
Abstract
Microtubule-associated TAU protein is a pathological hallmark in Alzheimer’s disease (AD), where hyperphosphorylation of TAU generates neurofibrillary tangles. To investigate the effects of TAU in a regenerative adult vertebrate brain system, we generated a cre/lox-based transgenic model of zebrafish that chronically expresses human TAUP301L, which is a variant of human TAU protein that forms neurofibrillary tangles in mouse models and humans. Interestingly, we found that although chronic and abundant expression of TAUP301L starting from early embryonic development led to hyperphosphorylation, TAUP301L did not form oligomers and neurofibrillary tangles, and did not cause elevated apoptosis and microglial activation, which are classical symptoms of tauopathies in mammals. Additionally, TAUP301L neither increased neural stem cell proliferation nor activated the expression of regenerative factor Interleukin-4, indicating that TAUP301L toxicity is prevented in the adult zebrafish brain. By combining TAUP301L expression with our established Aβ42 toxicity model, we found that Aβ42 ceases to initiate neurofibrillary tangle formation by TAUP301L, and TAUP301L does not exacerbate the toxicity of Aβ42. Therefore, our results propose a cellular mechanism that protects the adult zebrafish brain against tauopathies, and our model can be used to understand how TAU toxicity can be prevented in humans.
Similar content being viewed by others

Introduction
Accumulation of microtubule-associated TAU protein is a pathological hallmark of Alzheimer’s disease (AD) and frontotemporal dementia (FTD)1,2,3,4. TAUP301L mutation leads to aggressive TAU aggregation and neurofibrillary tangle (NFT) formation in animal models of FTD3,4,5,6, whereas in AD, Aβ42 is believed to cause hyperphosphorylation of TAU, and subsequent aggregation7,8,9. In animal models of Tauopathies, motor deficits and neurodegeneration were observed10,11,12,13. Since Tauopathies are also seen in AD, transgenic models aiming to recapitulate the main symptoms of AD brains were generated using known familial mutations of AD and FTD, such as 3xTg14, TauPS2APP15, Tg257616, PLB1-triple17, and rTg2122118. Although these models mimic the pathological features of the human disease, the proposed causative link between Aβ42 and TAU aggregation is still to be clarified19,20,21. Majority of the studies suggested a link between TAU toxicity and Amyloid aggregation16,22,23,24,25,26,27. However, there are also conflicting studies where Amyloid toxicity is not dependent on TAU28,29,30, and a recent study suggested that TAU might even ameliorate Amyloid toxicity31, indicating that the effects of Amyloid toxicity on TAU-dependent tangle formation, and the role of TAU tangles on exacerbating the Amyloid toxicity is still to be clarified. Therefore, new animal models where the effects of Aβ42 and TAU can be addressed independently and in combination to increase our understanding of the relationship between Aβ42 and TAU.
Neurodegenerative diseases cause a progressive deterioration of neuronal circuits and pronounced hampering of the stem cell proliferation32. Therefore, a plausible way of circumventing these diseases is to design regenerative therapy options where neurons would survive the toxicity burden exerted by TAU and Aβ42, but also the neural stem cells would enhance their proliferation to supply more cells that could be replenishing the function of the lost ones33. Mammalian models are excellent to recapitulate the pathophysiology of neurodegenerative diseases, however, since mammals are poorly regenerating, basic and translational questions regarding the regenerative capacity cannot be addressed well. Zebrafish, as a vertebrate serves as an excellent tool for questions concerning regeneration because of its high regenerative ability.
In zebrafish, several Tauopathy models were generated before. Transient expression of human TAU fused to GFP under gata2 promoter was shown to cause TAU phosphorylation in zebrafish larvae using Western blot analyses34. Another study using transient expression of human TAU-GFP under neuronal HuC/D promoter has also shown that TAU can be hyperphosphorylated35. The first stable TAU transgenic was generated by expressing TAU (0N4R) under the control of enolase promoter36. TAU was expressed in adult fish brain; however, no phenotypic characterization was documented. The best-characterized zebrafish TAU model so far is Gal4/UAS-mediated expression of human TAUP301L under the control of HuC/D promoter37. This line shows motor neuron degeneration, hyperphosphorylation of TAU and tangle formation in the spinal cord. However, studies in this line only focused on the embryonic to juvenile spinal cord but not the brain. A recent study used a new allele of TAU with a mutation causing A152T conversion and showed hyperphosphorylation of TAU, motor neuron degeneration in the spinal cord, and cell death in larval zebrafish retina38. Therefore, these zebrafish models expressing human TAU have documented hyperphosphorylation of TAU and tauopathy in the spinal cord and the retina; however, the effects of TAU in the brain are understudied and therefore the effects are still unknown. In our study, we developed two conditional transgenic models of zebrafish, which expressed human TAUP301L chronically in radial glial cells and the neurons derived from these progenitors in early embryonic development (with her4.1 promoter) and directly in neurons (with neural beta tubulin promoter). Expression of human TAUP301L in adult zebrafish brain showed that human TAUP301L can be phosphorylated, but neither forms any aggregates nor causes neurodegeneration on its own. Additionally, by combining the transgenic model of TAUP301L expression with our recent Amyloid toxicity model39,40, we showed that Amyloid toxicity does not cause TAU aggregation, and TAUP301L expression does not exacerbate the effects of Amyloid toxicity and AD-like neurodegeneration in adult zebrafish brain. Our results indicate that in adult zebrafish brain, human TAUP301L can be phosphorylated but does not form aggregates and does not cause neurodegeneration, and Amyloid toxicity does not elicit Tauopathies.
Results
Conditional transgenic cassettes lead to strong neuronal expression of of TAUP301L in zebrafish brain
To investigate the chronic effects of human TAUP301L expression in zebrafish brain, we generated a cre/lox-based conditional double-transgenic zebrafish that expresses TAUP301L. The spatial control cassette drives the expression of inducible cre recombinase (creERT2) under the control of her4.1 promoter, which is active in neural stem/progenitor cells with radial glial identity (Fig. 1a, Tg(her4.1:mCherry-T2A-creERT2)). We specifically chose her4.1 promoter because we hypothesized that if the expression of TAUP301L started from the stem cell stage, all neurons produced by those stem cells after the recombination would express the protein. In contrast to conditional expression systems that use mature neuronal promoters such as HuC/D41, enolase36 or neural beta tubulin (NBT)42, using a stem cell promoter would increase the number of neurons expressing the transgene throughout the life of the fish. Additionally, since in familial forms of neurodegenerative diseases, mutant proteins are expressed in all cells – including the stem cells - of the patients during development, our approach would also help to address the effects of TAUP301L in neural stem cells of zebrafish brain in a way reminiscent of familial forms of Tauopathies in humans.

(a) Naming of transgenic constructs: sTg – effector transgenic, dTg – double transgenic. (b) Scheme for timing of recombination and analyses. (c) sTg and dTg animals (5 days post fertilization) treated with tamoxifen. DsRed expression indicates the presence of the effector cassette. Note the recombination in the central nervous system by GFP expression. (d) Immunohistochemistry (IHC) for GFP and TAUP301L on coronal sections of telencephalon of a 6 month-old sTg animal. Single channel images of the whole section for GFP (d’) and TAU (d”). (1) is the enlarged view of the inset in d. (e) IHC for GFP and TAUP301L on coronal sections of telencephalon of a 6-month old dTg animal. Single channel images of the whole section for GFP (e’) and TAU (e”). (2) is the enlarged view of the inset in f. (f) IHC for HuC/D and TAUP301L on coronal sections of telencephalon of an sTg animal. Single channel images of the whole section for TAU (f’) and HuC/D (f”). (3) is the enlarged view of the inset in f. (g) IHC for HuC/D and TAUP301L on coronal sections of telencephalon of a dTg animal. Single channel images of the whole section for TAU (g’) and HuC/D (g”). (4) is the enlarged view of the inset in g. (h) Western blot analyses for expression of TAUP301L (left) and hyperphosphorylated TAUP301L (right) in telencephalon. Beta actin is used as a loading control. (i) IHC for GFP and AT8 in a dTg animal. Individual channels are shown for GFP (i’) and AT8 (i”). Arrows represent the cytoplasmis signal. (j) IHC for TAUP301L and T205 in a dTg animal. Individual channels are shown for TAUP301L (j’) and T205 (j”). (k) Gallyas silver (black) and Hematoxylin (pink) staining in telencephalon of a dTg animal. (k’) Enlarged region in k’. Note the absence of Gallyas silver-positive cells. (l) Positive control for Gallyas silver staining in human neurons treated with Amyloid, showing neurofibrillary tangles. (m) Immunohistochemistry for TAUP301L (green) combined with TUNEL detection of apoptotic cells (red) in recombined 6 months old dTg animals. Individual fluorescent channels are shown in m’ and m”. (n–p) Higher magnification images from the insets in m. (q) Quantification of TUNEL-positive cells in the telencephalon in sTg and dTg animals. (r) Quantification of apoptotic cells containing hyperphosphorylated TAUP301L (T205-positive). Values represent mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.005. Scale bars equal 10 μm (i–j’) and 50 μm elsewhere. n = 6 fish and > 30 histological sections for every staining. Larvae are 5 days old, and adult animals are 6 months old.
The effector cassette is responsive to cre and expresses TAUP301L under the ubiquitous promoter after recombination (Fig. 1a, Tg(ubi:loxP-DsRed-STOP-loxP-GFP-T2A-TAUP301L). From here on, Tg(ubi:loxP-DsRed-STOP-loxP-GFP-T2A-TAUP301L) will be named as sTg, double transgenic (Tg(ubi:loxP-DsRed-STOP-loxP-GFP-T2A-TAUP301L) and Tg(her4.1:mCherry-T2A-creERT2) will be denoted as dTg). Since cre activity is induced by addition of Tamoxifen (TAM), we treated sTg and dTg animals with TAM from 16 hours to 48 hours post fertilization in order to recombine all radial glial cells (RGCs), and analyzed the adult animals at 1, 3, and 6 months post fertilization (Fig. 1b). While the recombination does not take place without TAM, after TAM treatment at 48 hours post fertilization dTg animals show GFP expression along the whole rostrocaudal axis of the central nervous system (Fig. 1c).
To determine how widespread TAUP301L is expressed by using her4.1 promoter, we performed immunostainings for GFP and TAUP301L on sagittal sections of the adult zebrafish brain (Supplementary Fig. 1). We observed a strong and reproducible expression of TAUP301L in adult zebrafish brain from olfactory bulb to the hindbrain (Supplementary Fig. 1 insets 1–9). These results indicate that TAUP301L is successfully expressed throughout the entire axis of zebrafish brain using a stem cell promoter. In order to further investigate whether the use of her4.1 promoter is appropriate to express TAU in maximum number of neurons, we expressed TAU by using the same recombination paradigm before but using a cre-driver line expressing cre recombinase under the control of neural beta-tubulin promoter (NBT): Tg(nbt:mCherry-T2A-creERT2) (designated hereafter as nTg). By performing recombination after crossing nTg to sTg, we found that nTg is leading to transgene expression along the rostrocaudal axis of the fish brain, and, compared to her4.1-driven TAU expression, nbt promoter also drives expression of the transgene in a similarly widespread manner (Supplementary Fig. 2), suggesting that her4.1 promoter can be used to target neurons in the adult zebrafish brain by cre-mediated recombination.
Based on the known pathological locations of TAU in human brains, we focused on the forebrain for the rest of our analyses. By performing immunostainings for GFP and TAUP301L on coronal sections of the zebrafish forebrain, we determined that compared to control animals (sTg), dTg animals show a specific pattern of recombination, which colocalizes with TAUP301L expression in the telencephalon (Fig. 1d–e”; Supplementary Fig. 3). To investigate whether TAUP301L is expressed in neurons, we performed immunostainings for TAUP301L and neuronal marker HuC/D in sTg (Fig. 1f–f’) and dTg animals (Fig. 1g–g”). We observed that neurons express TAUP301L (Fig. 1g4). To determine the percentage of neurons in the pallium that express TAUP301L, we quantified the number of cells that co-localize HuC/D and TAUP301L (Supplementary Fig. 4). We found that TAUP301L is expressed in 91.8% % of the neurons (Supplementary Fig. 4), indicating that in our model, majority of the neurons in the forebrain express TAUP301L.
TAUP301L is hyperphosphorylated in adult zebrafish brain
One of the hallmarks of Tauopathies in human brains is hyperphosphorylation of TAU and formation of neurofibrillary tangles, which can be detected by histological stainings. To analyze whether chronic expression of human TAUP301L in adult zebrafish brain leads to hyperphosphorylation and tangle formation, we analyzed the immunoreactivity of the adult fish brain to AT8, an indicator of late stage Tau hyperphosphorylation at the Serine-202 and Threonine-205 (Fig. 1h). Western blot analyses showed that human TAUP301L is specifically expressed only in dTg but not sTg animals, and when compared to the beta-actin loading control, the expression is strong and abundant (Fig. 1h). We also found that TAUP301L leads to AT8 immunoreactivity (~70 kDa expected size), which is suggestive of TAUP301L hyperphoshorylation (Fig. 1h). The band around 82 kDa is the fusion of GFP and TAU in the cre-effector cassette, and results from inefficient cleavage of t2a peptide in approximately 8% of the cases as documented before43. The strong band above 82 kDa is a non-specific recognition of AT8 in zebrafish in the nucleus (Supplementary Fig. 5). By performing quantification of the relative intensities of detected bands, we found that 3.8 ± 1.4% of the total TAU is AT8-positive in adult zebrafish brain. To confirm the results from western blot analyses, we performed immunohistochemical stainings for AT8 and TAUP301L (Fig. 1i–i”). We observed that a small portion of the TAUP301L-positive cells display AT8 immunoreactivity in the cytoplasm (Fig. 1i–i”). However, since AT8 staining leads to a nuclear background staining in adult zebrafish brain and therefore determining the cytoplasmic staining in adult brain is difficult (Supplementary Fig. 5). To further verify the phosphorylation status of TAU, we performed immunohistochemical staining with an antibody that specifically recognizes phosphorylation of Threonine-205 (T205) (Fig. 1j–j”’). We found that this staining does not cause background signals (Fig. 1j–j”), and approximately 76.1% of the TAUP301L-positive cells are also positive for T205 phosphorylation (Supplementary Fig. 6). These results indicate that chronic expression of TAUP301L can cause hyperphosyphorylation in adult zebrafish brain. We also analyzed the early phosphorylation of TAU by immunohistochemistry and western blot for AT180 (Threonine-231 and Serine-235) (Supplementary Fig. 7) and AT270 (Threonine-181) (Supplementary Fig. 7). We found that AT180 and AT270 give strong non-specific cross-reactivity in zebrafish in immunohistochemistry (Supplementary Fig. 7a,b”’, Supplementary Fig. 8a,b”’), and AT270 additionally show non-specific signal in western blots (Supplementary Fig. 8c), indicating that these antibodies cannot be reliably used. AT180, however, shows a specific band of phosphorylated TAU in dTg animals (Supplementary Fig. 7c), confirming the phosphorylation determined by AT8 and T205 antibodies. In overall, these results indicate that TAUP301L is hyperphosphorylated in adult zebrafish brain.
Hyperphosphorylation of TAUP301L is known to cause neurofibrillary tangle (NFT) formation1,2, and to test whether chronic expression of human TAUP301L would lead to NFTs, we histologically stained coronal sections of adult zebrafish telencephalon with Gallyas silver (Fig. 1k,k’). We did not observe any tangle formation, whereas a positive control from human neurons treated with Amyloid-β42 gave positive signal for Gallyas silver (Fig. 1l), indicating that despite its hyperphosphorylation, human TAUP301L does not form NFTs in adult zebrafish brain.
We hypothesized that if NFTs – as the toxic outcome of TAU – do not form in adult zebrafish brain, the pathological outcomes of neurodegeneration would not manifest in dTg animals. To test this hypothesis, we investigated the levels of apoptosis (Fig. 1m–q), microglia/macrophage activation as an indication of chronic inflammation (Fig. 2a–e) and proliferation of radial glial cells (RGCs) as a standard readout after neuronal loss in adult zebrafish brain39,44,45 (Fig. 2f–j).

(a) Immunohistochemistry (IHC) for L-Plastin (red) and TAUP301L (green) on coronal sections of telencephalon of a 6 month-old sTg animal. Single channel images of the whole section for TAUP301L (a’) and L-Plastin (a”). (b) The enlarged view of the inset in a. (c) IHC for L-Plastin (red) and TAUP301L (green) on coronal sections of telencephalon of a 6-month old dTg animal. Single channel images of the whole section for TAUP301L (c’) and L-Plastin (c”). (d) The enlarged view of the inset in c. (e) Quantification of round and ramified L-Plastin-positive cells in the telencephalon in sTg and dTg animals. (f) Immunohistochemistry (IHC) for S100β (red), PCNA (green) and TAUP301L (white) on coronal sections of telencephalon of a 6-month old sTg animal. Single channel images of the whole section for S100β (F’), PCNA (f”), and TAUP301L (f”’). (g) IHC for S100β (red), PCNA (green) and TAUP301L (white) on coronal sections of telencephalon of a 6-month old dTg animal. Single channel images of the whole section for S100β (g’), PCNA (g”), and TAUP301L (g”’). (h) The enlarged view of the inset in f. (i) The enlarged view of the inset in g. (j) Quantification of the total number of proliferating glial cells in the telencephalon of sTg and dTg animals. Values represent mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.005. Scale bars equal 50 μm (a–g”’) and 20 μm (h,i). n = 7 fish and > 30 histological sections for every staining. All animals are 6 months old.
Phosphorylated TAUP301L does not cause cell death, inflammation and stem cell activation in adult zebrafish brain
To determine whether the cells where TAU is hyperphosphorylated undergo apoptosis, we performed TUNEL staining and found that the overall levels of cell death in dTg animals is not significantly different than sTg animals (Fig. 1q), while our previously established AD model of adult zebrafish brain shows extensive cell death (Supplementary Fig. 9), which is also confirmed by unchanged relative expression levels of apoptotic regulator Caspase 3 (Supplementary Fig. 9i). We also observed that only a minor fraction of T205-positive cells containing phosphorylated-TAU are TUNEL positive (Fig. 1r). These results indicate that TAUP301L expression does not lead to cell death in adult zebrafish brain.
Tauopathies cause chronic inflammation by significantly inducing the numbers of macrophages/microglia as well as favoring for their activation state46. To test if chronic expression of human TAUP301L in adult zebrafish brain would alter the microglial dynamics, we performed immunostaining for L-Plastin, a marker for microglia and macrophages in adult zebrafish brain (Fig. 2a–d). Compared to sTg, dTg animals did not show a significant difference in the overall number and activation state of the microglia, as the number of ramified and round L-Plastin-positive cells remains unchanged (Fig. 2d). This result indicates that chronic expression of human TAUP301L does not induce chronic activation of microglia in adult zebrafish brain, and this suggests that chronic inflammation does not take place.
Zebrafish has an extensive regenerative ability in its brain, and loss of neurons causes radial glial cells to respond by increasing their proliferation39,44,45,47,48,49,50,51,52,53. To test if chronic expression of TAUP301L would induce the proliferation of neuronal progenitors, we performed immunostaining for S100β (a progenitor cell marker) and PCNA (proliferation marker) in sTg and dTg animals (Fig. 2f–i). Compared to sTg animals, chronic expression of TAUP301L did not alter the level of proliferation of radial glial cells (Fig. 2j). Additionally, similar to her4.1, nbt-driven TAU did not alter neural stem cell proliferation (S100β-PCNA stainings) or microglial activation in adult zebrafish brain (L-Plastin stainings) (Supplementary Fig. 2). These results indicate that despite the presence and hyperphosphorylation of human TAUP301L, zebrafish brain does not show any pathological hallmarks of Tauopathies, and major pathological hallmarks - cell death, inflammation and cell proliferation - are not affected. We also confirmed these findings by detecting the relative mRNA expression levels of pro-inflammatory cytokines tnfa, ifng, il1b, il6, and il12a (Supplementary Fig. 10), and observed that these genes do not change their expression levels.
Phosphorylated TAUP301L neither exacerbates the toxicity of Aβ42 nor initiates regeneration programs in adult zebrafish brain
In Alzheimer’s mouse models and cultures of human neurons, Amyloidβ42 (Aβ42) deposition leads to hyperphoyphorylated TAU and NFT formation54,55,56, and Tauopathies are suggested to mediate the Amyloid toxicity9,18,56,57,58. We have recently generated an Aβ42 toxicity model in adult zebrafish brain, where Aβ42 aggregation leads to cell death, inflammation and induction of radial glial cell proliferation and neurogenesis39. We hypothesized that by combining the AD model of adult zebrafish brain to the transgenic expression of human TAUP301L would help determining if chronic expression of TAUP301L would exacerbate the effects of Aβ42. Therefore, we analyzed the radial glial cell proliferation in sTg and dTg animals injected with PBS or Aβ42 as readout of pathology and regenerative response (Fig. 3a–e). In both sTg and dTg animals, injection of Aβ42 increased the cell proliferation to the same degree (Fig. 3e), and Aβ42 aggregation did not cause NFT formation (data not shown), indicating that TAUP301L does not exacerbate the pathological burden of Aβ42 in zebrafish.

Immunohistochemistry for PCNA (green), S100β (red), and TAUP301L (white) on coronal sections of telencephalon of a 6 month-old sTg animal injected with PBS (a), sTg animal injected with Aβ42 (b), dTg animal injected with PBS (c), and dTg animal injected with Aβ42 (d). (e) Quantification of the total number of proliferating radial glial cells in the telencephalon of sTg and dTg animals injected with PBS or Aβ42. Values represent mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.005. Scale bars equal 100 μm. n = 5 fish and > 20 histological sections for every staining. All animals are 6 months old.
A typical symptom of Amyloid toxicity is inflammation and activation of macrophages/microglia59,60,61. To determine whether TAUP301L expression would alter the inflammatory outcome exerted by Aβ42, we performed immunostaining for L-Plastin – a marker for macrophages/microglia – in sTg and dTg TAUP301L lines (Fig. 4a–d). We found that Aβ42 increases both the ramified and round macrophage/microglia in sTg animals as we have documented before39, and this increase is statistically not different in TAUP301L-expressing dTg animals (Fig. 4e). Additionally, sTg and dTg animals that are not injected with Aβ42 do not differ in the number and activation state of macrophages/microglia (Fig. 4e). These results suggest that chronic expression human of TAUP301L does not change the number and activation state of the microglia/macrophages as determined by L-Plastin staining, and does not exacerbate the effects of Aβ42 in adult zebrafish brain, and interestingly despite the widespread expression of TAU in adult zebrafish brain (Supplementary Figs 1 and 2), Aβ42 does not cause NFT formation from the TAU expressed thereof.

(a) Immunohistochemistry (IHC) for TAUP301L (red), GFP (green) and L-Plastin (white) on coronal sections of telencephalon of a 6 month-old sTg animal injected with PBS (a), sTg animal injected with Aβ42 (b), dTg animal injected with PBS (c), and dTg animal injected with Aβ42 (d). Insets below the panels show individual fluorescent channels for TAUP301L (red), GFP (green) and L-Plastin (white). Columns 1–4 are single and merged images of the regions indicated in (a–d), respectively. (e) Quantification of the total number of ramified and round L-Plastin-positive cells in the telencephalon of sTg and dTg animals injected with PBS or Aβ42. (f–g) Immunohistochemistry for Interleukin-4 (red) on coronal sections of telencephalon of a 6-month old dTg animal injected with PBS (f) or with Aβ42 (g). (h) Relative change in the expression levels of il4 after Aβ42 injection compared to control injection. Medial ventricular regions are shown. DAPI (cyan) marks the nuclei. Values represent mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.005. Scale bars equal 50 μm. n = 6 fish and > 30 histological sections for every staining. All animals are 6 months old.
In adult zebrafish brain, Aβ42 was shown to cause a specific neurodegeneration-induced regeneration response by activating the expression of Interleukin-4 (IL4) – a biomarker of neurodegeneration-induced regenerative response in adult zebrafish brain39. Since TAUP301L expression and its hyperphosphorylation did not lead to changes in cell death and stem cell proliferation, we hypothesized that in our TAU model, we would not see the neurodegeneration induced IL4 expression. To test this hypothesis, we performed immunohistochemical staining for IL4 in dTg animals injected with PBS or Aβ42 (Fig. 4f,g). We found that IL4 is not expressed in dTg animals with PBS injection (Fig. 4f), whereas Aβ42 injection leads to induction of IL4 expression that is detected by both immunostaining (Fig. 4g) and relative mRNA expression levels (upregulation by 39.5 ± 5.9 folds) (Fig. 4g,h). These results support our previous findings that TAUP301L hyperphosphorylation does not lead to neurodegeneration and subsequent regeneration response in adult zebrafish brain.
In several mouse models of TAUP301L, the phenotypes manifest in aged animals62. We have recently shown that aging exacerbates the neurodegeneration phenotypes in adult zebrafish brain40. Therefore, we hypothesized that chronic expression of TAUP301L in older zebrafish could manifest Tauopathies that we did not observe in younger fish (1–6 months of age). To test this hypothesis, we analyzed TAU expression (Supplementary Fig. 11a–f) and microglia activation that is indicative of chronic inflammation (Supplementary Fig. 11g–l) in 14 month-old adult zebrafish brains. We observed that although TAU is expressed strongly at 14 month-old brains of dTg animals (Supplementary Fig. 11d–f), this does not cause macrophage/microglia activation (Supplementary Fig. 11g–m), indicating that the lack of Tauopathies in adult zebrafish brain is not due to the late manifestation as seen in mouse models.
TAUP301L is phosphorylated in zebrafish larvae yet causing no overt behavioral phenotypes
Our TAU model of adult zebrafish brain does not form neurofibrillary tangles despite phosphorylation of the TAU protein. Previously, phosphorylated TAU was documented in zebrafish spinal cord and these fish showed defective escape response at 2 days of development, yet they can regain all behavioral functions at 4 days of development and afterwards37 suggesting that zebrafish can effectively overcome TAU-mediated pathology. To investigate whether in our zebrafish lines TAU is expressed and phosphorylated in larval stages, we performed immunohistochemical staining for TAU and western blot analysis for phosphorylated TAU markers AT8, AT180, and T205 (Supplementary Fig. 12). We found that TAU is widely expressed in zebrafish larvae along the whole rostrocaudal axis of the central nervous system, and it is phosphorylated (Supplementary Fig. 12). By band intensity quantifications, we found that 91.4 ± 4.7% of total TAU is AT180-positive and 70.3 ± 4.7% of total TAU is AT8-positive in the whole larvae.
In order to examine whether the larval fish shows escape response deficits, we performed a behavioral assay for 2- and 4 day-old larvae as described before37 (Supplementary Videos 1 and 2). We observed that dTg larvae show normal escape behavior as in their non-transgenic siblings and sTg counterparts (Supplementary Videos 1 and 2), indicating that the Tauopathy in the spinal cord of the larvae is causing the behavioral deficits. These results recapitulate what has been documented before37,63.
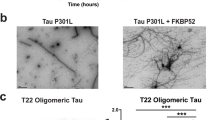
TAUP301L does not form oligomers in adult zebrafish brain
Our results showed that while TAUP301L is phosphorylated, neurofibrillary tangles do not form. We hypothesized that this could be because TAUP301L might not be forming oligomers that are the precursors of tangles. In order to test this hypothesis, we performed immmunohistochemical staining with T22 antibody that detects TAU oligomers. We found that TAUP301L does not form oligomers in adult zebrafish brain in the presence or absence of Amyloid-β42 (Supplementary Fig. 13). These results indicate that in adult zebrafish brain, despite being phosphorylated, TAUP301L does not form oligomeric forms of TAU, and this could be the reason for absence of neurofibrillary tangles.
Discussion
In this study, we generated two cre-lox-based conditional expression lines for adult zebrafish brain to study the effects of chronic expression of human TAUP301L and its relation to Aβ42 toxicity. We found that although chronically expressed TAU is hyperphosphorylated, it does not form oligomers and neurofibrillary tangles, and hence does not cause neurodegeneration in the adult zebrafish brain (Fig. 5).

Schematic comparison of the effects of TAU in mammalian and zebrafish brain.
Previous reports of TAU in zebrafish used larval and juvenile animals34,37,63 or did not thoroughly investigate the adult phenotypes36. Our study is therefore the first study investigating TAU in a chronic expression model of adult zebrafish brain, where we found that human TAUP301L was hyperphosphorylated as documented before in zebrafish36,64. However, hyperphosphorylation of TAU did not cause formation of oligomers and neurofibrillary tangles in adult zebrafish brain. We believe that this is not due to the inability of phosphorylated TAU to form neurofibrillary tangles per se, but because of a specific blockage of tangle formation in adult zebrafish brain.
The lack of tangle formation despite the presence of hyperphosphorylated form of TAU might be due to several reasons. First, zebrafish brain might not have the circuitries and neuronal subtypes that are susceptible to TAU aggregation in mammals, and therefore modeling Tauopathies in zebrafish might not be appropriate. We believe that this is not the case, because several studies have shown that adult zebrafish brain have analogous regions to the human brains65,66, and the majority of the neuronal subtypes present in our brains are existent in adult zebrafish brain45,67,68,69. Additionally, all the constituents of the machinery known to be required for TAU phosphorylation and aggregation is present in zebrafish37,63 as zebrafish proteome contains more than 80% of the proteins present in humans70 and the kinases that are implicated in TAU hyperphosphorylation (e.g.: GSK3β, Cdk5, MARK1 and CK1α71) are present in zebrafish, and are expressed (Supplementary Fig. 14). Second, the expression level of TAU is known to be a critical determinant of the aggregation dynamics62. In our experiments, we used ubiquitin promoter, which drives the expression of the downstream gene efficiently72. Also, our western blot results indicate that the levels of TAU are higher than the housekeeping gene beta-actin (Fig. 1h), and immunohistochemical stainings detect strong expression of TAU protein (Figs 1e, g and 2c). Based on these results, we believe that the lack of aggregation of TAU is not due to the levels of TAU protein. Third, our transgenic line may not be expressing TAU in the neurons that are susceptible to Tauopathies in humans. This is a challenging possibility, yet, as we determined by sagittal sections of the adult fish brain (Supplementary Figs 1 and 2), her4.1 and nbt promoters can lead to TAU expression in the majority of the neurons in the adult fish brain, and it is very unlikely that such a widespread expression would not include the neurons of the forebrain that are susceptible to Tauopathies. Therefore, we hypothesize that the lack of neurofibrillary tangles in our model is not due to targeting the right cell types but due to an active protective mechanism that prevents phosphorylated TAU from forming tangles. This is an interesting option suggesting that zebrafish brain could serve as an in vivo model for experimentation on how Tauopathies can be prevented by using endogenous cellular programs. Further research on why zebrafish does not form Tauopathies in the brain while it does in the larval spinal cord37,63 will constitute an exciting basic and pre-clinical research line. For instance, prevention of TAU dephosphorylation by Okadaic acid was shown to increase hyperphosphorylated state of TAU and subsequently leading to neurofibrillary tangles73. When we injected Okadaic acid to TAUP301L-expressing adult zebrafish brains, we still did not observe neurofibrillary tangle formation (data not shown), suggesting a currently unknown cellular mechanisms that might be utilized to prevent phosphorylated TAU from forming tangles in human brains. We also found that in the larvae of our zebrafish model, TAU is expressed and hyperphosphorylated (Supplementary Fig. 12) but does not show any behavioral phenotypes such as escape response (Supplementary Videos 1 and 2). Additionally, adult dTg zebrafish do not show any swimming defects or escape response (data not shown) suggesting that TAU hyperphosphorylation does not cause NFT formation in larval and adult stages in our model. Thus, using our zebrafish TAU model as a tool to uncover putative protective mechanisms that prevent phosphorylated TAU from forming tangles will be of great interest to the medical field.
It is still controversial whether neuroinflammatory events lead to TAU phosphorylation or phosphorylated-TAU precedes neuroinflammation46,57,74,75,76. Our results suggest that phosphorylated-TAU is not sufficient to induce microglial activation by itself, and that aggregated forms of TAU might be required for this. We found that in our model, oligomeric TAU is not formed (Supplementary Fig. 13), suggesting that TAU phosphorylation might not be caused by immune cell activity and more toxic forms of TAU (such as oligomers) are required to elicit neuroinflammation by TAU. Our model can help to investigate this interesting hypothesis by analyzing the effects of TAU on various immune cell types, not necessarily confined to microglia, but also other immune cells involved in adaptive immune responses.
We recently established an Amyloid-β42 toxicity model in zebrafish, and observed that Aβ42 aggregation causes cell death, inflammation, synaptic degeneration, and memory deficits, which collectively lead to regenerative response including stem cell activation and neurogenesis39,62, signifying the role of neuro-immune cross talk for efficient regeneration response in contrast to mammals77,78. When we combined amyloid toxicity model with chronic genetic expression of human TAUP301L, we found that TAU expression does not exacerbate the Amyloid phenotypes, and Amyloid toxicity does not cause TAU-mediated neurofibrillary tangle formation. Additionally, unlike Aβ42, TAUP301L does not induce the expression of neurodegeneration-induced factor Interleukin-439 (Fig. 4g), providing further evidence that TAUP301L expression fails to elicit a neurodegeneration-regeneration cascade in adult zebrafish brain. Since zebrafish brain is highly regenerative after various type of neuronal loss33,45,67,79, we believe that the lack of stem cell proliferation response and activation of regenerative programs is an indication that adult zebrafish brain can cope well with the phosphorylated forms of TAUP301L.
Several studies documented Tauopathy-related neurodegeneration phenotypes in the absence of tangle formation17,18,80,81,82. Conflicting with these findings, Tau aggregation was also proposed as the major cause of toxicity in animal models1,2,4,9,13,56,61,83. We have to note that in our genetic model, we have used the longest isoform of human TAU protein (2N4R). Different versions of TAU have been shown to have different levels of aggregation potential, and therefore, in future other human TAU versions could be tested in zebrafish to come to a more consolidated conclusion of a protective mechanism preventing tangle formation. Additionally, protein aggregation is temperature dependent, and zebrafish lives at 28 °C while our body temperature is 37 °C. Therefore, a possible link between the temperature and aggregation dynamics of TAU could also account for the observed differences, and this aspect of Tauopathies can also be studied in zebrafish. Given that keeping zebrafish at 37 °C has been used as a means of heat stress, the effects of temperature on folding, phosphorylation and aggregation dynamics and clearance of TAU are therefore interesting aspects of TAU toxicity that can be tested in vivo in zebrafish.
In overall, we believe that our new chronic conditional TAU expression model in zebrafish is a useful assay system where various neurodegeneration paradigms could be modularly combined, and the effects of any intervention on the TAU aggregation could be measured. For instance, our model would allow screening for molecules or pathways that could abrogate a putative “protective mechanism” that prevents phosphorylated TAU from getting aggregated. In such a case, adult zebrafish brain may potentially start generating neurofibrillary tangles. Alternatively, possible factors that are related to immune system could have effects on how TAU is phosphorylated and how phosphorylated forms of TAU proceed further to form oligomers and neurofibrillary tangles. Such “reverse” studies could uncover currently unknown mechanisms that may help us to further understand and design therapies for Tauopathies in humans.
Zebrafish brain is highly regenerative and responds to injuries by activating specific molecular programs33,45,67. A possible regenerative therapy in human brains could entail mobilizing the endogenous stem cells to form new neurons that replace the lost ones, and regulation of the synaptic plasticity to compensate for the lost circuit connections. Reductionist models of neurodegenerative diseases in zebrafish can therefore help us to understand which types pathogenicity would activate regenerative programs in stem cells. By cataloging the effects of individual pathogenic hallmarks of neurodegenerative diseases in zebrafish, this animal model can help clinical studies to design regenerative therapies or prevention strategies that are naturally occurring in a vertebrate brain. Since previous zebrafish models of Tauopathies did not focus on the regenerative response of the brain, our model is also a useful addition to the toolbox of regeneration research in order to understand how a vertebrate brain responds to toxic protein aggregation or even prevents the progression of disease pathology.
Materials and Methods
Ethics statement
All animal experiments were carried out in accordance with the animal experimentation permits of Referate 24 (Veterinärwesen, Pharmazie, und GMP) of the state administration office of Saxony, Germany (Landesdirektion Sachsen) and the ethical commission of TU Dresden (Kommission für Tierversuche). The approved experimental protocols are licensed under the permit numbers TVV-35/2016 and TVV-52/2015 through the regular official processes.
Generation of transgenic zebrafish lines and recombination
The longest isoform of human TAU (2N4R) with P301L mutation template was a gift from Bettina Schmid and Christian Haass37. The template was amplified by PCR using the forward (5′-atggctgagccccgccag-3′) and reverse (5′-tcacaaaccctgcttggccagg-3′) primer pairs that were flanked by XhoI and AscI restriction enzymes, respectively. The PCR product was subcloned into pGEM-T-Easy vector (Promega) and sequenced by M13 universal primers. The TAUP301L was released from pGEM-T-Easy by XhoI/AscI enzymes and cloned into pTol(Red-to-Green) (ubiquitin promoter-driven72 loxP-DsRed-loxP-GFP cassette-containing Tol2 transgenesis vector47,84). For generating neural beta tubulin (nbt) promoter-driven cre line, we amplified the nbt promoter using forward (5′-tgggccctctagaccctgtctg-3′) and reverse (5′-ggccggccgattgggttgagtc-3′) primers flanked by ApaI and FseI restriction enzyme sites. PCR product was subcloned into her4.1:mCherry-t2a-CreERT2 transgenesis plasmid containing Tol2 sites84,85 by using appropriate restriction enzymes.
For transgenesis, we used a protocol as described85 with minor modifications. Briefly, 1 nL of 30 ng/µL plasmid and Tol2 transposase mix in RNAse-free water were injected into fertilized zebrafish eggs (F0), the fish were raised to adulthood, and were outcrossed to wild type zebrafish (AB background) to establish the founder fish (F1). 6 founders were identified, and were raised to adulthood for Tg(ubi:loxp-DsRed-loxp-GFP-t2a-hTAUP301L). Tg(nbt:mCherry-t2a-CreERT2) founders were identified by outcrossing to wild type animals and screening for mCherry expression. Positive embryos were raised to adulthood and crossed to a control cre-effector line that shows fluorescence after recombination in order to test the effective recombination capacity. To identify a cre-effector TAU line with high efficiency of recombination, all F1 fish for TAU were outcrossed to Tg(her4.1:mCherry-t2a-CreERT2) or Tg(nbt:mCherry-t2a-CreERT2) cre-driver lines, and the F2 progeny were treated with freshly prepared 5 µM of Tamoxifen (TAM) in E3 medium between 16–32 hpf and 32–48 hpf. At 48hpf, the F2 progeny with high GFP expression were sorted from non-GFP siblings. The outcrosses were repeated to obtain a stably segregating transgene insertion (achieved after F4). The experiments and results given here are from F5 and later offspring.
Tissue preparation and immunohistochemical stainings
The fish were sacrificed, and the heads were fixed in 2% PFA overnight at 4 °C. After washing 3 times 10 min with Phosphate buffer, the heads were decalcified using 20% Sucrose/EDTA. Samples were mounted in 20% Sucrose/7.5% Gelatin as described39. Samples were cryosectioned at 12 µM thickness, and were stained with the following antibodies: chicken anti-GFP IgG (1:2000; Abcam), mouse anti-Tau13 IgG1 (IHC 1:1000, WB 1:500; Abcam), mouse anti-phospho-Tau (AT180) IgG1 (IHC 1:500, WB 1:1000; Pierce, Thermo Scientific), mouse anti-phospho-Tau (AT8) IgG1 (IHC 1:500, WB 1:1000; Pierce, Thermo Scientific), mouse anti-phospho-Tau (AT270) IgG1 (IHC 1:500, WB 1:1000; Pierce, Thermo Scientific), mouse anti-PCNA IgG2a (1:500; Dako), rabbit anti-S100β IgG (1:500; Dako), rabbit-anti-L-plastin IgG (1:5000) (gift from Michael Redd), rabbit anti-beta actin (WB 1:1000, Abcam), rabbit anti-synaptophysin (1:500, Abcam), mouse anti-HuC/D IgG2b (1:500, Life Technologies), mouse anti IL4 (1:500, R&D Systems), and rabbit anti-Tau (T22) (1:500, Merck).
Gallyas silver staining
Brain samples were fixed as described above. Heads were embedded in paraffin, sectioned at a thickness of 10 µM. Samples were pretreated with 0.25% potassium permanganate at room temperature for 15 min86, washed in 2% oxalic acid for 2 min, and incubated in 0.4% lanthanum nitrate/2% sodium acetate/3% H2O2 solution at room temperature for 60 min87. The following treatments were performed: 5% periodic acid (5 min), alkaline silver iodide solution (1 min), 0.5% acetic acid (10 min), developer solution containing tungsto-silicic acid (5–10 min), 0.5% acetic acid, 0.1% gold chloride (5 min), and 1% sodium thiosulfate (5 min). Samples were counterstained with 0.1% nuclear fast red (2 min), dehydrated using Methanol, and were mounted in DPX.
Western blotting
For western blotting, the telencephalon of 6 month-old fish were dissected or 9 day-old larvae were sacrificed according the local ethical regulations. The telencephalon or whole larvae were used for protein isolation using 200 µL RIPA buffer (Sigma, R0278) with addition of phosphatase (Roche, Catalog Number 04906837001) and protease inhibitors (Roche, Catalog number 04963132001). 5 µl of protein ladder (ThermoFisher, #26634) and 10 µl of total protein was loaded in 4–12% Bis-Tris precast gradient gels (NuPage, Catalog number NP0322BOX) and the gels were run in NuPAGE MES SDS running buffer (Novex, Life technologies, NP0006-1) at 200 V for 60 min. Blots were transferred to methanol activated PVDF (Novex, Life technologies, LC2002) membrane, and were blocked in 10% milk powder in 0.2% Tween in 1X-PBS for 1h at room temperature. The primary antibodies at appropriate dilutions in 2 ml 0.2% Tween in 1X-PBS were applied overnight at 4 °C in 15 or 50 ml plastic containers. Following the washing steps, secondary antibodies at 1:4000 dilutions (anti-rabbit IgG HRP (Santa Cruz, #sc-2004) or anti-mouse IgG peroxidase (Sigma, #A8924)) were applied at room temperature for 2 h. Gel images were acquired by ImageQuant LAS4000 (GE Healthcare) using Western BLoT Ultra Sensitive HRP Substrate (Takara, #T7104A).
Cerebroventricular microinjection
Zebrafish at the age of 6 months were injected with 20 µM Aβ42 as described previously39,88,89,90. For cerebroventricular microinjection, the fish were first anaesthetized in 0.001% MESAB, and a slit was generated in the skull over the optic tectum without damaging the brain. 1 µl of solution containing monomeric Amyloid-β4239,40 was injected into the cerebroventricular fluid (for control animals PBS was injected). The fish were returned to their containers and recovery was carefully observed. All the injected fish survived the procedure and did not show any defects that might be due to the injections. 25 µg of Okadaic acid (Sigma #O8010) was dissolved in 500 µl distilled 1x-PBS, and 5 ng/µL was injected into the fish brain as described previously89,90. 3 dpi fish were sacrificed and prepared for immunohistochemical stainings as described above.
Detection of mRNA expression levels
mRNA isolation and detection of relative mRNA expression levels were performed as described39,40.
Imaging, quantification, and statistical analyses
Images were acquired using structured illumination microscope (Zeiss AxioImager Z1) and scanning confocal microscope (Leica SP5 II, DM6000). In order to maintain the comparative intensities of the images and not to introduce bias, we used the same settings for all experimental groups of controls and transgenic animals per experiment. Images were taken using the tiling option of the Apotome microscope with no auto-correction to prevent inconsistencies in fluorescence intensity. Acquired images were stitched together using the Zeiss ZEN software installed to the Apotome configuration. Raw images are used to generate figures using Adobe Photoshop and Adobe Illustrator software. Cell counting was performed manually on the entire sections taken on a slide and stereological calculations were performed as described91. The statistical analyses were performed using GraphPad Prism (Version 6.02) for one-way ANOVA followed by a Tukey’s post-hoc test and for Student’s T-Test. Error bars shown are the s.d, and asterisks indicate significance: *p < 0.05, **p < 0.01, ***p < 0.001. p > 0.05 is not significant (n.s.). Student’s T-test was performed for paired samples, and a T-Test for independent measurements. All sample sizes and analyses powers were calculated using G*Power-Software, (http://www.gpower.hhu.de/en.html). No animals were excluded from the analyses. n = 5 fish for every experimental group, and in total more than 20 sections were used for stereological quantification. The ages of the animals used are denoted in the respective figure legends. Quantification of band intensities in western blots were performed as described92.
Behavioral analyses
Escape response assays were performed as described37. Tamoxifen was replaced with fresh E3 medium. The video and imaging taken by changing filter from bright field to DsRED and then to GFP. The escape response was recorded at GFP channel to localize the dTg fish. Escape response was measured by touching the tail using Olympus MVX10 stereo microscope and CellSens Dimension software.
Data Availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Iqbal, K., Liu, F. & Gong, C. X. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12, 15–27, https://doi.org/10.1038/nrneurol.2015.225 (2016).
Iqbal, K. et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739, 198–210, https://doi.org/10.1016/j.bbadis.2004.09.008 (2005).
Hutton, M. et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705, https://doi.org/10.1038/31508 (1998).
Spillantini, M. G. et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95, 7737–7741 (1998).
Poorkaj, P. et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43, 815–825, https://doi.org/10.1002/ana.410430617 (1998).
Matsuo, E. S. et al. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 13, 989–1002 (1994).
Lewis, J. et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25, 402–405, https://doi.org/10.1038/78078 (2000).
Duff, K. et al. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis 7, 87–98, https://doi.org/10.1006/nbdi.1999.0279 (2000).
Gotz, J. & Ittner, L. M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci 9, 532–544, https://doi.org/10.1038/nrn2420 (2008).
Allen, B. et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22, 9340–9351 (2002).
Andorfer, C. et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86, 582–590 (2003).
Tanemura, K. et al. Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol Dis 8, 1036–1045, https://doi.org/10.1006/nbdi.2001.0439 (2001).
Terwel, D. et al. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem 280, 3963–3973, https://doi.org/10.1074/jbc.M409876200 (2005).
Oddo, S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 (2003).
Grueninger, F. et al. Phosphorylation of Tau at S422 is enhanced by Abeta in TauPS2APP triple transgenic mice. Neurobiol Dis 37, 294–306, https://doi.org/10.1016/j.nbd.2009.09.004 (2010).
Lewis, J. et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491, https://doi.org/10.1126/science.1058189 (2001).
Platt, B. et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS One 6, e27068, https://doi.org/10.1371/journal.pone.0027068 (2011).
Jackson, R. J. et al. Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur J Neurosci 44, 3056–3066, https://doi.org/10.1111/ejn.13442 (2016).
Nisbet, R. M., Polanco, J. C., Ittner, L. M. & Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta neuropathologica 129, 207–220, https://doi.org/10.1007/s00401-014-1371-2 (2015).
Ittner, L. M. & Gotz, J. Amyloid-beta and tau–a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12, 65–72, https://doi.org/10.1038/nrn2967 (2011).
Bloom, G. S. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508, https://doi.org/10.1001/jamaneurol.2013.5847 (2014).
Gotz, J., Chen, F., van Dorpe, J. & Nitsch, R. M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495, https://doi.org/10.1126/science.1062097 (2001).
Hurtado, D. E. et al. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol 177, 1977–1988, https://doi.org/10.2353/ajpath.2010.100346 (2010).
Roberson, E. D. et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754, https://doi.org/10.1126/science.1141736 (2007).
Nussbaum, J. M. et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 485, 651–655, https://doi.org/10.1038/nature11060 (2012).
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P. & Ferreira, A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99, 6364–6369, https://doi.org/10.1073/pnas.092136199 (2002).
Vossel, K. A. et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science 330, 198, https://doi.org/10.1126/science.1194653 (2010).
Ramser, E. M. et al. Amyloid-beta oligomers induce tau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol Biol Cell 24, 2494–2505, https://doi.org/10.1091/mbc.E12-12-0858 (2013).
Reifert, J., Hartung-Cranston, D. & Feinstein, S. C. Amyloid beta-mediated cell death of cultured hippocampal neurons reveals extensive Tau fragmentation without increased full-length tau phosphorylation. J Biol Chem 286, 20797–20811, https://doi.org/10.1074/jbc.M111.234674 (2011).
Takach, O., Gill, T. B. & Silverman, M. A. Modulation of insulin signaling rescues BDNF transport defects independent of tau in amyloid-beta oligomer-treated hippocampal neurons. Neurobiol Aging 36, 1378–1382, https://doi.org/10.1016/j.neurobiolaging.2014.11.018 (2015).
Ittner, A. et al. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science 354, 904–908, https://doi.org/10.1126/science.aah6205 (2016).
Tincer, G., Mashkaryan, V., Bhattarai, P. & Kizil, C. Neural stem/progenitor cells in Alzheimer’s disease. Yale J Biol Med 89, 23–35 (2016).
Cosacak, M. I., Papadimitriou, C. & Kizil, C. Regeneration, Plasticity, and Induced Molecular Programs in Adult Zebrafish Brain. Biomed Res Int 2015, 769763, https://doi.org/10.1155/2015/769763 (2015).
Tomasiewicz, H. G., Flaherty, D. B., Soria, J. P. & Wood, J. G. Transgenic zebrafish model of neurodegeneration. J Neurosci Res 70, 734–745, https://doi.org/10.1002/jnr.10451 (2002).
Wu, B. K. et al. Multiple signaling factors and drugs alleviate neuronal death induced by expression of human and zebrafish tau proteins in vivo. J Biomed Sci 23, 25, https://doi.org/10.1186/s12929-016-0237-4 (2016).
Bai, Q., Garver, J. A., Hukriede, N. A. & Burton, E. A. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res 35, 6501–6516, https://doi.org/10.1093/nar/gkm608 (2007).
Paquet, D. et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest 119, 1382–1395, https://doi.org/10.1172/Jci37537 (2009).
Lopez, A. et al. A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 140, 1128–1146, https://doi.org/10.1093/brain/awx005 (2017).
Bhattarai, P. et al. IL4/STAT6 signaling activates neural stem cell proliferation and neurogenesis upon Amyloid-β42 aggregation in adult zebrafish brain. Cell Reports 17, 941–948, https://doi.org/10.1016/j.celrep.2016.09.075 (2016).
Bhattarai, P., Thomas, A. K., Zhang, Y. & Kizil, C. The effects of aging on Amyloid-β42-induced neurodegeneration and regeneration in adult zebrafish brain. Neurogenesis, https://doi.org/10.1080/23262133.2017.1322666 (2017).
Park, H. C. et al. Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev Biol 227, 279–293, https://doi.org/10.1006/dbio.2000.9898 (2000).
Peri, F. & Nusslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916–927, https://doi.org/10.1016/j.cell.2008.04.037 (2008).
Wang, Y., Wang, F., Wang, R., Zhao, P. & Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Sci Rep 5, 16273, https://doi.org/10.1038/srep16273 (2015).
Kizil, C. et al. Regenerative neurogenesis from neural progenitor cells requires injury-induced expression of Gata3. Dev Cell 23, 1230–1237, https://doi.org/10.1016/j.devcel.2012.10.014 (2012).
Kizil, C., Kaslin, J., Kroehne, V. & Brand, M. Adult neurogenesis and brain regeneration in zebrafish. Dev Neurobiol 72, 429–461, https://doi.org/10.1002/dneu.20918 (2012).
Jay, T. R. et al. TREM2 deficiency eliminates TREM2+inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295, https://doi.org/10.1084/jem.20142322 (2015).
Kroehne, V., Freudenreich, D., Hans, S., Kaslin, J. & Brand, M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development 138, 4831–4841, https://doi.org/10.1242/dev.072587 (2011).
Kizil, C. et al. The chemokine receptor cxcr5 regulates the regenerative neurogenesis response in the adult zebrafish brain. Neural Dev 7, 27, https://doi.org/10.1186/1749-8104-7-27 (2012).
Kyritsis, N. et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 338, 1353–1356, https://doi.org/10.1126/science.1228773 (2012).
Marz, M., Schmidt, R., Rastegar, S. & Strahle, U. Regenerative response following stab injury in the adult zebrafish telencephalon. Dev Dyn 240, 2221–2231, https://doi.org/10.1002/dvdy.22710 (2012).
Baumgart, E. V., Barbosa, J. S., Bally-Cuif, L., Gotz, M. & Ninkovic, J. Stab wound injury of the zebrafish telencephalon: a model for comparative analysis of reactive gliosis. Glia 60, 343–357, https://doi.org/10.1002/glia.22269 (2012).
Kishimoto, N., Shimizu, K. & Sawamoto, K. Neuronal regeneration in a zebrafish model of adult brain injury. Dis Model Mech 5, 200–209, https://doi.org/10.1242/dmm.007336 (2012).
Barbosa, J. S. et al. Neurodevelopment. Live imaging of adult neural stem cell behavior in the intact and injured zebrafish brain. Science 348, 789–793, https://doi.org/10.1126/science.aaa2729 (2015).
Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278, https://doi.org/10.1038/nature13800 (2014).
LaFerla, F. M. & Green, K. N. Animal models of Alzheimer disease. Cold Spring Harbor perspectives in medicine 2, https://doi.org/10.1101/cshperspect.a006320 (2012).
Goedert, M., Klug, A. & Crowther, R. A. Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimers Dis 9, 195–207 (2006).
Ghosh, S. et al. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 33, 5053–5064, https://doi.org/10.1523/JNEUROSCI.4361-12.2013 (2013).
Demars, M., Hu, Y. S., Gadadhar, A. & Lazarov, O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J Neurosci Res 88, 2103–2117, https://doi.org/10.1002/jnr.22387 (2010).
Heppner, F. L., Ransohoff, R. M. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372, https://doi.org/10.1038/nrn3880 (2015).
Wyss-Coray, T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 12, 1005–1015, https://doi.org/10.1038/nm1484 (2006).
Akiyama, H. et al. Inflammation and Alzheimer’s disease. Neurobiol Aging 21, 383–421 (2000).
Roberson, E. D. Mouse models of frontotemporal dementia. Ann Neurol 72, 837–849, https://doi.org/10.1002/ana.23722 (2013).
Paquet, D., Schmid, B. & Haass, C. Transgenic zebrafish as a novel animal model to study tauopathies and other neurodegenerative disorders in vivo. Neurodegener Dis 7, 99–102, https://doi.org/10.1159/000285515 (2010).
Sepulveda-Diaz, J. E. et al. HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 138, 1339–1354, https://doi.org/10.1093/brain/awv056 (2015).
Ganz, J. et al. Subdivisions of the adult zebrafish subpallium by molecular marker analysis. J Comp Neurol 520, 633–655, https://doi.org/10.1002/cne.22757 (2011).
Mueller, T. & Wullimann, M. F. An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav Evol 74, 30–42, https://doi.org/10.1159/000229011 (2009).
Alunni, A. & Bally-Cuif, L. A comparative view of regenerative neurogenesis in vertebrates. Development 143, 741–753, https://doi.org/10.1242/dev.122796 (2016).
Kaslin, J. et al. Stem cells in the adult zebrafish cerebellum: initiation and maintenance of a novel stem cell niche. J Neurosci 29, 6142–6153, https://doi.org/10.1523/JNEUROSCI.0072-09.2009 (2009).
Mueller, T., Vernier, P. & Wullimann, M. F. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res 1011, 156–169, https://doi.org/10.1016/j.brainres.2004.02.073 (2004).
Howe, K. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503, https://doi.org/10.1038/nature12111 (2013).
Cavallini, A. et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem 288, 23331–23347, https://doi.org/10.1074/jbc.M113.463984 (2013).
Mosimann, C. et al. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 138, 169–177, https://doi.org/10.1242/dev.059345 (2011).
Baker, S. & Gotz, J. A local insult of okadaic acid in wild-type mice induces tau phosphorylation and protein aggregation in anatomically distinct brain regions. Acta Neuropathol Commun 4, 32, https://doi.org/10.1186/s40478-016-0300-0 (2016).
Zilka, N. et al. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. Journal of neuroinflammation 9, 47, https://doi.org/10.1186/1742-2094-9-47 (2012).
Kitazawa, M., Yamasaki, T. R. & LaFerla, F. M. Microglia as a potential bridge between the amyloid beta-peptide and tau. Ann N Y Acad Sci 1035, 85–103, https://doi.org/10.1196/annals.1332.006 (2004).
Arnaud, L., Robakis, N. K. & Figueiredo-Pereira, M. E. It may take inflammation, phosphorylation and ubiquitination to ‘tangle’ in Alzheimer’s disease. Neurodegener Dis 3, 313–319, https://doi.org/10.1159/000095638 (2006).
Kizil, C., Kyritsis, N. & Brand, M. Effects of inflammation on stem cells: together they strive? EMBO reports 16, 416–426, https://doi.org/10.15252/embr.201439702 (2015).
Kyritsis, N., Kizil, C. & Brand, M. Neuroinflammation and central nervous system regeneration in vertebrates. Trends Cell Biol 24, 128–135, https://doi.org/10.1016/j.tcb.2013.08.004 (2014).
Fleisch, V. C., Fraser, B. & Allison, W. T. Investigating regeneration and functional integration of CNS neurons: lessons from zebrafish genetics and other fish species. Biochim Biophys Acta 1812, 364–380, https://doi.org/10.1016/j.bbadis.2010.10.012 (2010).
Espuny-Camacho, I. et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron 93(1066-1081), e1068, https://doi.org/10.1016/j.neuron.2017.02.001 (2017).
Maeda, S. et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO reports 17, 530–551, https://doi.org/10.15252/embr.201541438 (2016).
Umeda, T. et al. Neurodegenerative disorder FTDP-17-related tau intron 10+16C–> T mutation increases tau exon 10 splicing and causes tauopathy in transgenic mice. Am J Pathol 183, 211–225, https://doi.org/10.1016/j.ajpath.2013.03.015 (2013).
Kirkitadze, M. D., Bitan, G. & Teplow, D. B. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res 69, 567–577, https://doi.org/10.1002/jnr.10328 (2002).
Hans, S. et al. Generation of a non-leaky heat shock-inducible Cre line for conditional Cre/lox strategies in zebrafish. Dev Dyn 240, 108–115, https://doi.org/10.1002/dvdy.22497 (2011).
Hans, S., Kaslin, J., Freudenreich, D. & Brand, M. Temporally-controlled site-specific recombination in zebrafish. PLoS One 4, e4640, https://doi.org/10.1371/journal.pone.0004640 (2009).
Uchihara, T., Kondo, H., Ikeda, K. & Kosaka, K. Alzheimer-type pathology in melanin-bleached sections of substantia nigra. J Neurol 242, 485–489 (1995).
Gallyas, F. & Wolff, J. R. Metal-catalyzed oxidation renders silver intensification selective. Applications for the histochemistry of diaminobenzidine and neurofibrillary changes. J Histochem Cytochem 34, 1667–1672, https://doi.org/10.1177/34.12.3537114 (1986).
Kizil, C. et al. Efficient cargo delivery using a short cell-penetrating peptide in vertebrate brains. PLoS One 10, e0124073 (2015).
Kizil, C., Iltzsche, A., Kaslin, J. & Brand, M. Micromanipulation of gene expression in the adult zebrafish brain using cerebroventricular microinjection of morpholino oligonucleotides. J Vis Exp, e50415, https://doi.org/10.3791/50415 (2013).
Kizil, C. & Brand, M. Cerebroventricular microinjection (CVMI) into adult zebrafish brain is an efficient misexpression method for forebrain ventricular cells. PLoS One 6, e27395, https://doi.org/10.1371/journal.pone.0027395 (2011).
West, M. J. Introduction to stereology. Cold Spring Harb Protoc 2012, https://doi.org/10.1101/pdb.top070623 (2012).
Kizil, C. et al. smp is required for beta catenin nuclear translocation in zebrafish development. Development 141, 3529–3539 (2014).
Acknowledgements
This work was supported by German Center for Neurodegenerative Diseases (DZNE) Dresden, Helmholtz Association (VH-NG-1021), Center for Regenerative Therapies Dresden (CRTD), TU Dresden, and Deutsche Forschungsgemeinschaft (DFG) (KI 1524/6-1) to C.K. We would like to thank Bettina Schmid and Christian Haass for the plasmids containing human TAUP301L, Stefan Hans and Michael Brand for her4.1 cre-driver zebrafish line and the plasmid containing the cre-effector cassette pTol(R2G), Michael Redd for L-Plastin antibody, Alvin Thomas and Yixin Zhang for Amyloid-β42 peptide, and Gerd Kempermann for advice and comments on the results.
Author information
Authors and Affiliations
Contributions
M.I.C. and C.K. conceived and designed the experiments, M.I.C., V.M., H.H. and K.B. generated the Tg(ubi:loxP-DsRed-loxP-GFP-t2a-hTAUP301L) cre-effector TAU expression transgenic line, C.A., M.I.C. and C.K. generated Tg(nbt:mCherry-t2a-creERT2) cre-driver line, M.I.C. characterized the transgenic lines, performed tissue preparation, sectioning, stainings and quantifications, L.B. and T.D. helped the stainings and quantifications, P.B. performed Amyloid injections, C.P. prepared human neuronal cultures and Gallyas silver staining. M.I.C. and C.K. analyzed the data and prepared the figures, C.K and M.I.C. wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cosacak, M.I., Bhattarai, P., Bocova, L. et al. Human TAUP301L overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci Rep 7, 12959 (2017). https://doi.org/10.1038/s41598-017-13311-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13311-5
This article is cited by
-
Zebrafish as a Model Organism for Studying Pathologic Mechanisms of Neurodegenerative Diseases and other Neural Disorders
Cellular and Molecular Neurobiology (2023)
-
Cannabinoid receptor CB2 ablation protects against TAU induced neurodegeneration
Acta Neuropathologica Communications (2021)
-
Mechanisms of Pathology-Induced Neural Stem Cell Plasticity and Neural Regeneration in Adult Zebrafish Brain
Current Pathobiology Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
