
Abstract
Hybridogenesis is a special mode of hybrid reproduction where one parental genome is eliminated and the other is transmitted clonally. We propose that this mechanism can perpetuate the genome of extinct species, based on new genetic data from Pelophylax water frogs. We characterized the genetic makeup of Italian hybridogenetic hybrids (P. kl. hispanicus and esculentus) and identified a new endemic lineage of Eastern-Mediterranean origin as one parental ancestor of P. kl. hispanicus. This taxon is nowadays extinct in the wild but its germline subsists through its hybridogenetic descendant, which can thus be considered as a “semi living fossil”. Such rare situation calls for realistic efforts of de-extinction through selective breeding without genetic engineering, and fuels the topical controversy of reviving long extinct species. “Ghost” species hidden by taxa of hybrid origin may be more frequent than suspected in vertebrate groups that experienced a strong history of hybridization and semi-sexual reproduction.
Similar content being viewed by others

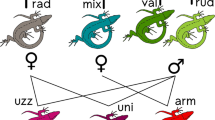

Introduction
In a context of the worldwide mass extinction of biodiversity, the hopes for rediscovering or reviving extinct wildlife have become a popular scientific fantasy1,2. So-called “de-extinction” programs, like the “mammophant” (mammoth × elephant in vitro hybrid) or the Lazarus project (cloning of the extinct platypus frog from frozen specimens) are taking this fantasy to the experimental level, following recent advances in cloning biotechnology3,4,5,6,7. Here we present a new opportunity: the preservation of the genome of an extinct species through hybridogenesis, a special reproductive mode of hybrids. In this mode, the parental genomes do not recombine in F1s: one is eliminated before meiosis, and the other is solely transmitted in a clonal manner8 (Fig. 1). Such case of semi-sexual reproduction is rare, but found in several amphibians and fishes9. It is particularly famous of European water frogs, where pool frogs (P. lessonae sensu lato) hybridize with marsh frogs (P. ridibundus s. l.) to form hybridogenetic hybrid species, aka kleptons, (P. kl. esculentus s. l.), in which the P. lessonae s. l. genome is excluded from the germline8. This group thus makes a fascinating model system to study hybridogenesis, where it can have far-reaching consequences: invasive marsh frogs are currently replacing pool frogs across Western Europe10; human-mediated introductions have led to the formation of novel hybridogenetic systems from naturally allopatric taxa, involving new klepton species11. As such, and while they benefit from a specific status, kleptons usually have a poor conservation value as they act as genetic parasites. In this report, we propose that hybridogenesis has perpetuated the lineage of an extinct clade of water frogs in Italy, and that it could be regarded as a potential mean to resurrect such extinct species.

Hybridogenesis in the P. kl. hispanicus complex. Pelophylax kl. hispanicus originates from hybridization between P. bergeri (B genome) and a new lineage P. n. t. 1 (E genome, as a reference to “Extinct”). It eliminates its P. bergeri germline and thus only contributes P. n. t. 1 gametes. This klepton requires P. bergeri to reproduce and their crosses yield klepton offspring only. These natural mechanisms allow to preserve the P. n. t. 1 germline in the wild.
The Apennine Peninsula (Italy and Swiss Ticino) is widely inhabited by pool frogs and their associated kleptons: P. lessonae with P. kl. esculentus north of the Apennine Mountains and its sister species P. bergeri with P. kl. hispanicus in the Apennines and in Sicily12 (Fig. 2). Yet, marsh frogs have never been reported over the Peninsula12,13, suggesting that either they did not contribute to its colonization, or they became extinct since. Their origin and nature have thus remained a mystery. By parsing nuclear sequence data from two independent intronic regions (abbreviated SAI-1 and CMI-2) together with microsatellite data, we characterized these ancestral marsh frog germlines in a comprehensive phylogeographic framework including most Pelophylax taxa known from the Western Palearctic. We made three striking discoveries.

(A) Nuclear phylogeny of Western Palearctic water frogs based on two nuclear loci combined (~1,600 bp, SAI-1 + CMI-2). Branch support are shown for internal nodes. (B) Distribution of Italian lineages from our sampling. (C) Distribution of water frog species, according to IUCN red list and our study (dash line: extinct range in Italy). Pelophylax c. f. bedriagae is a species complex involving multiple cryptic taxa summarized as P. bedriagae s. s., cf. 1 and cf. 2, based on SAI-1 and mtDNA variation18. For a mitochondrial phylogeny, see refs11,16. For information on the reference sequences, see Table S1 and references therein. Nearby Swiss Ticino localities were pooled for visibility. The map was built using ArcGIS 9.3 (http://www.esri.com/arcgis/about-arcgis).
First, we identified the marsh frog ancestor of P. kl. hispanicus as a new monophyletic lineage that we refer to as Pelophylax n. t. 1 (Fig. 2). This “ghost” species, widespread south of the Apennines, is supported by the SAI-1 phylogeny, which distinguishes all Western-Palearctic species (Fig. S1), and is represented by specific haplotypes for the shorter CMI-2 (Fig. S2). Some of the latter were also sampled north of the Apennines, as a probable result of past hybridization or incomplete lineage sorting among marsh frogs for this less polymorphic marker (Fig. S2; as also illustrated by unresolved P. ridibundus/P. cypriensis alleles). Potential signs of ancient admixture also arise from the microsatellite data: P. n. t. 1 is distinguished from the North-Italian marsh frog germline (see below), but few individuals have intermediate clustering (Fig. S3). Phylogenetic analyses show that P. n. t. 1 was most closely related to Eastern-Mediterranean water frogs, namely the Cyprian-endemic P. cypriensis, as well as Western Anatolian lineages of the P. c.f. bedriagae complex (Fig. 2), from which it diverged during the Pleistocene (1.5 Mya, 95% HPD 0.6-2.6; Fig. S4). This timing parallels the split between the pool frogs P. lessonae and P. bergeri north and south of the Apennines during the Calabrian (estimated at ~1.3 Mya14), a Pleistocene period characterized by extreme cold and desiccation. Nowadays, P. n. t. 1 appears to be extinct as such, and its genome only persists in the hybridogenetic hybrid P. kl. hispanicus (Fig. 1).
Second, we identified the marsh frog ancestor of kleptons P. kl. esculentus from N-Italy/S-Switzerland as the European marsh frog P. ridibundus, that currently inhabits most of the Western Palearctic12,13 (Fig. 2, Fig. S1, Fig. S2). The presence of different marsh frog lineages south and north of the Apennines is in clear accordance with well-established biogeographic paradigms, notably for amphibians, and especially pool frogs14.
Third, we unexpectedly found a new cryptic clade of pool frogs restricted to NW-Italy and S-Switzerland (Ticino; Fig. 2, Fig. S1, Fig. S2), diverged as early as the Late-Pliocene (3.0 Mya, 95% HPD 1.2-4.9) that we refer to as Pelophylax n. t. 2. It appears deeply admixed and panmictic with P. lessonae, as also suggested by the microsatellites (Fig. S3). This lineage might stem from long term isolation in the remote Alpine valleys (as in other herps15), before secondary contact and introgression by P. lessonae. Alternatively, it could be of hybrid origin resulting from leaky hybridogenesis between pool and marsh frogs, which would explain the intermediate phylogenetic position (Fig. 2), together with the absence of reciprocal mitochondrial divergence16.
Although our sequence markers discriminate all known species17,18, the current data is insufficient to accurately reconstruct the biogeography of marsh frogs of the Apennine Peninsula. Notably, it is unclear whether Pelophylax. n. t. 1 have speciated in the Italian Peninsula or colonized it from Central/Eastern Europe, where it would have become extinct as well. It is however unlikely that the divergences took place solely in the kleptons after hybridogenesis was initiated: P. n. t. 1 would otherwise cluster as a sister species of P. ridibundus found north of the Apennines, which we can reject from the phylogeny (Fig. 2). The contrasted genetic structures between P. bergeri and P. kl. hispanicus (and their marsh frog germlines) also argue for an independent evolution of P. n. t. 1 (Fig. S3). However, clonality may have boosted the evolutionary rate of this lineage and overestimated our dating estimates. In addition, the intronic markers used could yield biased phylogenetic signals due to specific features, like retrotransposons17. Given the complex evolutionary history of Western Palearctic water frogs, involving frequent events of hybridization between species19,20, robust nuclear phylogenies and admixture analyses will require high-throughput genomic data.
If it was ever present in Italy, why this taxon disappeared remains a mystery. Water frogs fossils from the middle Pleistocene to the Holocene are found throughout Italy21 but distinguishing between potential Pelophylax residents is challenging with current biometric methods22. Yet, many of these records were attributed to marsh frogs “P. ridibundus”23 and could thus belong to P. n. t. 1.
The persistence of the genetic legacy of an extinct taxon provides exciting insights. By carrying this lineage, P. kl. hispanicus can thus be considered as a “semi-living fossil” and deserves a strong conservation value. The same is true for P. bergeri on which P. kl. hispanicus relies for reproduction. Beyond its fascinating mechanism of genetic preservation, this system fuels the fantasy for de-extinction. Thanks to the hybridogenetic context, we envisage simple ways that would allow to resuscitate P. n. t. 1 without genetic engineering. Theoretically, crossings between P. kl. hispanicus individuals would produce pure P. n. t. 1 offspring. However, most of these crossings are sterile, otherwise marsh frogs would be naturally found in Central and South-Italy. Like in other water frog hybridogenetic systems, the P. n. t. 1 germline likely degenerated following many generations of clonal transmission (ref.24 and references therein). However, intense efforts of multiple controlled crossing experiments may allow to obtain few viable offspring, i.e. those where different deleterious mutations were fixed in their parental germlines. Although audacious, these might be rewarding given the flexibility of amphibian development; e.g. eggs can sometimes still develop under artificial haploid parthenogenesis (notably in Pelophylax25). An alternative approach would be to cross P. kl. hispanicus with its closest living relatives, e.g. the Anatolian lineage P. bedriagae c. f. 2. Subsequent backcrossing of the resulting F1 P. n. t. 1 × P. c. f. bedriagae hybrids with P. kl. hispanicus might allow to dilute the P. c. f. bedriagae genome while simultaneously purging deleterious mutations accumulated on the P. n. t. 1 germline. Selective breeding is a common de-extinction practice, e.g. envisaged to resurrect the aurochs26 and applied to recreate the quagga (an extinct subspecies of zebra), although the resulting individuals could not be genetically identical27. The implementation of such approaches would contribute to the ethical debate of reviving long extinct species. De-extinction is questionable when it pursues a technological achievement28. While this would not be the case here, the responsibility of mankind in restoring a biodiversity that “naturally” vanished prior to the sixth mass extinction still remains a topic of extreme controversy28. More generally, the de-extinction possibilities offered by scientific advances should not defuse the dramatic measures required to protect wildlife on the verge of extinction.
This study calls for the tracking of “ghost” vertebrate lineages that remained hidden and protected in other species of hybrid origins, through hybridogenesis or other mechanisms with potentially similar consequences (e.g. allopolyploidisation29,30). Unknown lineages underlie the genetic makeup of some polyploid plants31 and fishes32. Such taxa may be more frequent than imagined in vertebrates, especially in amphibians and fishes where these reproductive modes have been well-documented9,33.
Methods
DNA Sampling
We included 77 individuals sampled across Italy and S-Switzerland (Ticino), representing 23 localities (Table S1). Eleven samples from the different Western-Palearctic species of water frogs were also included for sequencing, to complement available data (see below). DNA was obtained from non-invasive buccal swabs or ethanol-preserved tissues, and was extracted with the Qiagen Blood & Tissue DNA extraction kit (Qiagen, Netherlands). Procedures were approved by the local and national ethics committees for animal experiments (karch Ticino) and performed in accordance with their guidelines and regulations. The mitochondrial lineage of all but three individuals was available from16.
Microsatellite analyses
Given the uncertainty to identify water frogs by morphology16, individuals from the study area were genotyped at microsatellite loci with diagnostic alleles to distinguish between pool (here P. bergeri /lessonae) from marsh frogs (P. ridibundus s. l.) frogs, as well as their kleptons (here P. kl. esculentus/hispanicus)10,16,34. This also confirmed the expected absence of marsh frogs (P. ridibundus s. l.).
To this purpose and to infer population structure, we analyzed nine polymorphic microsatellites in 72 individuals of pool and edible frogs from the study area (loci and methods16). Furthermore, we were able to parse the genotypes of 8 markers in 26 kleptons and reconstructed their marsh frog haplotypes, as previously done in Western Switzerland10. Both datasets (pool frogs/kleptons and phased marsh frog haplotypes) were analyzed by Principal Component Analyses (PCA) on individual genotypes, using the ade4 and adegenet packages in R. The differences between pool frogs and their associated kleptons stem from their marsh frog germlines. The genetic structure of kleptons should thus mirror the genetic structure of pool frogs if their marsh frog germlines only co-evolved in the kleptons after hybridogenesis was initiated, but not necessarily if the divergence predate hybridogenesis.
Sequencing of nuclear introns
We amplified and sequenced intronic portions of the nuclear genes Serum albumin (intron 1, abbreviated SAI-1) and Cellular myelocytomatosis (intron 2, abbreviated CMI-2) in respectively 45 (34 pool frogs and 11 kleptons) and 60 (49 pool frogs and 11 kleptons) individuals from the study area. These two markers were chosen as they have been widely used for phylogeography of water frogs and were consistently shown to discriminate between currently recognized taxa16,18,35. PCR conditions and primers are available in Table S2. We first attempted direct sequencing and then cloned heterozygous individuals. For SAI-1, we developed a set of primers specific to P. bergeri/lessonae alleles and a set of primers specific to the alleles of other taxa (Fig. S5). This allowed to independently amplify and sequence the two alleles of kleptons that harbored a P. bergeri or a P. lessonae allele (i.e. P. bergeri/P. n. t. 1 and P. lessonae/P. ridibundus). However, kleptons with a P. n. t. 2 allele, as well as heterozygous pool frogs had to be cloned. Cloning was performed with the TOPO-cloning kit (Invitrogen), and a minimum of eight clones per individual was sequenced.
Phylogenetic analyses
We complemented our new data with published sequences available for these two genes. For SAI-1, this included the haplotypes found across the ranges of all species16,17,18 (see Table S1 for details). For CMI-2, this included haplotypes from P. lessonae, P. bergeri and a P. ridibundus × P. bedriagae c. f. 1 hybrid from E-Turkey35. GenBank accession numbers of haplotypes analyzed in this study are provided in Table S3. Sequences were manually aligned in Seaview36. Phylogenetic analyses were performed on 59 SAI-1 and 31 CMI-2 haplotypes from 15 taxa (including P. n. t. 1 and P. n. t. 2), with the Asian P. nigromaculatus and the early-diverged European P. perezi as outgroups. Phylogenetic Bayesian inferences were performed with MrBayes37, with 10 million iterations and 2 chains. Evolutionary models were chosen according to JModelTest38 (SAI-1: GTR + G; CMI-2: HKY + G). We also computed haplotype networks using TCS39 to visualize haplotype variation, including indels. In addition to marker-specific trees, we built a phylogeny based on concatenated sequences representative of the variation of each species for the two markers.
We performed molecular dating by calibrating the phylogeny to the known split of P. cretensis at the end of the Messinian salinity crisis (~5.3-5.5 Mya)40,41. To this purpose, we estimated the mutation rates of our nuclear markers beforehand from the evolution of the mitochondrial genomes published for ten species of European and Asian water frogs, which provides a well-resolved phylogeny42. Analyses were conducted in BEAST using a Yule-process prior, an uncorrelated lognormal relaxed clock43 and different partitions for each sequence set. This calibration involved a normally distributed prior at 5.4 Mya (95% HDP 4.8-5.9) for the split of P. cretensis. Then, we applied the molecular clock on the nuclear dataset using the estimated mutations rates with normally-distributed priors covering their 95% HDP (SAI-1: 0.0056 (0.0027-0.0112); CMI-2: 0.0113 (0.0026-0.0286)). We ran two independent chains of 20 million iterations each (with the first 2 million excluded as burnin) and used the Tracer module to check for convergence and effective sample size of parameters. We built a time-calibrated phylogeny from the BEAST runs with the module TreeAnnotator and the R package phyloch44.
Change history
12 April 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Kumar, S. Extinction need not be forever. Nature. 492, 9 (2012).
Biton, R. et al. The rediscovered Hula painted frog is a living fossil. Nat. Comm. 4, 1959 (2013).
Lanza, R. et al. Cloning of an endangered species (Bos gaurus) using interspecies nuclear transfer. Cloning. 2, 79–84 (2000).
Loi, P. et al. Genetic rescue of an endangered mammal by cross-species nuclear transfer using post-mortem somatic cells. Nat. Biotechnol. 10, 962–964 (2001).
Clarke, S. Bizarre extinct frog brought back to life. ABCNews. http://www.abc.net.au/news/2013–03–16/bizarre-extinct-frog-brought-back-to-life/4575916 (2013).
Shapiro, B. Mammoth 2.0: will genome engineering resurrect extinct species. Genome Biol. 16, 228 (2016).
Reardon, S. Welcome to the CRISPR zoo. Nature. 531, 160–163 (2016).
Graf, J. D. & Müller, W. P. Experimental gynogenesis provides evidence of hybridogenetic reproduction in the Rana esculenta complex. Experientia. 35, 1574–1576 (1979).
Avise, J. C. Reproduction by the Semichaste: Gynogenesis, Hybridogenesis and kleptogenesis. In Clonality: the genetics, ecology, and evolution of sexual abstinence in vertebrate animals. (Oxford, 2008).
Leuenberger, J., Gander, A., Schmidt, B. R. & Perrin, N. Are invasive marsh frogs (Pelophylax ridibundus) replacing the native P. lessonae/P. esculentus hybridogenetic complex in Western Europe? Genetic evidence from a field study. Conserv. Genet. 15, 869–878 (2014).
Dufresnes, C., Denöel, M., di Santo, L. & Dubey, S. Triple invasion of Pelophylax water frogs in southern France, potentially inducing a new hybridogenetic complex. Sci. Rep. 7, 6506 (2017).
Nöllert, A. & Nöllert, C. (2003). Rana ridibunda. In Guide des amphibiens d’Europe 341–346 (Delachaux et Niestlé, 2003).
Kuzmin, S. et al. “Pelophylax ridibundus”. The IUCN Red List of Threatened Species. e.T58705A11825745 (2009).
Canestrelli, D. & Nascetti, G. Phylogeography of the pool frog Rana (Pelophylax) lessonae in the Italian Peninsula and Sicily: multiple refugia, glacial expansions and nuclear-mitochondrial discordance. J. Biogeogr. 35, 1923–1936 (2008).
Ghielmi, S., Menegon, M., Marsden, S. J., Laddaga, L. & Ursenbacher, S. A new vertebrate for Europe: The discovery of a range-restricted relict viper in the western Italian Alps. J. Zoolog. Syst. Evol. Res. 54, 161–173 (2016).
Dufresnes, C. et al. Cryptic invasion of Italian pool frogs (Pelophylax bergeri) across Western Europe unraveled by multilocus phylogeography. Biol. Invasions. 19, 1407–1429 (2017).
Plötner, J. et al. Evolution of serum albumin intron-1 is shaped by a 5′ truncated non-long terminal repeat retro- transposon in western Palearctic water frogs (Neobatrachia). Mol. Phylogenet. Evol. 53, 784–791 (2009).
Plötner, J. et al. Genetic data reveal that water frogs of Cyprus (genus Pelophylax) are an endemic species of Messinian origin. Zoosyst. Evol. 88, 261–283 (2012).
Herczeg, D., Vörös, J., Christiansen, D. G., Benovics, M. & Mikulicek, P. Taxonomic composition and ploidy level among European water frogs (Anura: Ranidae: Pelophylax) in eastern Hungary. J. Zoolog. Syst. Evol. Res. 55, 129–137 (2017).
Kolenda, K., Pietras-Lebioda, S., Hofman, M., Ogielska, M. & Pabijan, M. (2017). Preliminary genetic data suggest the occurrence of the Balkan water frog, Pelophylax kurtmuelleri, in southwestern Poland. Amphib-reptil. 38, 187–196 (2017).
Holman, J. A. In Pleistocene amphibians and reptiles in Britain and Europe 63-66 (Oxford, 1998).
Blain, H. A., Lozano-Fernandez, I. & Böhme, G. Variation in the ilium of central European water frogs Pelophylax (Amphibia, Ranidae) and its implications for species-level identification of fragmentary anuran fossils. Zool. Stud. 54, 5 (2015).
Böhme, M. & Ilg, A. fosFARbase, www.wahre-staerke.com [accessed July 2017] (2003).
Vorburger, C., Schmeller, D. S., Hotz, H., Guex, G. D. & Reyer, H. U. Masked damage: mutational load in hemiclonal water frogs. in Lost sex: the evolutionary biology of parthenogenesis 433–446 (Springer, 2009).
Mittwoch, U. Parthenogenesis. J. Med. Genet. 15, 165–181 (1972).
Sinding, M. S. & Gilbert, M. T. P. The draft genome of extinct European aurochs and its implications for de-extinction. Open Quaternary. 2, 7 (2016).
Harley, E. H., Knight, M. H., Lardner, C., Wooding, B. & Gregor, M. The Quagga project: progress over 20 years of selective breeding. S. Afr. J. Wildl. Res. 39, 155–163 (2009).
Sandler, R. The ethics of reviving long extinct species. Conserv. Biol. 28, 354–360 (2013).
Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6, 836–846 (2005).
Janko, K., Bohen, J., Lamatch, D. & Flajshans, M. The gynogenetic reproduction of diploid and triploid hybrid spined loaches (Cobitis: Teleostei), and their ability to establish successful clonal lineages – on the evolution of polyploidy in asexual vertebrates. Genetica. 131, 185–194 (2007).
Luo, X. et al. Chasing ghosts: allopolyploid origin of Oxyria sinensis (Polygonaceae) from its only diploid congener and an unknown ancestor. Mol. Ecol. 26, 3037–3049 (2017).
Crespo-López, M. E., Pala, I., Duarte, T. L., Dowling, T. E. & Coelho, M. M. Genetic structure of the diploid-polyploid fish Squalius alburnoides in southern Iberian basins Tejo and Guadiana, based on microsatellites. J. Fish. Biol. 71, 423–436 (2007).
Schmid, M., Evans, B. J. & Bogart, J. P. Polyploidy in Amphibia. Cytogenet. Genome Res. 145, 315–330 (2015).
Hotz, H. et al. Microsatellites: a tool for evolutionary genetic studies of western Palearctic water frogs. Mitt. Mus. Naturkunde Berl. Zoolog. Reihe 77, 43–50 (2001).
Komaki, S. et al. Robust molecular phylogeny and palaeodistirbution modelling resolve a complex evolutionary history: glacial cycling drove recurrent mtDNA introgression among Pelophylax frogs in East Asia. J. Biogeogr. 42, 2159–2171 (2015).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 (2010).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19, 1562–1574 (2003).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 9, 772 (2012).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Beerli, P., Hotz, H. & Uzzel, T. Geological dated sea barriers calibrate a protein clock for Aegean water frogs. Evolution 50, 1676–1687 (1996).
Kriigsman, W., Hilgen, F. J., Raffi, I., Sierro, F. J. & Wilson, D. S. Chronology, causes and progression of the Messinian salinity crisis. Nature. 400, 652–655 (2000).
Hofman, S., Pabijan, M., Osikowski, A., Litvinchuk, S. N. & Szymura, J. M. Phylogenetic relationships among four new complete mitogenome sequences of Pelophylax (Amphibia: Anura) from the Balkans and Cyprus. MDN. 27, 3434–3437 (2016).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 4 (2007).
Heibl, C. PHYLOCH: R language tree plotting tools and interfaces to diverse phylogenetic software packages. http://www.christophheibl.de/Rpackages.html (2013).
Acknowledgements
We thank Glib Mazepa and Daniele Canestrelli for providing samples. This study was funded by the Federal Office for the Environment (FOEN/OFEV/BAFU; Francis Cordillot), the karch Ticino (Tiziano Maddalena), Hintermann & Weber SA, Fondazione Bolle di Magadino and Museo cantonale di storia naturale, Lugano.
Author information
Authors and Affiliations
Contributions
S.D. and C.D. designed the study, performed labwork and drafted the manuscript. S.D. performed fieldwork. C.D. conducted the analyses.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dubey, S., Dufresnes, C. An extinct vertebrate preserved by its living hybridogenetic descendant. Sci Rep 7, 12768 (2017). https://doi.org/10.1038/s41598-017-12942-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12942-y
This article is cited by
-
Far from home: tracing the origin of non-native water frogs (genus Pelophylax) in Malta by molecular markers
Biological Invasions (2024)
-
Hybrid swarm as a result of hybridization between two alien and two native water frog species (genus Pelophylax) in Central Croatia: Crna Mlaka fishpond acting as a species melting pot?
Biological Invasions (2022)
-
Population genomics of an exceptional hybridogenetic system of Pelophylax water frogs
BMC Evolutionary Biology (2019)
-
Perspectives on the clonal persistence of presumed ‘ghost’ genomes in unisexual or allopolyploid taxa arising via hybridization
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
