
Abstract
Seven silicon(IV) phthalocyanine carboxylate esters (SiPcs, 1–7) with non-, partially- and per-fluorinated aliphatic (linear or branched at the alpha-carbon) and aromatic ester groups have been synthesized, their solid-state structures determined and their optoelectronic properties characterized. The SiPcs exhibit quasi-reversible oxidation waves (vs. Fc+/Fc) at 0.58–0.75 V and reduction waves at −0.97 to −1.16 V centered on the phthalocyanine ring with a narrow redox gap range of 1.70–1.75 V. Strong absorbance in the near-infrared (NIR) region is observed for 1–7 with the lowest-energy absorption maximum (Q band) varying little as a function of ester between 682 and 691 nm. SiPcs 1–7 fluorescence in the near-infrared with emission maxima at 691–700 nm. The photoluminescence quantum yields range from 40 to 52%. As a function of esterification, the SiPcs 1–7 exhibit moderate-to-good solubility in chlorinated solvents, such as 1,2-dichlorobenzene and chloroform.
Similar content being viewed by others


Introduction
Phthalocyanines (Pcs) are one of the most extensively studied classes of organic functional macrocycles, having four nitrogen-bridged isoindole units1,2. Pcs are known for their robust thermal and chemical properties, and their diverse optoelectronic and magnetic properties, especially their strong absorption in the near Infrared (λabs ~ 700 nm). These attractive optoelectronic properties have catalysed the use of Pcs as dyes and pigments3, and as components in electrochromic4 and optical limiting devices5,6, organic thin-film transistors7,8, organic light emitting diodes (OLEDs)9, multistage-redox-dependent fluorophores10, organic (OSC)11,12 and dye-sensitized solar cells (DSSCs)13,14, and also in NIR fluorescence imaging12,15,16.
Many of the solid-state properties of Pcs are highly dependent on the extent of intermolecular π-π stacking interactions, especially in aggregates in polar protic solvents such as water or ethanol17. Pcs are normally characterised by an intense absorption of the Q-bands in the near infrared (NIR) region of ~700 nm18. Dimerization or aggregation between neighbouring Pc molecules generally leads to fluorescence quenching and a broadened, red-shifted principal Q-band due to electronic/exciton coupling interactions19,20 that can normally be observed even in very dilute solutions. Aggregation is an unfavorable property of Pcs since it can not only cause difficulties in purification and characterization21 but also result in the quenching of luminescence due to enhanced non-radiative excited state decay22, and therefore lowers the photoluminescence quantum yields, ΦPL. In the context of achieving non-aggregated Pc derivatives, several approaches have been adopted. These include introduction of bulky peripheral substituents such as tert-butyl groups14,23,24 and rigid spirocyclic fused rings25, and the introduction of metal ions such as Sn(IV) and Ln(III) to break the planarity of the Pc-containing compounds26,27 such that the metal ion sits outside the plane of the Pc macrocycle. Another viable approach is to introduce elements such as Ru(II), Si(IV) or Ge(IV), that possess more than four valencies. Many Pc derivatives have been explored including main group metals and 1st–3rd row transition metals such as Al(III), Cu(II), Ni(II), Co(II), Zn(II), Ga (III), In(III), Sn(IV), Ge(IV), Ln(III), Pt(II) exhibiting square-planar, square-pyramidal, or octahedral coordination geometries18. Pcs containing Si(IV) are an attractive subclass of Pcs because of the elemental abundance and very low toxicity levels of silicon coupled with their low band gap (~1.7 eV). The SiPcs with axial oxygen-terminated ligands (alkoxide, siloxide, aryloxide, and carboxylate) have been known since the 1960s28,29 and they have been applied as photosensitizing agents in photodynamic therapy30. Recently, SiPcs with siloxide and aryloxide ligands have found use as chromophores and fluorophores in DSSCs and OLEDs31, the latter with external quantum efficiencies of up to 4.5%13. In contrast, to the best of our knowledge, SiPcs with carboxylate ligands have rarely been used in organic electronics. Considering the strong nature of Si-O bond, the wide range of commercially-available carboxylic acids and their capacity to confer increased solubility, silicon phthalocyanine carboxylates are an attractive target for the design of solution-processable electron-accepting materials for organic electronics.
In a recent study, we investigated the optoelectronic properties of two SiPcs bearing axial alkyl/aryl-carboxylate ligands (R1 and R2, Fig. 1) and explored their use in OLED and OSC devices32. Herein, we report a systematic study of the synthesis and optoelectronic characterization of a series of axially substituted SiPc carboxylates, 1–7, bearing perfluorinated (1 and 2) and partially fluorinated (4) carboxylates, alkyl carboxylates (3 and 5) and aryl carboxylates (6 and 7) (Fig. 1). The new SiPcs were comprehensively characterized by 1H and 19F NMR spectroscopy, NSI+ high-resolution or MALDI mass spectrometry, melting point determination, elemental analysis and single crystal XRD. The effect of different axial substituents on the optoelectronic properties of these complexes is discussed, supported by Density Functional Theory (DFT) calculations, with the results compared to our previously reported reference compounds R1 and R2.

Synthesis of SiPc complexes 1–7 and benchmark complexes R1 and R2. Proton labelling scheme (α and β protons) for the macrocycle ring is also shown.
SiPcs 1–7 were synthesized by nucleophilic substitution reactions of commercially available silicon phthalocyanine dichloride, SiPcCl2 with an excess of the corresponding carboxylic acids, either in DMF (SiPc 3) or in diglyme (SiPcs 1, 2, 4–7) at 140–165 °C (Fig. 1) in poor to good yields (3–64%) based on the carboxylic acids. The SiPcs were purified by column chromatography on silica. The product typically elutes as the first blue band from the column and it is usually well separated from the other fractions. Compounds 1–7 were obtained as air and moisture stable blue solids. It was found that the choice of solvent plays a crucial role in the synthesis of the SiPcs. For instance, 1 and 6 can be prepared from diglyme, but not from DMF. The aliphatic carboxylates were either linear at the alpha-carbon (1–2) or branched (3–5). The aromatic carboxylates possessed solubilizing groups (6 and 7) that were either electron-withdrawing (trilfuoromethyl in 6) or electron-donating (octyloxy in 7).
All the compound characterization data are consistent with the proposed SiPc structures and further confirmed by single crystal data. Compounds 2–7 show melting points >300 °C while 1, possessing a long-chain aliphatic carboxylate C19H39CO2 − ligands shows a significantly lower melting point of 168–170 °C. Fluorinated SiPcs 1, 4, and 6 exhibit low solubility of <5 mg mL−1 in chlorinated solvents, such as 1,2-dichlorobenzene and chloroform while the other SiPcs are soluble in those solvents (>10–30 mg mL−1) but are insoluble in acetonitrile and alkanes. The solubility of SiPcs 2, 3, 5 and 7 in 1,2-dichlorobenzene are comparable to the same reported for reference complexes R1 (>10 mg mL−1) and R2 (>35 mg mL−1) in 1,2-dichlorobenzene32.
The axially-substituted SiPcs have characteristic 1H NMR spectra (Fig. 2). The 1H resonances of the axial substituents close to the phthalocyanine ring are strongly shifted upfield as they experience a large diamagnetic ring-current shielding effect induced by the macrocyclic ring. This effect is more pronounced in 3–5 where some of the aliphatic protons appear ≤ −1 ppm. The phthalocyanine ring 1H resonances appear as two downfield-shifted unresolved multiplets integrating each as 8 H at around 9.8 and 8.5 ppm, for the α and β protons, respectively26. The two axially substituted carboxylate ligands are symmetric on the NMR timescale and exhibit a single set of NMR resonances. This observation in conjunction with the presence of the two sets of phthalocyanine ring 1H resonances suggest a local D 4h symmetry in 1–7 in solution state, irrespective of ester ligand. Electrospray ionization mass spectrometry showed the expected isotope distribution pattern for both the [M + H]+ and [M + Na]+ ions for 1–7.

Stacked 1H NMR spectra of 1–7 in CDCl3 at 400 MHz at r.t. (the 1H NMR spectrum of 4 was recorded in CD2Cl2 at 400 MHz at r.t.).
Crystal structures
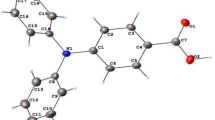
The crystal structures of 1–7 were determined. Details of structure refinements and some crystallographic parameters are collected in Supplementary Table S1 in the ESI. Selected bond distances of SiPcs 1–7, R1 and R2 are gathered in Supplementary Table S2. The structure of 6 shows two independent molecules in the asymmetric unit, as well as a co-crystallized molecule of acetonitrile. The flexible polyfluorinated chains of the carboxylate ligand in 2 result in a lower quality, but unambiguous, determination of its structure.
In all seven structures, the hypervalent silicon(IV) adopts the expected distorted octahedral SiN4O2 coordination environment (Fig. 3)32. Except in the case of 6, the SiPc has a crystallographic inversion center at the silicon atom; therefore, the SiN4 fragment is planar, irrespective of any distortions within the phthalocyanine ring itself, the SiO2 fragment is linear, and only two of the Si–N and one of the Si–O bonds are unique. In 6, the same planar SiN4 fragment (root-mean-squared deviation of the SiN4 fragment from planarity, 0.0019 and 0.0003 Å, for Si1 and Si51, respectively), and linear SiO2 fragment [O–Si–O angles, 179.59(9) and 179.40(8)°, for Si1 and Si51, respectively] are seen, despite the lack of a center of symmetry at Si. The Si–N bonds are significantly longer [1.8897(18)–1.9203(11) Å] than the Si–O bonds [1.7358(9)–1.7793(15) Å]. The Si–N bond lengths decrease while the Si–O bond lengths increase when the carboxylate ester becomes more electron-withdrawing from a non-fluorinated to a (per-)fluorinated one (Supplementary Table S2). All Si–N and Si–O bond distances fell within the range of distances for six-coordinate SiPc compounds with axial O-donor ligands, found in a search of the Cambridge Structural Database (CSD version 5.37)33,34. The Si–N distances were seen to lie below the middle of the range of distances (1.89–1.95 Å), whereas the Si–O distances were at the upper end of the range (1.65–1.78 Å).

X-ray structures of 1–7, with ellipsoids drawn at the 50% probability level. Hydrogen atoms, solvent molecules, minor disorder components, and additional independent molecules are omitted for clarity.
The complexes show a limited selection of both π···π and C–H···π interactions; however, these do not involve the aryl groups of the carboxylate ligands in 6 and 7, which is likely due to the surrounding steric bulk. In contrast, the phenyl rings of the phthalocyanines do participate in π–stacking, with centroid···centroid distances ranging from 3.5361(11) to 3.7795(19) Å. The exceptions to this occur in 1 and 5, which show no centroid···centroid distances less than 3.8 Å, although there are phenyl rings positioned appropriately at distances slightly greater than this that show patterns of interactions in line with the other structures. This π–π stacking occurs through either the two trans-benzenes to give an infinite single chain (3 and 4, Fig. 4) or through the two pairs of cis-benzenes to give an infinite double-chain (1, 2, 5, 6, and 7, Fig. 5). As well as these π···π interactions, compounds 5 and 7 also show weak C–H···π interactions between aliphatic C–H groups in the axial ligands and aromatic rings in either the phthalocyanine, or the axial ligand, at distances of 3.00 and 2.71 Å, respectively. The interaction in 5 is at the conventional van der Waals limit, but C–H···π interactions have been suggested to be effective at distances beyond this value35.

Views of sections of the structure of 3 showing (a) the single-chain mode of π···π interactions and (b) the resulting herringbone packing motif. Hydrogen atoms are omitted for clarity.

Views of sections of the structure of 6 showing (a) the double-chain mode of π···π interactions and (b) the herringbone packing motif that results. Hydrogens Hydrogen atoms are omitted for clarity.
All the complexes, apart from 5, exhibit weak intermolecular C–H···O hydrogen bonds to the carbonyl oxygen of the carboxylate ligand, which acts as acceptor for two C–H···O interactions simultaneously. These predominantly involve aromatic phthalocyanine C–H groups, but also aliphatic C–H groups in some cases. The H···O separations vary between 2.37 and 2.55 Å, with corresponding C···O separations of 3.265(3) to 3.445(13) Å. For 5, the steric bulk of the adamantyl group may restrict some close-packing interactions with the adjacent carboxylates. The SiPcs with fluorinated carboxylate ligands (1, 2, 4, and 6) additionally exhibit weak intermolecular C–H···F interactions. These show a similar distance spread to the C–H···O hydrogen bonds, with H···F separations varying between 2.36 and 2.62 Å, and C···F separations between 3.169(4) to 3445(3) Å. Such interactions would generally be considered too weak to make a significant contribution to the packing, except that they are seen to occur in cooperative pairs; working in conjunction with other C–H···F interactions. The exception to this pattern is in 4, where no other interactions are seen to be operating cooperatively with the C–H···F interactions, suggesting that in this case the close H···F contact may just be an artifact of packing.
The presence of the axial ligands can lead to disruption of intermolecular interaction in the solid state. The linear alkyl chains of the carboxylates in 1 and 2 are arranged parallel to the plane of the phthalocyanine ring (Fig. 6), whereas all the other carboxylate ligands are arranged nearly orthogonal to it, even in the case of 7, containing an aryl octyl ether (Fig. 3). This arrangement of axial ligands is mirrored in other SiPcs, where the presence of aryl carboxylate ligands show positioning orthogonal to the SiPc32,36,37, while alkyl carboxylates show a parallel position26,32,37,38,39,40. Potentially arising from these differing chain orientations, and the resulting presence or absence of steric bulk across the face of the SiPc, the phthalocyanine rings pack with the ring-planes arranged in a parallel fashion in 1, 2 and 5, whereas in the rest of the SiPcs they pack in a herringbone pattern, with SiPc planes approximately orthogonal. This pattern of arrangements is not mirrored as closely in other known structures; although those with aryl carboxylates do show packing with orthogonal SiPcs37, those with alkyl carboxylates show a mixture of packing motifs, most showing a parallel arrangement of SiPcs37,39, but some showing herringbone packing37,40, or other motifs with orthogonally arranged SiPcs36,39. The intermolecular separation was estimated using the shortest Si···Si distance in each structure. Surprisingly, the longest separation is found to be in 4 (Table 1), which has the highest solubility in the series, and the shortest in 7.

View of a section of the structure of 1 showing the all-parallel packing motif. Hydrogen atoms are omitted for clarity.
Electrochemistry
The cyclic voltammograms of compounds 1 and 3–7 were obtained in dichloromethane at room temperature and are shown in Fig. 7 and the electrochemical data collated in Table 1. Due to poor yield, the electrochemical measurement of 2 was not possible. At positive potential, the SiPcs exhibit mono-electronic quasi-reversible oxidations in the range 0.58–0.75 V vs. Fc/Fc+. DFT calculations indicate that the highest occupied molecular orbitals (HOMOs) are distributed on the isoindole moieties of the macrocycles (Fig. 8). Based on the DFT calculations, and in line with the literature data, the first oxidation processes of 1 and 3–7 are assigned to the removal of one electron from the isoindole moiety31. The lower energies calculated for the HOMOs of 1 (EHOMO = −5.40 eV), 3 (EHOMO = −5.27 eV) and 4 (EHOMO = −5.30 eV) compared to that of R1 (EHOMO = −5.12 eV) are in good agreement with their more positive anodic potentials (Table 1). The slightly shallower HOMO of 5 (EHOMO = −5.09 eV) bearing the adamantyl moiety compared to that of R1 (EHOMO = −5.12 eV) is in line with the 10 mV cathodic shift of its oxidation potential compared to that of R1. The energy calculated for the HOMO (EHOMO = −5.35 eV) of bis(trifluoromethyl)phenyl carboxylate-substituted compound 6 is significantly lower than that of R2 (EHOMO = −5.07 eV), aligning with the 100 mV anodic shift of the oxidation potential of 6 compared to that of R2. However, the trend observed in the lower HOMO energy calculated for 7 (EHOMO = −5.25 eV) compared to that of R2 (EHOMO = −5.07 eV) is not in line with the observed 200 mV cathodic shift in its oxidation potential compared to that of R2. So, in general, when electron-withdrawing fluorinated groups are added to the carboxylate ligand, the redox potentials are shifted positively, the oxidation becomes harder; e.g., compare (1 and 4) vs. (3 and 5) or 6 vs. 7 (Table 2). A similar anodic shift of 30 mV is also observed on substitution of one methyl group in the tBu group in 3 with a -CF3 group in 4. Compared to R1, all the fluorinated SiPcs, 1, 4, and 6 exhibit anodically shifted oxidation potentials as a function of incorporation of electron-withdrawing -CF3 groups (Table 1). The similar argument is also valid for the observed 100 mV anodic shift in the oxidation potential of SiPC 6 compared to that of R2 (Table 1). The most positive oxidation potential in the series is observed for 1, which contains the perfluoroheptanoate axial ligands.

Cyclic voltammograms of 1 and 3–7 in degassed dichloromethane, recorded at a scan rate of 100 mV/s.

Calculated frontier MO energies of 1–7, obtained from DFT [(B3LYP/SBKJC-VDZ for Si(IV)) and (6–31 G** for C,H,N,O,(F)] with CPCM(MeCN) and 0.5 eV threshold of degeneracy (orbitals are isocontoured at 0.03). Kohn-Sham MOs of 1–7 are also shown.
Upon scanning to negative potentials, the SiPcs exhibit one or two quasi-reversible monoelectronic reductions, the first at −0.97 to −1.16 V and the second at −1.47 to −1.63 V, which are typical of Pcs41. For all the SiPcs, DFT calculations point to a lowest unoccupied molecular orbital (LUMO) that has isoindole character coupled with the contribution from the bridging N-atoms. Therefore, and in line with the literature data, the first reduction can be assigned to reduction of the macrocycle itself. Similar to the trend observed in the oxidation of (1 and 4) vs. (3 and 5), the presence of the electron-withdrawing fluorinated groups anodically shifts the first reduction potential of (1 and 4) by 60–190 mV compared to those of (3 and 5) (Table 2). This trend is in good agreement with the more stabilized computed LUMOs of 1 and 4 (ELUMO = −3.33 eV for 1 and −3.21 eV for 4) compared to those of 3 and 5 (ELUMO = −3.17 eV for 3 and −2.97 eV for 5). The computed and experimentally determined LUMO energies of the aryl-substituted SiPcs 6 and 7 containing either electron-withdrawing -CF3 or electron-donating octyloxy groups, respectively, also follow the expected trend. The first reduction potential of 6 is 80 mV anodically shifted compared to that of 7; the LUMO of 6 (ELUMO = −3.27 eV for 6) is likewise more stabilised compared to the LUMO of 7 (ELUMO = −3.15 eV for 7) (Table 2). DFT calculations demonstrate that the LUMO + 1 is centred on the isoindole moiety in all the compounds. The observed quasi-reversible reductions of 3–7 in the range of −1.47 to −1.63 V are assigned to the reduction of the macrocyclic ring system (Fig. 7). Except for 1, the first reduction potentials of 3–7 are cathodically shifted by 40–170 mV compared to axially alkoxy-disubstituted SiPcs (Ered = −990 mV)42.
The redox gap, ΔEredox, spans only 60 mV across the series of SiPcs and remains nearly constant at 1.69–1.75 V since axial substitution modulates these potentials in the same direction (Table 1). A similar behavior of the redox potentials and the near-constancy of the redox gap have been noted previously for other series of Pc complexes26.
Photophysics
The UV-vis absorption spectra of all the SiPcs were recorded in dichloromethane and values are collated in Table 2. An overlay of the experimental UV-vis absorption spectra of 1 and 7 with their predicted transitions calculated by time-dependent DFT (TD-DFT) at room temperature in dichloromethane is shown in Fig. 9 and similar overlays of the experimental and TD-DFT predicted UV-vis profiles for SiPcs 1–7 are shown in Supplementary Figure S1. The absorption spectra exhibit profiles that are typical of the phthalocyanine chromophore; a B (or Soret) band at 355–360 nm, an intense and sharp near-infrared Q band at 682–691 nm, together with two vibronic bands at 613–621 nm and 651–659 nm. The TD-DFT-assigned characterisations of these bands are tabulated in Table 3. The Q-bands have molar absorption extinction coefficients, ε, of (26–29) × 104 M−1 cm−1 (Table 3). For all the SiPcs the absorptivity of the Q band strictly follows the Beer-Lambert law as a function of concentration, thereby indicating that the compounds are essentially free from aggregation in dichloromethane43. Furthermore, the absence of either a red-shifting of the Q band at ca. 750 nm and a blue-shifting Q band at ca. 630 nm, excludes the formation of J and H aggregates, respectively. The retention of monomeric behaviour even at 10–4 M demonstrates that the axial ligands are very effective in sterically isolating the chromophoric Pc rings, by preventing intermolecular π stacking interactions between the macrocycles in solution38. The absorption spectra have a window between 420–560 nm, which is typical of phthalocyanines. The absorption cut-off (optical band gap) of the SiPcs is at 720–730 nm (1.72–1.70 eV). The TD-DFT calculations of 1–7 predict that the bands found between 257–350 nm are principally π → π* locally excited (LE) transitions within the conjugated macrocycle, whereas the π → π* LE bands of the aromatic carboxylates in 6 and 7 are superimposed with those of the phthalocyanine. The B bands at ca. 355 nm for 1 and 2 are due to the π → π* based transitions within the isoindole moieties of the macrocycle, whereas, these same bands in 3–7 also contain small contributions from n → π* transitions from the bridging N-atoms connecting the four isoindole moieties to the π* orbital of the isoindole (Table 3). For 6, this band contains an additional π → π* based ligand-to-ligand charge transfer (LLCT) transition emanating from the 3,5-bis(trifluoromethyl)phenylcarboxylate to the macrocycle. The Q-band for all the SiPcs is predicted to originate from π → π* LE transitions among the isoindole moieties of the macrocycle. Fluorination of the carboxylate ligand from L3 to L4 leads to a modest red-shift of 3 nm in the Q band of 4 compared to that of 3; similar behavior exists between 6 and 7. The position of the Q band, which relates to the HOMO-LUMO gap, remains nearly constant at 682–691 nm across the series. Therefore, the HOMO-LUMO energy gap is invariant to the original of the axial substitution of the carboxyaltes, an observation corroborated by their electrochemistry. However, the lowest energy absorption maxima of these complexes are red-shifted by ca. 10–20 nm compared to those of alkoxy- and siloxy-substituted SiPcs43,44,45.

Typical overlays of experimental UV-vis absorption spectra (curved lines) of 1 and 7 with their predicted transitions (vertical bars) calculated by TD-DFT at room temperature in dichloromethane.
The SiPcs 1–7 were also characterized by photoluminescence spectroscopy in dilute and degassed dichloromethane solutions at ambient temperature. Relevant photophysical data are shown in Table 4 and the photoluminescence spectra of SiPcs 1 and 7 are shown in Fig. 10, whereas the photoluminescence spectra of all the SiPcs in dichloromethane solutions are shown in Supplementary Figure S2. All the SiPcs emit strongly in the near-infrared (NIR) region with λ em ranging narrowly from 688–700 nm. Two lower intensity bands are observed at between 710–790 nm. The photoluminescence spectrum of each of the SiPcs is a mirror image of the absorption spectrum. The λ em values are typical of carboxy-substituted SiPc compounds and the emission is modestly red-shifted by ca. 8–30 nm compared to alkoxy- and silyloxy-disubstituted SiPcs. All the SiPcs exhibit short mono-exponential photoluminescence decay kinetics (τ) in the range 6.7–7.3 ns and high photoluminescence quantum yields (ΦPL = 40–52%) in dichloromethane solution. The calculated radiative lifetime, τrad = τ × ΦPL −1, ranges from 13.8–18 ns and the radiative, k r, and non-radiative, k nr, decay constants, [k r = ΦPL/τε and k nr = (k r/ΦPL) − k r] are in the range of (55.5–72.2) × 106 and (66.7–83.3) × 106 s−1 for all the compounds. The mirror image behavior between absorption and emission spectra, small Stokes shift (λ abs − λ em, of 6–10 nm), short excited-state decay and radiative lifetimes are all consistent with a spin-allowed fluorescence process from the S1 state. Unlike the strong emission quenching observed for amino-alkoxy-substituted SiPcs, produced by intramolecular photoinduced electron transfer (PET)44,46, the presence of the carboxylate ligands is benign to the radiative decay of the excited state. In general, for the SiPcs with electron-donating groups, higher fluorescence intensity was observed compared to those with electron-withdrawing groups, Table 4)47. The higher ΦPL values of 3 vs. 4 and 6 vs. 7 are the result of a decrease in k nr of the respective non-fluorinated SiPcs, 3 and 6 vs. the fluorinated SiPcs, 4 and 7.

Typical normalized emission spectra of complexes 1 and 7 in degassed dichloromethane solutions at room temperature (λexc = 360 nm).
Thin-film photoluminescence studies of the SiPcs 1–7 were performed in a PMMA host and the corresponding PL emission maxima, photoluminescence quantum yields and excited-state lifetime values are compiled in Table 5. The concentration of the SiPcs in the PMMA matrix were maintained at an optical density below 0.1 to avoid any concentration quenching effects. The PL emission maxima of SiPcs 1–7 in doped PMMA overlap nicely with their respective solution PL maxima, suggesting that in both thin film and solution states aggregation induced red-shifting of the PL maximum is not evident. The film PL quantum yield values of the SiPcs remain very similar to those observed in solution, except for SiPc 6 which shows higher PLQY in the film. The higher PLQY of SiPc 6 seems to be associated with a higher rate of radiative decay constant (k r ) of 81.8 × 106 s−1 compared to the k r values of the other SiPcs (66.1–80.0 × 106 s−1).
Conclusions
In summary, a series of SiPcs bearing axially substituted carboxylate ligands have been synthesized and structurally and optoelectronically characterized. Their structures were unambiguously characterized by 1H NMR spectroscopy, ESI-MS, elemental analyses and single crystal XRD analyses. The presence of electron-withdrawing or donating groups on the carboxylate ligands does not exert any significant influence on the optoelectronic properties of the SiPcs as the electron-donating/withdrawing groups exert similar effects upon the phthalocyanine ring based HOMO and LUMO. The oxidation and the reduction processes of the complexes were both found to be quasi-reversible. All the SiPcs exhibit NIR fluorescence with high photoluminescence quantum yields and no sign of aggregation in dilute solution. This study demonstrates the wide selection of carboxylate ligands available to render the SiPc compounds solution-processable and thus amenable for use in OLED and OPV devices.
Experimental Section
General Synthetic Procedure
Commercial chemicals were used as supplied. Chromatography was performed on columns with an i.d. of 25–30 mm on silica gel. The reaction was performed under nitrogen using standard Schlenk techniques. Purification and handling of the products were carried out under air. The progress of reactions and the elution of products were followed on TLC plates (silica gel on aluminum sheets, 250 µm with indicator F-254). Compounds were visualized under UV light. 1H and 19F NMR spectra were recorded with Bruker AVANCE 300 (300 MHz for 1H; 282 MHz for 19F) and Bruker AVANCE II 400 spectrometers (400 MHz for 1H; 377 MHz for 19F). The following abbreviations have been used for multiplicity assignments: “s” for singlet, “d” for doublet, “t” for triplet, “m” for multiplet, and “br” for broad. 1H NMR spectra were referenced to the solvent peak. Melting points (m.p.) were recorded in open-end capillaries on an Electrothermal melting point apparatus IA9200 and are uncorrected. The heating rate was 0.5 or 1.0 °C/min. High-resolution nanospray ionization (HR NSI+) and MALDI+ mass spectra, were recorded at the EPSRC National Mass Spectrometry Service Centre, Swansea University. Elemental analyses were performed by Mr. Stephen Boyer, London Metropolitan University.
Silicon phthalocyanine dichloride (SiPcCl2; dye content ~85%) and carboxylic acid (excess) were stirred in 3–4 mL of dry and degassed DMF or diglyme (degassed by bubbling with nitrogen for 5 min) at 140–165 °C overnight to give dark green-blue solution or suspension. The volume of the solvent was chosen to be as small as possible, while still allowing the efficient mixing of the reagents. The temperature of the reaction was as high as possible to help dissolve the starting materials; however, the temperature was lowered for the fluorinated acids to prevent side-reactions involving the electron-deficient C–F bonds. The molar ratio of SiPcCl2 to carboxylic acid ranged from 3.9–7.4. The reaction mixture was cooled to room temperature. Water (50 mL or more) was added to precipitate the product (blue solid). For 2, addition of water precipitated a dark oil; the supernatant solution was decanted and the oil was crystallized on mixing with methanol/water (1/1, v/v). The solid product was filtered. The product was washed on the filter with water for 6; with water and hexane (5–10 mL) for 3; or with water and methanol/water (1/1, v/v) for 1, 2, 4, 5 and 7. The washing was performed to remove the reaction solvents and the excess of the acid. The product was purified by column chromatography on 10–22 g of silica. The elution was performed with dichloromethane (except for 4) or with 0–0.3% methanol in dichloromethane (for 4) to give green-blue eluate of the product as the first band. The combined product fractions were evaporated to dryness. The product was dried under vacuum.
The products are air- and moisture stable blue solids that are soluble in chlorinated solvents (chloroform, dichloromethane, 1,2-dichlorobenzene) and are insoluble in acetonitrile. The exact shade, brightness, and depth of the blue color of the product mainly depend on whether the product is crystalline or not. The crystals of the product may look red in reflection. The products were stored in the dark. Further details are provided below.
-
1.
SiPcCl2 (100 mg, 0.16 mmol, 1 equiv.) and perfluoroheptanoic acid (286 mg, 0.79 mmol, 4.94 equiv.) in diglyme at 140 °C gave 87 mg (0.069 mmol, 43%) of 1. M.p.: 314 °C (dec.). 1H NMR (300 MHz, CDCl3): δ = 9.72 (m, 8 H), 8.45 (m, 8 H) ppm. 19F NMR (377 MHz, CDCl3): δ = −81.01 (m, 6 F), −121.22 (t, J = 11 Hz, 4 F), −123.77 (m, 8 H), −125.55 (m, 4 H), −126.80 (m, 4 H) ppm. HR NSI+ MS: m/z 1267.0721 ({M + H}+, 100%); theor. C46H17F26N8O4Si+ m/z 1267.0721. Anal. Calcd for C46H16F26N8O4Si (MW 1266.73): C, 43.62; H, 1.27; N, 8.85. Found: C, 43.64; H, 1.22; N, 8.76.
-
2.
SiPcCl2 (100 mg, 0.16 mmol, 1 equiv.) and perfluoro-3,7-dimethyloctanoic acid (351 mg, 0.68 mmol, 4.25 equiv.) in diglyme at 140 °C gave 7 mg (0.004 mmol, 3%) of 2. 1H NMR (300 MHz, CDCl3): δ = 9.71 (m, 8 H), 8.44 (m, 8 H) ppm. 19F NMR (282 MHz, CDCl3): δ = −72.36 (m, CF3), −113.75 (m, CF2), −115.55 (m, CF2), −121.05 (m, CF2), −186.72 (m, CF), −187.20 (m, CF) ppm. HR NSI+ MS: m/z 1567.0519 ({M + H}+, 100%); theor. C52H17F38N8O4Si+ m/z 1567.0530.
-
3.
SiPcCl2 (200 mg, 0.33 mmol, 1 equiv.) and pivalic acid (250 mg, 2.45 mmol, 7.42 equiv.) in N,N′-DMF gave 151 mg (0.20 mmol, 59%) of 3. M.p.: >380 °C. 1H NMR (400 MHz, CDCl3): δ = 9.69 (m, 8 H), 8.38 (m, 8 H), −1.30 (s, 18 H) ppm. 1H NMR (400 MHz, CD2Cl2): δ = 9.72 (m, 8 H), 8.43 (m, 8 H), −1.32 (s, 18 H) ppm. HR NSI+ MS: m/z 765.2363 ({M + Na}+, 6%), 743.2546 ({M + H}+, 1%); theor. C42H34N8O4SiNa+ m/z 765.2364; theor. C42H35N8O4Si+ m/z 743.2545. Anal. Calcd for C42H34N8O4Si·1.5H2O (MW 769.90): C, 65.52; H, 4.84; N, 14.55. Found: C, 65.69; H, 4.57; N, 14.24.
-
4.
SiPcCl2 (100 mg, 0.16 mmol, 1 equiv.) and 3,3,3-trifluoro-2,2-dimethylpropionic acid (163 mg, 1.04 mmol, 6.5 equiv.) in diglyme at 160 °C gave 87 mg (0.10 mmol, 64%) of 4. M.p.: >380 °C. 1H NMR (300 MHz, CD2Cl2): δ = 9.74 (m, 8 H), 8.46 (m, 8 H), −0.98 (s, 12 H) ppm. 19F NMR (282 MHz, CD2Cl2): δ = −77.9 ppm. HR NSI+ MS: m/z 851.1982 ({M + H}+, 100%); theor. C42H29F6N8O4Si+ m/z 851.1980. Anal. Calcd for C42H28F6N8O4Si (MW 850.81): C, 59.29; H, 3.32; N, 13.17. Found: C, 59.61; H, 3.20; N, 12.97.
-
5.
SiPcCl2 (100 mg, 0.16 mmol, 1 equiv.) and 1-adamantanecarboxylic acid (133 mg, 0.98 mmol, 6.12 equiv.) in diglyme at 160 °C gave 52 mg (0.058 mmol, 36%) of 5. M.p.: >380 °C. 1H NMR (300 MHz, CDCl3): δ = 9.69 (m, 8 H), 8.37 (m, 8 H), 0.76 (m, 12 H), 0.39 (m, 6 H), −0.95 (s, br, 12 H) ppm. HR NSI+ MS: m/z 921.3306 ({M + Na}+, 12%); theor. C54H46N8O4SiNa+ m/z 921.3303. Anal. Calcd for C54H46N8O4Si (MW 899.10): C, 72.14; H, 5.16; N, 12.46. Found: C, 72.11; H, 4.95; N, 12.37.
-
6.
SiPcCl2 (100 mg, 0.16 mmol, 1 equiv.) and 3,5-bis(trifluoromethyl)benzoic acid (169 mg, 0.65 mmol, 4.06 equiv.) in diglyme at 160 °C gave 72 mg (0.068 mmol, 43%) of 6. M.p.: >350 °C (dec.).1H NMR (300 MHz, CDCl3): δ = 9.74 (m, 8 H), 8.43 (m, 8 H), 7.09 (s, br, 2 H), 5.47 (d, J = 1.0 Hz, 4 H) ppm. 19F NMR (282 MHz, CDCl3): δ = −64.4 ppm. 1H NMR (300 MHz, CD2Cl2): δ = 9.75 (m, 8 H), 8.47 (m, 8 H), 7.12 (s, 2 H), 5.48 (s, 2 H) ppm. 19F NMR (282 MHz, CD2Cl2): δ = −64.8 ppm. HR NSI+ MS: m/z 1077.1239 ({M + Na}+, 30%); theor. C50H22F12N8O4SiNa+ m/z 1077.1234. Anal. Calcd for C50H22F12N8O4Si (MW 1054.84): C, 56.93; H, 2.10; N, 10.62. Found: C, 56.55; H, 2.08; N, 10.51.
-
7.
SiPcCl2 (100 mg, 0.16 mmol, 1 equiv.) and 4-octyloxybenzoic acid (164 mg, 0.66 mmol, 4.12 equiv.) in diglyme at 160 °C gave 41 mg (0.039 mmol, 25%) of 7. M.p.: 307–309 °C. 1H NMR (400 MHz, CDCl3): δ = 9.70 (m, 8 H), 8.37 (m, 8 H), 5.69 (d, J = 9.2 Hz, 4 H), 5.04 (d, J = 9.0 Hz, 4 H), 3.34 (t, J = 6.6 Hz, 4 H), 1.33 (m, 4 H), 1.14 (m, 4 H), 1.06 (m, 16 H), 0.76 (t, J = 6.9 Hz, 6 H) ppm. MALDI+ MS: m/z 1038.4 (M+, 90%); theor. C62H58N8O6Si+ m/z 1038.4. Anal. Calcd for C62H58N8O6Si (MW 1039.28): C, 71.65; H, 5.63; N, 10.78. Found: C, 71.77; H, 5.53; N, 10.78.
Photophysical measurements
The solutions for photophysical investigation were prepared in HPLC-grade dichloromethane. Electronic absorption spectra were recorded with a Shimadzu UV-1800, UV-VIS Spectrophotometer in optical quartz cells of 10 mm path length. The absorption spectra were recorded on a PerkinElmer LAMBDA 950 UV/Vis/NIR spectrophotometer. The photoluminescence spectra were recorded on an Edinburgh Instruments FLS-980 fluorescence spectrometer and were corrected for the spectral sensitivity of the detection system. In dilute solutions used in the photophysical measurements, the photoluminescence spectra were independent of the excitation wavelength. In our earlier work, the absolute photoluminescence quantum yields (PLQY) of SiPc carboxylates R1 and R2 were determined using an integrating sphere on a Hamamatsu C9920–02 absolute PLQY measurement system32. Photoluminescene quantum yields for the SiPcs in this work were determined by a relative method using R2 as the standard. The photoluminescence lifetime was measured using the time-correlated single-photon-counting technique integrated in the FLS-980 fluorescence spectrometer. The samples were excited at 375 nm by pico-second pulsed diode laser head Picoquant LDH-D-C-375. The luminescence decay was measured at a wavelength corresponding to the emission peak. The emitted light was dispersed in a monochromator with an entrance slit of 2–4 nm and was detected on a microchannel plate detector (<100 ps response). The luminescence decay was fitted to a mono-exponential function with aχ2 value in the range of 1–1.25.
Electrochemistry
Electrochemical experiments were conducted in HPLC grade dichloromethane with 0.1 M tetra-n-butylammonium hexafluorophosphate as the supporting electrolyte, with a PC controlled electrochemical workstation (Electrochemical Analyzer/Workstation CHI600C, CH Instruments, Inc.) at room temperature.
The experiments were carried out under nitrogen in an electrochemical cell through which a stream of nitrogen was passed. The volume of the solvent was 2–3 mL. Glassy-carbon disk (3 mm diameter), platinum spiral, and platinum wire served as working, counter, and quasi-reference electrodes, respectively. Estimated error: ±30 mV. Cyclic voltammetry was performed at scan rates of 0.1 V/s and 1 V/s. The potentials of reversible redox processes were independent of the scan rate.
DFT calculations
All calculations were performed with the Gaussian09, revision D.0148 suite of programs employing the DFT method, the Becke three-parameter hybrid functional49, and Lee-Yang-Parr’s gradient-corrected correlation functional (B3LYP)50. Singlet ground state geometry optimizations and single point energy calculations for 1–7, R1 and R2 were carried out at the (R)B3LYP levels, using their respective crystallographic structures as starting points. All elements except Silicon were assigned the 6–31 G(d,p) basis set51. The double-ζ quality SBKJC VDZ ECP basis set52 with an effective core potential was employed for the Si(VI)-ion. Vertical electronic excitations based on (R)B3LYP-optimized geometries were computed for 1–7, R1 and R2 using the TD-DFT formalism53,54 in dichloromethane using conductor-like polarizable continuum model (CPCM)55,56,57. Vibrational frequency calculations were performed to ensure that the optimized geometries represent the local minima and there are only positive eigenvalues. The electronic distribution and localization of the singlet excited states were visualized using the electron density difference maps (ED-DMs)58. Gausssum 2.2 and Chemissian v3.859 were employed to visualize the absorption spectra (simulated with Gaussian distribution with a full-width at half maximum (fwhm) set to 3000 cm−1) and to calculate the fractional contributions of various groups to each molecular orbital. All calculated structures and Kohn-Sham orbitals were visualized with ChemCraft60.
X-ray Crystallography
Diffraction data for compounds 1 and 3–7 were collected by using a Rigaku FR-X Ultrahigh brilliance Microfocus RA generator/confocal optics and Rigaku XtaLAB P200 system, using Mo Kα radiation (λ = 0.71073 Å). Data for compound 2 were collected by using a Rigaku MM-007HF High brilliance RA generator/confocal optics and Rigaku XtaLAB P100 system, using Cu Kα radiation (λ = 1.54187 Å). Intensity data for all structures were collected at 173 K, using either ω, or both ω and φ steps accumulating area detector images spanning at least a hemisphere of reciprocal space. All data were corrected for Lorentz polarization effects. A multiscan absorption correction was applied by using CrystalClear61. Structures were solved by direct (SIR200462 or SIR201163) methods and refined by full-matrix least-squares against F2 (SHELXL-2013)64. Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were refined using a riding model. All calculations were performed using the CrystalStructure65 interface. Data for 2 showed positional disorder throughout the perfluoroalkyl chain that could not be modelled. This resulted in the unusually large thermal ellipsoids seen for these atoms, and necessitated the use of a range of restraints to bond distances and thermal ellipsoids. Crystallographic data are presented in Table 1.
Data availability
The data supporting this study are available at: http://dx.doi.org/10.17630/ea33220b-53f6-4503-84d0-ae052914ecff.
References
Reetz, M. T. & Jiao, N. Copper-phthalocyanine conjugates of serum albumins as enantioselective catalysts in Diels-Alder reactions. Angew. Chem. Int. Ed. 45, 2416–2419, https://doi.org/10.1002/anie.200504561 (2006).
Shankar, K., Feng, X. & Grimes, C. A. Enhanced Harvesting of Red Photons in Nanowire Solar Cells: Evidence of Resonance Energy Transfer. ACS Nano 3, 788–794, https://doi.org/10.1021/nn900090x (2009).
de la Torre, G., Claessens, C. G. & Torres, T. Phthalocyanines: old dyes, new materials. Putting color in nanotechnology. Chem. Commun. 2000–2015, doi:https://doi.org/10.1039/B614234F (2007).
Mortimer, R. J. Electrochromic materials. Chem. Soc. Rev. 26, 147–156, https://doi.org/10.1039/CS9972600147 (1997).
de la Torre, G., Vázquez, P., Agulló-López, F. & Torres, T. Phthalocyanines and related compounds:organic targets for nonlinear optical applications. J. Mater. Chem. 8, 1671–1683, https://doi.org/10.1039/A803533D (1998).
Emmelius, M., Pawlowski, G. & Vollmann, H. W. Materials for Optical Data Storage. Angew. Chem. Int. Ed. Engl. 28, 1445–1471, https://doi.org/10.1002/anie.198914453 (1989).
Melville, O. A., Lessard, B. H. & Bender, T. P. Phthalocyanine-Based Organic Thin-Film Transistors: A Review of Recent Advances. ACS Appl Mater Interfaces 7, 13105–13118, https://doi.org/10.1021/acsami.5b01718 (2015).
Guillaud, G., Simon, J. & Germain, J. P. Metallophthalocyanines: Gas sensors, resistors and field effect transistors. Coord. Chem. Rev. 178–180, Part 2, 1433–1484, doi:https://doi.org/10.1016/S0010-8545(98)00177-5 (1998).
Hohnholz, D., Steinbrecher, S. & Hanack, M. Applications of phthalocyanines in organic light emitting devices. J. Mol. Struct. 521, 231–237, https://doi.org/10.1016/S0022-2860(99)00438-X (2000).
Loosli, C. et al. Synthesis and Electrochemical and Photophysical Studies of Tetrathiafulvalene-Annulated Phthalocyanines. J. Org. Chem. 70, 4988–4992, https://doi.org/10.1021/jo0501801 (2005).
Mishra, A. & Bäuerle, P. Small Molecule Organic Semiconductors on the Move: Promises for Future Solar Energy Technology. Angew. Chem. Int. Ed. 51, 2020–2067, https://doi.org/10.1002/anie.201102326 (2012).
Martín-Gomis, L., Fernández-Lázaro, F. & Sastre-Santos, A. Advances in phthalocyanine-sensitized solar cells (PcSSCs). J. Mater. Chem. A 2, 15672–15682, https://doi.org/10.1039/C4TA01894J (2014).
Lim, B. et al. Silicon-Naphthalo/Phthalocyanine-Hybrid Sensitizer for Efficient Red Response in Dye-Sensitized Solar Cells. Org. Lett. 15, 784–787, https://doi.org/10.1021/ol303436q (2013).
Ragoussi, M.-E., Ince, M. & Torres, T. Recent Advances in Phthalocyanine-Based Sensitizers for Dye-Sensitized Solar Cells. Eur. J. Org. Chem. 2013, 6475–6489, https://doi.org/10.1002/ejoc.201301009 (2013).
Li, H., Jensen, T. J., Fronczek, F. R. & Vicente, M. G. H. Syntheses and Properties of a Series of Cationic Water-Soluble Phthalocyanines. J. Med. Chem. 51, 502–511, https://doi.org/10.1021/jm070781f (2008).
Taratula, O. et al. A Multifunctional Theranostic Platform Based on Phthalocyanine-Loaded Dendrimer for Image-Guided Drug Delivery and Photodynamic Therapy. Mol. Pharmaceutics 10, 3946–3958, https://doi.org/10.1021/mp400397t (2013).
Ciliberto, E. et al. The.pi.-.pi. interactions and bandwidths in molecular metals. A chemical, structural, photoelectron spectroscopic, and Hartree-Fock-Slater study of monomeric and cofacially joined dimeric silicon phthalocyanines. J. Am. Chem. Soc. 106, 7748–7761, https://doi.org/10.1021/ja00337a018 (1984).
Nyokong, T. Effects of substituents on the photochemical and photophysical properties of main group metal phthalocyanines. Coord. Chem. Rev. 251, 1707–1722, https://doi.org/10.1016/j.ccr.2006.11.011 (2007).
Mizuguchi, J. & Matsumoto, S. Molecular Distortion and Exciton Coupling Effects in β Metal-Free Phthalocyanine. J. Phys. Chem. A 103, 614–616, https://doi.org/10.1021/jp9836168 (1999).
Farren, C., FitzGerald, S., Beeby, A. & Bryce, M. R. The first genuine observation of fluorescent mononuclear phthalocyanine aggregates. Chem. Commun. 572–573, doi:https://doi.org/10.1039/B110876J (2002).
Lukyanets, E. A. Phthalocyanines as photosensitizers in the photodynamic therapy of cancer. J. Porphyrins Phthalocyanines 3, 424–432, doi:https://doi.org/10.1002/(SICI)1099-1409(199908/10)3:6/7424::AID-JPP1513.0.CO;2-K (1999).
Darwent, J. R., Douglas, P., Harriman, A., Porter, G. & Richoux, M.-C. Metal phthalocyanines and porphyrins as photosensitizers for reduction of water to hydrogen. Coord. Chem. Rev. 44, 83–126, https://doi.org/10.1016/S0010-8545(00)80518-4 (1982).
Graham, K. R. et al. Extended Conjugation Platinum(II) Porphyrins for use in Near-Infrared Emitting Organic Light Emitting Diodes. Chem. Mater. 23, 5305–5312, https://doi.org/10.1021/cm202242x (2011).
Chen, Y.-C., Hsu, C.-Y., Lin, R. Y.-Y., Ho, K.-C. & Lin, J. T. Materials for the Active Layer of Organic Photovoltaics: Ternary Solar Cell Approach. ChemSusChem 6, 20–35, https://doi.org/10.1002/cssc.201200609 (2013).
Makhseed, S., McKeown, N. B., Msayib, K. & Bumajdad, A. Inducing solid-state isolation of the phthalocyanine macrocycle by its incorporation within rigid, randomly shaped oligomers. J. Mater. Chem. 15, 1865–1870, https://doi.org/10.1039/B416387G (2005).
Silver, J. et al. Novel Seven Coordination Geometry of Sn(IV): Crystal Structures of Phthalocyaninato Bis(undecylcarboxylato)Sn(IV), Its Si(IV) Analogue, and Phthalocyaninato Bis(chloro)silicon(IV). The Electrochemistry of the Si(IV) Analogue and Related Compounds. Inorg. Chem. 40, 5434–5439, https://doi.org/10.1021/ic001120a (2001).
Ishikawa, N., Sugita, M. & Wernsdorfer, W. Nuclear Spin Driven Quantum Tunneling of Magnetization in a New Lanthanide Single-Molecule Magnet: Bis(Phthalocyaninato)holmium Anion. J. Am. Chem. Soc. 127, 3650–3651, https://doi.org/10.1021/ja0428661 (2005).
Joyner, R. D., Cekada, J., Linck, R. G. & Kenney, M. E. Diphenoxysilicon phthalocyanine. J. Inorg. Nucl. Chem. 15, 387–388, https://doi.org/10.1016/0022-1902(60)80072-3 (1960).
Krueger, P. C. & Kenney, M. E. Dialkoxyphthalocyaninosilicon Derivatives. J. Org. Chem. 28, 3379–3381, https://doi.org/10.1021/jo01047a023 (1963).
Bonnett, R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem. Soc. Rev. 24, 19–33, https://doi.org/10.1039/CS9952400019 (1995).
Flora, W. H., Hall, H. K. & Armstrong, N. R. Guest Emission Processes in Doped Organic Light-Emitting Diodes: Use of Phthalocyanine and Naphthalocyanine Near-IR Dopants. J. Phys. Chem. B 107, 1142–1150, https://doi.org/10.1021/jp021368g (2003).
Zysman-Colman, E. et al. Solution-Processable Silicon Phthalocyanines in Electroluminescent and Photovoltaic Devices. ACS Appl. Mater. Interfaces 8, 9247–9253, https://doi.org/10.1021/acsami.5b12408 (2016).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The Cambridge Structural Database. Acta Cryst. B72, 171–179, https://doi.org/10.1107/S2052520616003954 (2016).
Allen, F. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Cryst. B58, 380–388, https://doi.org/10.1107/S0108768102003890 (2002).
Umezawa, Y., Tsuboyama, S., Honda, K., Uzawa, J. & Nishio, M. CH/π Interaction in the Crystal Structure of Organic Compounds. A Database Study. Bull. Chem. Soc. Jpn. 71, 1207–1213, https://doi.org/10.1246/bcsj.71.1207 (1998).
Silver, J., Sosa-Sanchez, J. L. & Frampton, C. S. Structure, Electrochemistry, and Properties of Bis(ferrocenecarboxylato)(phthalocyaninato)silicon(IV) and Its Implications for (Si(Pc)O)n Polymer Chemistry. Inorg. Chem. 37, 411–417, https://doi.org/10.1021/ic970934x (1998).
Farren, C., FitzGerald, S., Bryce, M. R., Beeby, A. & Batsanov, A. S. Synthesis, structure and optical characterisation of silicon phthalocyanine bis-esters. J. Chem. Soc., Perkin Trans. 2, 59–66, https://doi.org/10.1039/B108778A (2002).
Barker, C. A. et al. Synthesis of new axially-disubstituted silicon-phthalocyanine derivatives: optical and structural characterisation. Tetrahedron 62, 9433–9439, https://doi.org/10.1016/j.tet.2006.07.046 (2006).
Sosa-Sánchez, J. L. et al. Novel Phthalocyaninatobis(alkylcarboxylato)silicon(IV) Compounds: NMR Data and X-ray Structures To Study the Spacing Provided by Long Hydrocarbon Tails That Enhance Their Solubility. Chem. Eur. J. 11, 4263–4273, https://doi.org/10.1002/chem.200500003 (2005).
Sosa-Sánchez, J. L. et al. Synthesis and characterization of a new (phthalocyani-nato)bis(carboxylate) silicon(IV) compound with increased solubility. J. Porphyrins Phthalocyanines 06, 198–202, https://doi.org/10.1142/s1088424602000221 (2002).
Xu, H., Wada, T., Ohkita, H., Benten, H. & Ito, S. Dye sensitization of polymer/fullerene solar cells incorporating bulky phthalocyanines. Electrochim. Acta 100, 214–219, https://doi.org/10.1016/j.electacta.2012.05.155 (2013).
Lever, A. B. P. & Minor, P. C. Electrochemistry of main-group phthalocyanines. Inorg. Chem. 20, 4015–4017, https://doi.org/10.1021/ic50225a088 (1981).
Shen, X.-M., Jiang, X.-J., Huang, C.-C., Zhang, H.-H. & Huang, J.-D. Highly photostable silicon(IV) phthalocyanines containing adamantane moieties: synthesis, structure, and properties. Tetrahedron 66, 9041–9048, https://doi.org/10.1016/j.tet.2010.09.007 (2010).
Jiang, X.-J., Yeung, S.-L., Lo, P.-C., Fong, W.-P. & Ng, D. K. P. Phthalocyanine−Polyamine Conjugates as Highly Efficient Photosensitizers for Photodynamic Therapy. J. Med. Chem. 54, 320–330, https://doi.org/10.1021/jm101253v (2011).
Sholto, A. & Ehrenberg, B. Hydrophobicity, topography in membranes and photosensitization of silicon phthalocyanines with axial ligands of varying lengths. Photochem. Photobiol. Sci 7, 344–351, https://doi.org/10.1039/B716377K (2008).
Jiang, X.-J. et al. Phthalocyanine–Polyamine Conjugates as pH-Controlled Photosensitizers for Photodynamic Therapy. Chem. Eur. J. 16, 4777–4783, https://doi.org/10.1002/chem.200903580 (2010).
Chen, X. et al. Effect of axial ligands on the photophysical properties of new silicon(IV) phthalocyanines. J. Coord. Chem. 68, 732–740, https://doi.org/10.1080/00958972.2014.998658 (2015).
Frisch, M. J. et al. Gaussian 09, Revision D.01 (Gaussian Inc., Wallingford, CT, 2013).
Becke, A. D. Density-functional Thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652, https://doi.org/10.1063/1.464913 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789, https://doi.org/10.1103/PhysRevB.37.785 (1988).
McLean, A. D. & Chandler, G. S. J. Chem. Phys. 72, 5639–5648, https://doi.org/10.1063/1.438980 (1980).
Binkley, J. S., Pople, J. A. & Hehre, W. J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 102, 939–947, https://doi.org/10.1021/ja00523a008 (1980).
Casida, M. E., Jamorski, C., Casida, K. C. & Salahub, D. R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 108, 4439–4449, https://doi.org/10.1063/1.475855 (1998).
Stratmann, R. E., Scuseria, G. E. & Frisch, M. J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 109, 8218–8224, https://doi.org/10.1063/1.477483 (1998).
Cossi, M., Rega, N., Scalmani, G. & Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681, https://doi.org/10.1002/jcc.10189 (2003).
Cossi, M. & Barone, V. Time-dependent density functional theory for molecules in liquid solutions. J. Chem. Phys. 115, 4708–4717, https://doi.org/10.1063/1.1394921 (2001).
Barone, V. & Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 102, 1995–2001, https://doi.org/10.1021/jp9716997 (1998).
Browne, W. R., O’Boyle, N. M., McGarvey, J. J. & Vos, J. G. Elucidating excited state electronic structure and intercomponent interactions in multicomponent and supramolecular systems. Chem. Soc. Rev. 34, 641–663, https://doi.org/10.1039/B400513A (2005).
Chemissian v4.42 © Lenoid, S. http://www.chemissian.com (2005–2016).
ChemCraft 1.5 (San Diego, CA), https://www.chemcraftprog.com.
CrystalClear-SM Expert v2.1. Rigaku Americas, Rigaku Americas, The Woodlands, Texas, USA and Rigaku Corporation, Tokyo, Japan, 2010–2014.
Burla, M. C. et al. SIR2004: an improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 38, 381–388, https://doi.org/10.1107/S002188980403225X (2005).
Burla, M. C. et al. SIR2011: a new package for crystal structure determination and refinement. J. Appl. Crystallogr. 45, 357–361, https://doi.org/10.1107/S0021889812001124 (2012).
Sheldrick, G. M. Sheldrick, G. Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8, https://doi.org/10.1107/S2053229614024218 (2015).
CrystalStructure v4.2. Rigaku Americas, Rigaku Americas, The Woodlands, Texas, USA and Rigaku Corporation, Tokyo, Japan, 2013.
Acknowledgements
EZ-C acknowledges the University of St Andrews for financial support. We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for analytical services. We are grateful to the EPSRC for financial support from grant EP/M02105X/1 and the European Research Council for financial support from grant 321305. We thank Dr. Nail Shavaleev for the syntheses of 1–7 and their optoelectronic characterization in this study. I.D.W.S. acknowledges a Royal Society Wolfson Research Merit Award.
Author information
Authors and Affiliations
Contributions
A.K. Pal conducted the DFT Calculations. S. Varghese conducted the photophysical measurements. D.B. Cordes and A.M.Z. Slawin solved the X-ray structures. A.K. Pal, S. Varghese, D. B. Cordes, A.M.Z. Slawin, I.D.W. Samuel and E. Zysman-Colman contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pal, A.K., Varghese, S., Cordes, D.B. et al. Near-Infrared Fluorescence of Silicon Phthalocyanine Carboxylate Esters. Sci Rep 7, 12282 (2017). https://doi.org/10.1038/s41598-017-12374-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12374-8
This article is cited by
-
Synthesis, characterization, thermal and photophysical properties of novel strontium (II) phthalocyanine
Journal of Inclusion Phenomena and Macrocyclic Chemistry (2022)
-
Near-infrared benzodiazoles as small molecule environmentally-sensitive fluorophores
Frontiers of Chemical Science and Engineering (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
