool. First, we searched for articles that mentioned the predicted miRNA- mRNA pairs in the corresponding cancer type, e.g., let-7a-5p-HMGA1 in breast cancer. Then we examined all 27,586 returned articles to confirm if the experimental evidence for a direct interaction is strong or not based on the experimental assays that have been used. Note that the negative expression correlation is not considered as strong evidence of direct interaction in our analysis. As a result, 43 miRNA-mRNA interactions associated with the nine types of cancer have been validated (Table 4). It is not surprising that those interactions are related to a few well-known cancer-associated miRNAs such as let-7a, miR-181a, and 200c, which reflects the reality that most of current experimental efforts are still on the assessment of well-reported targets in various cancer conditions. We believe computational prediction like ours can bring more new targets in this direction.
Model Quality Assessment
Robustness of the proposed method was assessed based on the consistency of regulator detection across datasets with various sample sizes. Take the 251 samples from stage I of KIRC as an example. For each size i (5<=i<= 251), we randomly generated or each integer i from 5 to 251 (maximum sample size), we randomly generated 1000 testing data sets with the sample size. We conducted regulator selection for each gene based on the final model in the meta-Lasso regression analysis on each dataset. The consistency across models with different sample sizes was evaluated by comparing the similarity of the selected regulators. Figure 7A shows the comparisons on these selected regulators based on three genes (CARD11, JAK2, and HMGA1), and a high-level stability of our model with the increasing sample size was observed. Moreover, for miRNA regulator identification (Fig. 7B), we found that our method has reached the high-level consistency for CARD11 and HMGA1 (JAK2 were not regulated by miRNAs in this stage).

Consistency assessments of regulator selection regarding difference sample sizes of three genes: CARD11 (red, 28 potential regulators), JAK2 (blue, 73), and HMGA1 (green, 109). The lines denote the transition Jaccard’s similarity of the regulators selected at different sample sizes of these genes. (A) Consistency of all regulator selection across difference sample sizes. (B) Consistency of microRNA regulator selection across difference sample sizes.
To further demonstrate the performance of miRDR, we also conducted a peer comparison with two other well-established methods: RACER57 and MCMG58, on miRNA target prediction using expression data. The test dataset was collected from 332 patients in LIHC, available at the TCGA website59, along with 34,986 experimental miRNA-target interactions detected by CLEAR-CLIP in Hepatocellular carcinoma cell (Huh-7.5)22. We utilized three methods to predict the miRNA-target interactions in each stage of LIHC. First, we found that RACER and MCMG predicted significant more interactions than miRDR (average number of predicted interactions: miRDR 1,634; RACER 10,371; MCMG 212,171) but less experimental validated interactions among the top ranked ones. When taking a close look at each stage, we ranked the predicted interactions from RACER and MCMG according to the score and compare with top-n predictions from miRDR at 100-intervals (Fig. 8A). It is clear that miRDR outperformed two other methods by identifying significantly more validated interactions in each stage (p-value of one-sided Wilcoxon singed rank test is shown in the figure). Moreover, unlike RACER and MCMG, miRDR’s performance was not affected by the input sample size. For example, RACER and MCMG predicted only half validated targets in smaller sets (in stage II (82), III (80) and IV (6)) than what they did in a bigger set (stage I (164)) among top-1000 predictions. At another evaluation, we compared the distribution of miRNA target count predicted by three methods, using the experimental data from CLEAR-CLIP as reference. As Fig. 8B shows, the distribution of miRDR is very close to the experimental one, while RACER and MCMG tend to predict too many more targets for most miRNAs.

(A) Method comparison on the identified miRNA-target interactions on the test set. MiRDR, RACER and MCMG were applied on the Hepatocellular carcinoma (LIHC) data. The x-axis represents the top ranked interaction predicted by miRDR while the y-axis represents the count of validated interactions. The p-values were calculated by one-sided Wilcoxon singed rank test; (B) Method comparison based on the counts of identified miRNA targets. X-axis: number of target genes; y-axis: miRNA percentage. Outer plot shows the overall distribution and the inner shows the zoom-in view of partial distribution in the range of [0, 200].
Although Lasso regression method is capable of handling the problem of more variable than samples60, we still recommend a minimum number of samples for each gene for using our meta-Lasso regression model in order to reach high confidence in the inferred miRNA regulators. Note these thresholding values also depend on the numbers of possible regulators each gene can have (Supplementary Table S8).
Software and database access
We have developed a microRNA Dynamic Regulation Database (miRDR) that archives all the identified miRNA regulatory modules in each cancer online at http://sbbi.unl.edu/miRDR. An R package implementing the proposed method is available for download from our miRDR webpage.
Discussion
In this study, we presented two important characteristics of miRNA regulation. Dynamic regulation refers to the fact that the same miRNAs interact with different targets under different conditions. Theoretically speaking, factors contributing to the dynamic property of miRNA regulation may include the distinct expression of miRNA and their target, potential SNPs involved in the binding site, DNA translocation in the gene region, competitive binding between miRNA and other RNA binding proteins. On the other hand, modularized regulation describes that miRNAs may cooperate when they target to the same gene or same functional process across different conditions. Intuitively speaking, when multiple miRNAs target the same process, especially when multiple genes are controlled (Fig. 6, Table 5 and Supplementary Table S6), they render higher efficacy in terms of functional regulation.
Through heterogeneous genomic data integration, modeling, and adjustments of gene expression based on other regulatory mechanisms such as TF and CNV, we have detected 14,554 conditional miRNA-gene interactions in 9 types of cancer. The discrepancy between the inferred interactions from our pipeline and the CLASH-reported interactions may be explained by the fact that the former reported conditional interactions highly associated with cancers while the latter reported interactions associated with AGO1 protein in HEK293 cell. To generalize the CLASH reported miRNA-gene interactions to different cell types, we also designed a filtering process to minimize the possibility of overuse. Gene expression and miRNA expression have been assessed to ensure both molecules have a meaningful expression in the cell of interest. We also checked if any somatic mutation occurs at the CLASH reported binding site on mRNA sequence in the new cell line. The design of miRNA regulatory score enables an efficient differentiation among miRNA-mRNA interactions. In contrast to the existing methods that evaluate the interactions based on simple features such as binding affinity or complementary sequences, our miRNA scoring system learned and considered more features directly from experimental data, which is expected to be more reliable.
Comparing with two leading algorithms for miRNA target prediction based on expression profiles, miRDR has showed its advantageous predictive power with two major improvements. First, comparing with other methods, miRDR’s predictions tend to include more validated interactions while not introducing significant high false prediction. This is an important aspect given the widely-accepted critique on high false positive rate in current target prediction. Another advantage was the stable performance of miRDR on fairly limited sample size. The meta-analysis design minimized the impacts of sample variation on the target prediction, which seems promising to facilitate the detection of weak signals between miRNA and targets with insufficient samples.
Overall, our knowledge of modularized miRNA regulation is still very limited. Along this line, Ding et al. identified 181 modules based on a simple statistical analysis on CLASH reported interactions24. In this study, we have identified 4,134 functional miRNA modules based on the consistent miRNA co-regulation across different cancer conditions, where all previously reported modules are covered. Note the previous study focused on miRNAs that target the same gene while our method assesses comprehensively all modules involved in similar functional processes, which therefore can be more general. For example, one novel 5-miRNAs module, namely miR-196a-5p, -196b-5p, -365a-3p, -378a-5p and -30b-5p, has been identified to regulate cell cycle and MAPK/Wnt singling pathways in kidney and uterine cancers in our study. It was also conceived as a functional module by Trajkovski et al. where they uncovered that these miRNAs cooperatively promote the brown adipogenesis pathway in mice61.
In addition, since new sequencing technologies such as CLASH and CLIP can produce non-overlapped data on miRNA interactome, future inclusion of interactions detected in different platforms and cell types is expected. The concern of minimum sample size for reliable detection of regulators for each gene using our meta-lasso regression approach is actually a common concern of all similar analysis. We envision controlling the initial set of regulators including both TFs and miRNAs may alleviate this concern.
Conclusion
In summary, we proposed an integrative computational method to identify reliable miRNA-gene interactions in a given biological condition based on the integration of heterogeneous genomic data. In order to stratifying complex gene regulation networks, our approach considers other gene-expression regulatory factors including TF regulation, CNV, and DNA methylation. Applying the meta-lasso regression approach to 9 types of cancer, we have examined in-depth all possible miRNA-mRNA interactions and regulatory modules that are involved in different phase of cancer progression. The observed properties of the identified interactions further demonstrated the dynamic feature of miRNA regulation during tumor progression. We hope the method developed in this study can provide the community a promising solution to overcoming scientific limitations in the study of dynamic miRNA regulation in complex systems by generating novel insights in dynamic targeting and dramatically reducing the extensive lab-load in related research areas.
Methods
Data preparation
Data used in this study are all publically available, including (1) raw sequencing reads of miRNA-mRNA interactions from the CLASH experiment21 (GSE50452); (2) DNA binding data of 150 TFs from large-scale ChIP-Seq analysis by the Encyclopedia of DNA Elements (ENCODE) Consortium45; (3) multi-level genomic data from 4,206 patients and 9 types of cancer from Cancer Genome Atlas (TCGA)62 including miRNA and mRNA expressions, CNVs, and DNA methylation data (Table 5). In addition, for each cancer patient, stage and subtype information was also retrieved from TCGA.
CLASH data reanalysis
To assess the detection confidence level of CLASH reported binding sites, we first reanalyzed the raw sequencing data from CLASH and performed reads mapping against human reference genome (HG19) and human mature miRNA sequences database (miRBase63, version 21). Then, three types of reads were examined for each miRNA-mRNA binding site in the CLASH experiment including (1) chimeric reads that represent miRNA-mRNA binding sites; (2) miRNA single reads that represent free miRNAs, which are unbound with any mRNAs; (3) mRNA single reads that represent unbound mRNA sequences, which are unbound with any miRNAs.
Binomial test was applied on each binding site to infer the detection confidence level of a specific binding site k between miRNA i and the mRNA j. Specifically, the chimeric reads count of interactions between miRNA i and mRNA j at the binding site k, C ijk was assumed to follow a binomial distribution \({C}_{ijk}\sim Binomial\,({C}_{all},\,{P}_{ijk})\) under the null hypothesis, where P ijk (equation (2)) represents the probability of a binding occurred between miRNA i and mRNA j at the binding site k. Binomial test was performed and the p-value for each miRNA-mRNA binding site was calculated by the formula
where C all is the count of chimeric reads supporting all interactions and the probability of a binding occurred between miRNA i and mRNA j at the binding site k is estimated by
where Sum i and Sum j represent the total reads counts (chimeric reads and single reads) associated with miRNA i and mRNA j, respectively. Binding sites with Bonferroni-multiple-test-adjusted p values less than 0.05 were considered to be significant and included in the further analysis.
Regulatory score (RS) calculation of each miRNA-mRNA pair
To assess the likelihood of interactions between a potential target mRNA and a miRNA, we designed a probability-based regulatory score. As introduced above, we first estimated the probability of an interaction event between miRNA i and mRNA j at the binding site k by the equation (2), which we denoted as P ijk .
Then, raw RS of each miRNA-mRNA pair was calculated by the aggregation of the binding probability and the binding affinity, e.g., represented by the MFE51 of the corresponding interactions, as follows:
where MFE k represents the minimum free energy of the binding site k and K is the total number of different binding sites between miRNA i and mRNA j.
To mimic the situation of competition among mRNAs on the same miRNA, raw RS was normalized by the total number of potential targets associated with miRNA i:
where RS i . denotes the set of raw RS between all target j and miRNA i.
Regulatory score (RS) calculation of each TF-gene pair
Similar to miRNAs, a TF may also have more than one binding site on a target gene. The RS for the TF t and gene j based on the binding site k was estimated based on the distance (d tjk ) between the binding site k and the transcription start site (TSS)47,64, as follows:
Because a smaller distance (d tjk ) is associated with a higher regulatory potential of TF, RS for all binding sites of a TF and gene pair can be aggregated as follows47:
To mimic the situation that the same TF can bind to more than one gene, raw RS was normalized by the formula \(R{{S}_{tj}}^{^{\prime} }=R{S}_{tj}/R{S}_{t}.\) where \(R{S}_{t}.\) is the sum of \(R{S}_{tj}\) for a fixed t over all j’s.
Identification of the Conditional Gene Regulators through the Lasso Regression
In this study, we considered each stage of the nine types of cancer as an individual condition D sc (stage: s = 1,2,3,4; cancers: c = 1,2,…,9). For each gene under a specific condition, four matrices were constructed for a lasso regression approach to identify the conditional TF and miRNA regulators (Fig. 9) including the following:
-
The expression vector Y of size (n × 1) represents the expression changes of gene j in each of the n cancer patients associated with this condition versus healthy control. Here, in sample n, the expression changes of gene j and miRNA i were calculated by comparing with the average expression in the normal control group as follows:
$${\rm{\Delta }}{g}_{jn}={\mathrm{log}}_{2}({g}_{jn}/\,\overline{{g}_{jcontrol}})$$(7)$${\rm{\Delta }}{m}_{in}={\mathrm{log}}_{2}({m}_{in}/\,\overline{{m}_{icontrol}})$$(8)-
where \(\,\overline{{g}_{jcontrol}}\) and \(\,\overline{{m}_{icontrol}}\) denote the average expression levels of g j and m i among the normal control samples.
-
-
The background matrix X 1 of size n × 2 includes values of the copy number change and methylation of the gene j in each cancer compared to the healthy control;
-
The regulator matrix P = [P 1,…, P n]T of size n × (r 1 + r 2) lists the expression changes of all regulators, including r 1 TFs and r 2 miRNAs, that interact with gene j, in each patient compared to the normal control;
-
The regulatory score matrix R of size 1 × (r 1 + r 2) lists the RS of each corresponding regulator to the gene j derived from equations (4) and (6).

Four matrices constructed for each gene j and each condition, i.e., a specific cancer stage. Y represents the vector of expression changes of gene j in the corresponding cancer patients under this condition; X 1 represents the matrix of background adjustment factors (CNV and DNA methylation); R represents a RS vector that contains the regulatory scores of all r 1 TF- and r 2 miRNA- regulators of gene j; P is the matrix that includes all the expression changes of each regulator in each patient under the corresponding condition.
Based on the four matrices, a linear system is created as equation (9), where X 2 = [P 1 × R, …, P n × R]T (inner products of R and each row in P) represents the regulatory effects by TFs and miRNAs. The regulatory scores in X 2 denote the impacts of corresponding regulator (column) to the gene in a specific sample (row). Before we solve the linear system in equation (9), the Frisch–Waugh–Lovell (FWL) method47,65,66 was applied to adjust for the gene expression change caused by the background factors (CNV and methylation) through a linear transformation, and convert the linear system into equation (10). Then, Lasso regression60,67 was used to identify the TFs and miRNAs that regulated the gene under the given condition.
Meta-Lasso regression for robust regulator identification
To minimize the bias caused by limited cancer samples from TCGA and enhance the robustness in regulator selection, meta-Lasso regression68 was adopted on regulator identification of each gene under each condition. The workflow is shown in Fig. 1B where a hierarchical sampling and model integration was performed. Li et al. demonstrated that the meta-Lasso method has the good consistency of selection and low false predication rate on several high-dimensional gene expression data68. Here, we adopted the objective function of their meta-Lasso regressions to detect the most likely regulators of each gene with the consideration of consistency across of multiple “individual” dataset:
where \({l}_{m}({\beta }_{{m}_{0}},g,{\zeta }_{m})\) is the log-likelihood function of the m-th dataset; M denotes the number of individual datasets; g p is the effect of the p-th regulator (out of P regulators) at the overall condition; and ζ mp is the effect of the p-th regulator at the m-th dataset (out of M datasets).
Specifically, among the samples under one cancer condition, two rounds of 5-fold data separation were performed to generate 25 individual subsets that each contains a randomly selected 64% (80% × 80%) of the entire samples (Fig. 1B). The final LASSO regression model was achieved by applying the meta-LASSO method to the hierarchical sampled subsets. Any none-zero regression coefficients in the final LASSO model indicate the interactions occurred between the corresponding regulators and genes.
Subsequently, the same analysis was conducted for every human gene j in 36 conditions across all cancers (4 × 9). As shown in the workflow (Fig. 1), we followed a hierarchical framework to identify cancer-specific regulations (based on the consistency across different stages of that cancer) and common cancer regulation (based on the consistency across all cancers) through similar model selection procedure as described above.
Assessment of the model stability through a robustness test
To assess the robustness of this model, particularly its dependence on the sample size, we performed regulator identification with different sample sizes from p (the total number of potential regulators) to the total number of patients under that condition. We randomly generated 1,000 sets of data to perform the analysis at each sample size n, then evaluate the consistency of the derived models by calculating the Jaccard’s similarity coefficient between selected regulators (L) between model F n (regulator selection using n samples) and its neighbor F n-1 (using n-1 samples), as follows:
Assessment of potential miRNA-gene interactions in a specific cancer
Although the reported miRNA-gene interactions, e.g., 37,751 from miRTarBase56 database were validated in a particular experiment on a certain type of cell, the same physical binding between the participating miRNA and gene can still be valid in other cells as long as their sequences and expressions are conserved. With appropriate processing, we believe the interactions obtained from one experiment can be conditionally generalized to other cell types. Specifically, for each interaction, we conducted the following checkup when applying it to a new cell type:
-
both miRNA and gene must have meaningful expressions in the cell type of interest to maintain the activity level of the interaction;
-
the annotated binding sites shouldn’t show any mutations in the new cancer cell. Otherwise, we would no longer consider this binding site in the regulatory score calculation step.
In this proof-of-concept study, we focused on the experimental-validated binding sites from CLASH due to the availability of raw sequencing read information that is required in our model for regulatory score calculation.
Detections of miRNA regulatory module based on functional involvement in cancers
Based on the conditional miRNA-gene interactions derived from all cancers and stages, we explored the modularized miRNA regulation by examining their involvement in known functional pathways, i.e., 171 fundamental pathways from the KEGG database69. We exhaustively searched for all k-miRNA (k = 1–5) candidate modules where miRNAs consistently regulated the same set of pathways across multiple conditions and evaluated the significance in the following steps:
-
i.
Under each condition, the miRNAs were linked to a set of KEGG pathways through their target genes. Then, the miRNAs were grouped by the commonality on the same pathway as candidate modules (k = 1–5);
-
ii.
For each candidate module under one condition, a pathway enrichment analysis was conducted to ensure the involvement of each pathway is statistically significant through a test based on the hypergeometric distribution70:
$$\Pr ({r}_{w}|{N}_{D},{p}_{w},n)=\frac{(\begin{array}{c}{p}_{w}{N}_{D}\\ {r}_{w}\end{array})(\begin{array}{c}(1-{p}_{w}){N}_{D}\\ n-{r}_{w}\end{array})}{(\begin{array}{c}{N}_{D}\\ n\end{array})}$$(13)where N D denotes the total number of miRNA-regulated genes in the condition D, P w represents the percentage of human genes associated with pathway w, n indicates the total number of target genes associated with the module and r w is the number of genes belong to pathway w.
-
iii.
A Binomial test was applied to assess the statistical significance of the involvement of |w|-pathways in a miRNA module M
$${w}_{MD}\sim Binomial(|W|,\,{P}_{WD})$$(14)$${P}_{WD}=\frac{\sum _{w=1}^{171}{(TotalnumberofModuleswithsizek)}_{wD}}{|W|(\begin{array}{c}K\\ k\end{array})}$$(15)where k denotes the number of miRNAs in the module while K is the total number of miRNAs in this analysis, i.e., 371; P WD denotes the overall probability of a k-miRNA group co-involves a common pathway under condition D; w MD represents the number of enriched pathways associated with the k-miRNA module M under condition D; | W | is the total number of functional pathways.
-
iv.
Another Binomial test was utilized to examine the statistical significance of the recurrence of the candidate module M across conditions.
where k and K are defined in (14); P k denotes the overall probability of a k-miRNA module associated with common pathways across conditions; d M represents the number of conditions where the k-miRNA module M is associated with common pathways; |D| is the total number of conditions.
Finally, we calculated False Discovery Rate (FDR) q-values from p-values using Benjamini’s method71. Module candidates with q-values < 0.05 were reported as miRNA regulatory modules, i.e. we controlled FDR at 0.05 in our integrative report of all miRNA module candidates with different k’s.
References
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Friedman, R. C., Farh, K. K., Burge, C. B. & Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105 (2009).
Khalid, U., Bowen, T., Fraser, D. J. & Jenkins, R. H. Acute kidney injury: a paradigm for miRNA regulation of the cell cycle. Biochem. Soc. Trans. 42, 1219–23 (2014).
Xiong, X. et al. Down-regulated miRNA-214 induces a cell cycle G1 arrest in gastric cancer cells by up-regulating the PTEN protein. Pathol. Oncol. Res. 17, 931–937 (2011).
Bueno, M. J. & Malumbres, M. MicroRNAs and the cell cycle. Biochimica et Biophysica Acta - Molecular Basis of Disease 1812, 592–601 (2011).
Pogue, A. I. et al. Micro RNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci. Lett. 476, 18–22 (2010).
Luo, Z., Zhao, Y. & Azencott, R. Impact of miRNA sequence on miRNA expression and correlation between miRNA expression and cell cycle regulation in breast cancer cells. PLoS One 9, (2014).
Böhlig, L., Friedrich, M. & Engeland, K. P53 activates the PANK1/miRNA-107 gene leading to downregulation of CDK6 and p130 cell cycle proteins. Nucleic Acids Res. 39, 440–453 (2011).
Zhao, Y. et al. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 129, 303–317 (2007).
Ye, X. M. et al. Epigenetic silencing of miR-375 induces trastuzumab resistance in HER2-positive breast cancer by targeting IGF1R. BMC Cancer 14, 134 (2014).
Zhao, X. et al. Identification of miRNA-miRNA synergistic relationships in colorectal cancer. Int J Biol Macromol 55, 98–103 (2013).
Qin, Q., Furong, W. & Baosheng, L. Multiple functions of hypoxia-regulated miR-210 in cancer. J Exp Clin Cancer Res 33, 50 (2014).
Garofalo, M., Leva, G. D. & Croce, C. M. MicroRNAs as anti-cancer therapy. Curr. Pharm. Des. 20, 5328–5335 (2014).
Chitkara, D., Mittal, A. & Mahato, R. I. miRNAs in pancreatic cancer: therapeutic potential, delivery challenges and strategies. Adv Drug Deliv Rev 81, 34–52 (2015).
Betel, D., Wilson, M., Gabow, A., Marks, D. S. & Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 36, (2008).
Miranda, K. C. et al. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 126, 1203–1217 (2006).
Maragkakis, M. et al. DIANA-microT web server: Elucidating microRNA functions through target prediction. Nucleic Acids Res. 37, (2009).
Lewis, B. P., Shih, I. H., Jones-Rhoades, M. W., Bartel, D. P. & Burge, C. B. Prediction of Mammalian MicroRNA Targets. Cell 115, 787–798 (2003).
Ritchie, W., Flamant, S. & Rasko, J. E. J. Predicting microRNA targets and functions: traps for the unwary. Nat. Methods 6, 397–398 (2009).
Witkos, T. M., Koscianska, E. & Krzyzosiak, W. J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 11, 93–109 (2011).
Helwak, A., Kudla, G., Dudnakova, T. & Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013).
Moore, M. J. et al. miRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat. Commun. 6, 8864 (2015).
Bartel, D. P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 116, 281–297 (2004).
Ding, J., Li, X. & Hu, H. MicroRNA modules prefer to bind weak and unconventional target sites. Bioinformatics 31, 1366–1374 (2015).
Rinck, A. et al. The human transcriptome is enriched for miRNA-binding sites located in cooperativity-permitting distance. RNA Biol 10, 1125–1135 (2013).
Song, R., Catchpoole, D. R., Kennedy, P. J. & Li, J. Identification of lung cancer miRNA-miRNA co-regulation networks through a progressive data refining approach. J Theor Biol 380, 271–279 (2015).
Jia, W., Chen, W. & Kang, J. The Functions of MicroRNAs and Long Non-coding RNAs in Embryonic and Induced Pluripotent Stem Cells. Genomics. Proteomics Bioinformatics 11, 275–283 (2013).
Xia, T. et al. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 4, 271–282 (2014).
Yoon, J.-H., Abdelmohsen, K. & Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 34, 9–14 (2014).
Jiang, P. & Coller, H. Functional interactions between microRNAs and RNA binding proteins. MicroRNA (Shāriqah, United Arab Emirates) 1, 70–9 (2012).
Gardiner, A. S., Twiss, J. L. & Perrone-Bizzozero, N. I. Competing Interactions of RNA-BindingProteins, MicroRNAs, and Their Targets Control Neuronal Development and Function. Biomolecules 5, 2903–18 (2015).
Tay, Y., Rinn, J. & Pandolfi, P. P. The multilayered complexity of ceRNA crosstalk and competition. Nature 505, 344–52 (2014).
Zeitlinger, J. et al. Program-specific distribution of a transcription factor dependent on partner transcription factor and MAPK signaling. Cell 113, 395–404 (2003).
Harbison, C. T. et al. Transcriptional regulatory code of a eukaryotic genome. Nature 431, 99–104 (2004).
Chin, L. J. et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res 68, 8535–8540 (2008).
Jiang, S. et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res 70, 3119–3127 (2010).
Takamizawa, J. et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64, 3753–3756 (2004).
Mayr, C., Hemann, M. T. & Bartel, D. P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science (80-.). 315, 1576–1579 (2007).
Poliseno, L. et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033–1038 (2010).
Mayr, C. & Bartel, D. P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684 (2009).
Sood, P., Krek, A., Zavolan, M., Macino, G. & Rajewsky, N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. 103, 2746–2751 (2006).
Lin, Y. et al. Transcription factor and miRNA co-regulatory network reveals shared and specific regulators in the development of B cell and T cell. Sci. Rep. 5, 15215 (2015).
Peng, C., Wang, M., Shen, Y., Feng, H. & Li, A. Reconstruction and analysis of transcription factor-miRNA co-regulatory feed-forward loops in human cancers using filter-wrapper feature selection. PLoS One 8, e78197 (2013).
Delfino, K. R. & Rodriguez-Zas, S. L. Transcription factor-microRNA-target gene networks associated with ovarian cancer survival and recurrence. PLoS One 8, e58608 (2013).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Quah, S. & Holland, P. W. H. The Hox cluster microRNA miR-615: a case study of intronic microRNA evolution. Evodevo 6, 31 (2015).
Jiang, P., Freedman, M. L., Liu, J. S. & Liu, X. S. Inference of transcriptional regulation in cancers. Proc Natl Acad Sci USA 112, 7731–7736 (2015).
Hausser, J. & Zavolan, M. Identification and consequences of miRNA-target interactions–beyond repression of gene expression. Nat Rev Genet 15, 599–612 (2014).
Salas, E. N., Shu, J., Cserhati, M. F., Weeks, D. P. & Ladunga, I. Pluralistic and stochastic gene regulation: examples, models and consistent theory. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw042 (2016).
Kim, J. et al. The co-regulation mechanism of transcription factors in the human gene regulatory network. Nucleic Acids Res. 40, 8849–61 (2012).
Rehmsmeier, M., Steffen, P., Hochsmann, M. & Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–17 (2004).
Vogelstein, B. et al. Cancer Genome Landscapes. Science (80-.). 339, 1546–1558 (2013).
Cancer Genome Atlas Research Network et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–9 (2013).
Cancer Genome Atlas Research Network et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 374, 135–45 (2016).
Dennis, G. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003).
Chou, C. H. et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44, D239–47 (2016).
Li, Y., Liang, M., Zhang, Z., Luo, J. & Zhang, Z. Regression analysis of combined gene expression regulation in acute myeloid leukemia. PLoS Comput Biol 10, e1003908 (2014).
Chen, X., Slack, F. J. & Zhao, H. Joint analysis of expression profiles from multiple cancers improves the identification of microRNA-gene interactions. Bioinformatics 29, 2137–2145 (2013).
Ally, A. et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169(1327–1341), e23 (2017).
Tibshirani, R. J. The lasso problem and uniqueness. Electron. J. Stat. 7, 1456–1490 (2013).
Trajkovski, M. & Lodish, H. MicroRNA networks regulate development of brown adipocytes. Trends in Endocrinology and Metabolism 24, 442–450 (2013).
Chang, K. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120 (2013).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42, D68–73 (2014).
Wang, S. et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc. 8, 2502–2515 (2013).
Melessa, S. & Chen, W. Two-Stage Regression Analysis and Biased Estimates in Accounting Research: An Application of The Frisch-Waugh-Lovell Theorem. Work. Pap. 1–34 https://doi.org/10.2139/ssrn.2597429 (2014).
Lovell, M. C. A Simple Proof of the FWL Theorem. J. Econ. Educ. 39, 88–91 (2008).
Tibshirani, R. Regression Selection and Shrinkage via the Lasso. Journal of the Royal Statistical Society B 58, 267–288 (1996).
Li, Q., Wang, S., Huang, C.-C., Yu, M. & Shao, J. Meta-analysis based variable selection for gene expression data. Biometrics 70, 872–880 (2014).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Li, G., Ma, Q., Tang, H., Paterson, A. H. & Xu, Y. QUBIC: A qualitative biclustering algorithm for analyses of gene expression data. Nucleic Acids Res. 37, (2009).
Strimmer, K. fdrtool: A versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462 (2008).
Shannon, P. et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Nishida, K., Ono, K., Kanaya, S. & Takahashi, K. KEGGscape: a Cytoscape app for pathway data integration. F1000Research https://doi.org/10.12688/f1000research.4524.1 (2014).
Sun, X. et al. MiR-208a stimulates the cocktail of SOX2 and β-catenin to inhibit the let-7 induction of self-renewal repression of breast cancer stem cells and formed miR208a/let-7 feedback loop via LIN28 and DICER1. Oncotarget 6, 32944–54 (2015).
Liu, K. et al. Let-7a inhibits growth and migration of breast cancer cells by targeting HMGA1. Int. J. Oncol. 46, 2526–34 (2015).
Yan, Y. et al. MicroRNA-10a is involved in the metastatic process by regulating Eph tyrosine kinase receptor A4-mediated epithelial-mesenchymal transition and adhesion in hepatoma cells. Hepatology 57, 667–77 (2013).
Li, H., Bian, C., Liao, L., Li, J. & Zhao, R. C. miR-17-5p promotes human breast cancer cell migration and invasion through suppression of HBP1. Breast Cancer Res. Treat. 126, 565–75 (2011).
Yu, Z. et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J. Cell Biol. 182, 509–517 (2008).
Zhang, Y., Han, T., Wei, G. & Wang, Y. Inhibition of microRNA-17/20a suppresses cell proliferation in gastric cancer by modulating UBE2C expression. Oncol. Rep. 33, 2529–36 (2015).
Panda, H., Chuang, T.-D., Luo, X. & Chegini, N. Endometrial miR-181a and miR-98 expression is altered during transition from normal into cancerous state and target PGR, PGRMC1, CYP19A1, DDX3X, and TIMP3. J. Clin. Endocrinol. Metab. 97, E1316–26 (2012).
Ikeda, Y., Tanji, E., Makino, N., Kawata, S. & Furukawa, T. MicroRNAs associated with mitogen-activated protein kinase in human pancreatic cancer. Mol. Cancer Res. 10, 259–69 (2012).
Wang, L. et al. MicroRNA-193b inhibits the proliferation, migration and invasion of gastric cancer cells via targeting cyclin D1. Acta Histochem. 118, 323–30 (2016).
Guerriero, I. et al. Analysis of miRNA profiles identified miR-196a as a crucial mediator of aberrant PI3K/AKT signaling in lung cancer cells. Oncotarget. https://doi.org/10.18632/oncotarget.13432 (2016).
Schimanski, C. C. et al. High miR-196a levels promote the oncogenic phenotype of colorectal cancer cells. World J. Gastroenterol. 15, 2089–2096 (2009).
Li, Y. et al. Ratio of miR-196s to HOXC8 messenger RNA correlates with breast cancer cell migration and metastasis. Cancer Res. 70, 7894–904 (2010).
Bai, W.-D. et al. MiR-200c suppresses TGF-β signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int. J. cancer 135, 1356–68 (2014).
Wang, X. et al. microRNA-200c modulates the epithelial-to-mesenchymal transition in human renal cell carcinoma metastasis. Oncol. Rep. 30, 643–650 (2013).
Hur, K. et al. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 62, 1315–26 (2013).
Hurteau, G. J., Carlson, J. A., Spivack, S. D. & Brock, G. J. Overexpression of the MicroRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res. 67, 7972–7976 (2007).
Ma, C. et al. MicroRNA-200c overexpression plays an inhibitory role in human pancreatic cancer stem cells by regulating epithelial-mesenchymal transition. Minerva Med. 106, 193–202 (2015).
Zhou, X. et al. The downregulation of miR-200c/141 promotes ZEB1/2 expression and gastric cancer progression. Med. Oncol. 32, 428 (2015).
Chuang, T.-D., Panda, H., Luo, X. & Chegini, N. miR-200c is aberrantly expressed in leiomyomas in an ethnic-dependent manner and targets ZEBs, VEGFA, TIMP2, and FBLN5. Endocr. Relat. Cancer 19, 541–56 (2012).
Liu, Y. et al. Precise Regulation of miR-210 is Critical for the Cellular Homeostasis Maintenance and Transplantation Efficacy Enhancement of Mesenchymal Stem Cell in Acute Liver Failure Therapy. Cell Transplant. https://doi.org/10.3727/096368916X694274 (2016).
White, K. et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol. Med. 7, 695–713 (2015).
Ke, J. et al. Role of microRNA221 in regulating normal mammary epithelial hierarchy and breast cancer stem-like cells. Oncotarget 6, 3709–21 (2015).
Cheng, F., Pan, Y., Lu, Y.-M., Zhu, L. & Chen, S. RNA-Binding Protein Dnd1 Promotes Breast Cancer Apoptosis by Stabilizing the Bim mRNA in a miR-221 Binding Site. Biomed Res. Int. 2017, 9596152 (2017).
Ye, Z., Hao, R., Cai, Y., Wang, X. & Huang, G. Knockdown of miR-221 promotes the cisplatin-inducing apoptosis by targeting the BIM-Bax/Bak axis in breast cancer. Tumour Biol. 37, 4509–15 (2016).
Panneerselvam, J. et al. IL-24 modulates the high mobility group (HMG) A1/miR222 /AKT signaling in lung cancer cells. Oncotarget 7, 70247–70263 (2016).
Zhang, Y. et al. High-mobility group A1 proteins enhance the expression of the oncogenic miR-222 in lung cancer cells. Mol. Cell. Biochem. 357, 363–71 (2011).
Ell, B. et al. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J. Biol. Chem. 289, 21888–95 (2014).
Tan, S. et al. Identification of miR-26 as a key mediator of estrogen stimulated cell proliferation by targeting CHD1, GREB1 and KPNA2. Breast Cancer Res. 16, R40 (2014).
Suh, S.-S. et al. FHIT suppresses epithelial-mesenchymal transition (EMT) and metastasis in lung cancer through modulation of microRNAs. PLoS Genet. 10, e1004652 (2014).
Zhong, K., Chen, K., Han, L. & Li, B. MicroRNA-30b/c inhibits non-small cell lung cancer cell proliferation by targeting Rab18. BMC Cancer 14, 703 (2014).
Jeon, M. J. et al. MicroRNA-30d and microRNA-181a regulate HOXA11 expression in the uterosacral ligaments and are overexpressed in pelvic organ prolapse. J. Cell. Mol. Med. 19, 501–9 (2015).
Xie, F. et al. miRNA-320a inhibits tumor proliferation and invasion by targeting c-Myc in human hepatocellular carcinoma. Onco. Targets. Ther. 10, 885–894 (2017).
Li, Q.-Q. et al. CREB1-driven expression of miR-320a promotes mitophagy by down-regulating VDAC1 expression during serum starvation in cervical cancer cells. Oncotarget 6, 34924–40 (2015).
Chen, B., Duan, L., Yin, G., Tan, J. & Jiang, X. Simultaneously expressed miR-424 and miR-381 synergistically suppress the proliferation and survival of renal cancer cells–Cdc2 activity is up-regulated by targeting WEE1. Clinics (Sao Paulo). 68, 825–33 (2013).
Martin, J. et al. Post-transcriptional regulation of Transforming Growth Factor Beta-1 by microRNA-744. PLoS One 6, e25044 (2011).
Acknowledgements
The authors would like to thank all SBBI members who have been involved in this work for providing helpful discussions and technical assistance. Particularly, we are thankful to Dr. Stephen Kachman at the Department of Statistics, UNL, for his advices on the statistical analysis. We also appreciate the UNL Holland Computing Center and the Open Science Grid for providing the computational facility. This research is support by the National Institutes of Health funded COBRE grant (1P20GM104320), UNL Food For Health seed grant and the Tobacco Settlement Fund as part of the Cui’s start-up grant support.
Author information
Authors and Affiliations
Contributions
J.S. and J.C. conceived and designed the experiments; J.S., B.S., Z.X., and T.G. performed the experiments, analyzed the data. J.S., Z.X. and J.C. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shu, J., Silva, B.V.R.e., Gao, T. et al. Dynamic and Modularized MicroRNA Regulation and Its Implication in Human Cancers. Sci Rep 7, 13356 (2017). https://doi.org/10.1038/s41598-017-13470-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13470-5
This article is cited by
-
Identification of novel miRNAs potentially involved in the pathogenesis of adult T-cell leukemia/lymphoma using WGCNA followed by RT-qPCR test of hub genes
Infectious Agents and Cancer (2023)
-
Discerning the Prospects of miRNAs as a Multi-Target Therapeutic and Diagnostic for Alzheimer’s Disease
Molecular Neurobiology (2023)
-
Host miRNA and immune cell interactions: relevance in nano-therapeutics for human health
Immunologic Research (2022)
-
Identifying miRNA synergism using multiple-intervention causal inference
BMC Bioinformatics (2019)
-
MicroRNA signature of human blood mononuclear cells
Molecular and Cellular Biochemistry (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
