
Abstract
Chromosomal microarray (CMA) is now recognized as the first-tier genetic test for detection of copy number variations (CNVs) in patients with autism spectrum disorder (ASD). The aims of this study were to identify known and novel ASD associated-CNVs and to evaluate the diagnostic yield of CMA in Thai patients with ASD. The Infinium CytoSNP-850K BeadChip was used to detect CNVs in 114 Thai patients comprised of 68 retrospective ASD patients (group 1) with the use of CMA as a second line test and 46 prospective ASD and developmental delay patients (group 2) with the use of CMA as the first-tier test. We identified 7 (6.1%) pathogenic CNVs and 22 (19.3%) variants of uncertain clinical significance (VOUS). A total of 29 patients with pathogenic CNVs and VOUS were found in 22% (15/68) and 30.4% (14/46) of the patients in groups 1 and 2, respectively. The difference in detected CNV frequencies between the 2 groups was not statistically significant (Chi square = 1.02, df = 1, P = 0.31). In addition, we propose one novel ASD candidate gene, SERINC2, which warrants further investigation. Our findings provide supportive evidence that CMA studies using population-specific reference databases in underrepresented populations are useful for identification of novel candidate genes.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder characterized by impairments in social interaction and communication, as well as stereotyped behaviors and restricted interests. For approximately 80% of cases, the cause of ASD is still unknown. Due to clinical diagnostic criteria of ASD with broader spectrum of symptoms, the prevalence of ASD is increasing worldwide. The estimated ASD prevalence in the US was 1 in 150 children in 2000, but by 2012 that had increased to 1 in 68 children1. In Thailand, the incidence rate of severe ASD was estimated at 9.9 per 10,000 in children aged 1–5 years2. Up to 70% of individuals with ASD also have intellectual disability/developmental delay (ID/DD). Genetic causes, including chromosomal abnormalities, copy number variations (CNVs), and single nucleotide variants are recognized as causative in 10–20% of ASD cases3. Knowing the genetic cause of ASD can help with genetic counseling and evaluation of recurrence risk in the families.
Since 2010, chromosomal microarray (CMA) has been recommended as the first-tier clinical diagnostic test for detection of CNVs in patients with ASD, ID, DD and multiple congenital abnormalities (MCA) of unknown causes4,5. CMA has a much higher diagnostic yield (10–20%) for these individuals than conventional cytogenetics (~3%)5,6,7. Most of CMA studies in these disorders have largely focused on Caucasian populations4,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26. To date only a few non-Caucasian studies have been reported27,28,29,30,31,32. In addition, CMA studies in specific populations have been shown to help widen the list of novel ASD candidate genes such as PTDSS1 and AGER as novel candidate genes of ASD in Lebanese subjects31, and population-specific CNVs in the YWHAE gene in Han Chinese subjects33. In our earlier study, we have categorized reference CNVs of more than 3,000 Thai individuals and created a CNV database that would facilitate the accurate clinical CNV interpretation for Thai patients34. In this study, we aimed to screen and identify known and novel CNVs associated with ASD in a large cohort of Thai patients with ASD. This is the first report demonstrating the utility of CMA for detection of CNVs in Thai patients referred with ASD of unknown cause. Our findings also provide supportive evidence that CMA studies in underrepresented populations can be useful for identification of novel candidate genes.
Results
Diagnostic yield of CMA in Thai patients with ASD
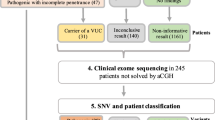
In our cohort of 114 Thai patients with ASD, a total of 742 CNVs was identified from all patients, ranging between 1 to 22 CNVs per patient. The size of the CNVs ranged from 1,632 bp to 18.9 Mb, with a median size of 86 kb. Among these CNVs, pathogenic CNVs and VOUS were identified in 7 (6.1%) and 22 (19.3%) patients, respectively. The overall diagnostic yield of CNVs in our cohort was 25.4% (29/114). The 29 patients with pathogenic CNVs and VOUS included 15 of 68 (22%) patients in group 1 and 14 of 46 (30.4%) patients in group 2. The summary and schematic diagrams of CMA results in the study are shown in Table 1, Figs 1 and 2. Although a higher frequency of both pathogenic CNVs and VOUS were identified in group 2 patients (30.4%) with the use of CMA as the first-tier test compared with patients in group 1 (22%) with the use of CMA as the second line test, no statistically significant differences were found in the frequencies between the 2 groups (Chi square = 1.02, df = 1, P = 0.31).

A Flowchart showing the patient characteristics and CMA analysis results in this study. *No statistically significant difference in frequencies of diagnostic yields between the 2 groups were observed using Chi-square test, P > 0.05 (Chi-squared = 1.02, df = 1, P = 0.31).

Pie charts summarizing the CNV classifications from the study. (a) Number of pathogenic CNVs and VOUS (likely pathogenic CNVs, likely benign CNVs, and VOUS with no subclassification); (b) Number of paternally or maternally inherited CNVs and de novo CNVs.
Parental testings were available for investigation of the inheritance status in 23 of 29 patients. Of these 23 patients, 5 de novo CNVs, 11 maternally inherited CNVs and 7 paternally inherited CNVs were identified (Fig. 2b). However, parental samples were not available in 3 pathogenic CNVs and 3 VOUS which was a limitation of our study. The clinical characteristics of patients and details of the pathogenic CNVs and VOUS are summarized in Table 2
.
Rare inherited CNVs and known syndromes
18 of the 23 CNVs in this study were transmitted from an asymptomatic parent. Most of the VOUS with inherited CNVs were below 1 Mb, but five CNVs were more than 1 Mb in size including four duplications and one deletion. Interestingly, one (TM54–3) was a 1.49 Mb deletion encompassing 2 OMIM genes (PTP4A3, GPR20), which was inherited from an asymptomatic father. Deletions in these regions have been reported as both pathogenic CNVs and VOUS in the ISCA/ClinGen and DECIPHER databases. Thus, it is difficult to interpret the significance of the deletion in this patient. However, we assumed that it was a VOUS, because genes in this region were not involved in neurological function which may have been associated with the phenotype of this patient.
Within the group of patients with pathogenic CNVs, there were patients with well-known microduplication or microdeletion syndromes, including the 1q21.1 duplication, 15q13.3 microdeletion, 16p13.11 microduplication, 22q13.3 deletion (Phelan-McDermid syndrome), 18q distal deletion, and 4p16.3 distal deletion (Wolf-Hirschhorn syndrome). Three of the pathogenic CNVs were inherited from a healthy parent. This is in line with the fact that some of these established syndromes are known to have incomplete penetrance and/or variable expressivity.
De novo CNVs and novel candidate genes for ASD
Among the de novo CNVs, one was a large deletion associated with the known 9q21.13 microdeletion syndrome, while the other 4 de novo VOUS were rare duplications of 127–220 Kb in size. A 127 kb duplication at 2q14.1 overlapping the DPP10 gene was identified in a female proband (TM18-3) and a 159 kb duplication at 3p26.3 overlapping CHL1 gene was identified in a male proband (TM4-3). Both the DPP10 and CHL1 genes have been previously reported as candidate genes of ASD. One male patient with non-syndromic ASD (TM41-3) had a 220 kb duplication at 1p35.2 including the FABP3, SNRNP40, NKAIN1 and SERINC2 genes. A de novo 217 kb duplication at 18q22.3 including the ZNF407, CNDP2 and CNDP1 genes was identified in a male proband with non-syndromic ASD (AR12-3). Table 3 provides details of all de novo CNVs and the mapping of the de novo duplications on chromosomes 1p35.2 and 18q22.3 in multiple CNV databases is shown in Supplementary Figure 1.
Incidental findings of common α-thalassemia mutations
In this study, we identified the deletions of α-globin genes, HBA1 and HBA2 as the most common incidental findings in our patient cohort. Chromosomal microarray was able to detect the common deletions of HBA1 and HBA2, leading to the identification of α-thalassemia mutations in 9 of the 114 ASD patients (7.89%). These α-thalassemia deletions were confirmed by single-tube multiplex-PCR assay as previously described35. Among the 9 patients, eight had available DNA for validation. The results confirmed 5 cases with heterozygous alpha-thalassemia-1 (–SEA/αα), one case with heterozygous alpha-thalassemia-2 (-α3.7/αα), one case with homozygous alpha-thalassemia-2 (-α3.7/-α3.7) and the other case with compound heterozygous alpha-thalassemia-1 and alpha-thalassemia-2 (-SEA /- α4.2), also known as Hb H disease (Supplementary Table S2).
Detection of absence of heterozygosity (AOH) regions
We also analyzed the AOH regions greater than 10 Mb in size in all patients. One case showed two large AOH regions of 40.6 Mb (15q11.1-15q22.2) and 12.4 Mb (15q26.1-15q26.3) interrupted by a region of heterozygosity (Supplementary Figure 2), a condition which was suspected to be from uniparental disomy (UPD). Methylation-specific polymerase chain reaction (MS-PCR) analysis of SNRPN gene was then performed using primers as previously described36,37, and the results confirmed that this patient had maternal uniparental isodisomy of the 15q11.2 region resulting in Prader-Willi syndrome (Supplementary Figure 3). Additionally, multiple large AOH regions (average 158.02 Mb) were also detected in another patient, encompassing 5.84% of the genome (data not shown), suggesting a consanguineous marriage between first cousin parents (coefficient of inbreeding (F) 1/16). Since this patient had an increased chance of having an autosomal recessive disease, it might be beneficial to further search for homozygous gene mutations by means of whole exome sequencing.
Discussion
We performed CMA in 114 Thai patients with ASD of unknown cause, resulting in an overall diagnostic yield of 25.4% composed of both pathogenic CNVs and VOUS. The diagnostic yields of only pathogenic CNVs and of VOUS alone are 6.1% and 19.3%, respectively. Our pathogenic CNV’s diagnostic yield is in line with those of other CMA studies in non-Asian patients with ASD, which range from 5.4% to 8.6%9,12,18,22. However, it is difficult to directly compare such diagnostic yields among studies due to the use of different inclusion criteria for diagnostic yields. Some studies report diagnostic yields from only pathogenic CNVs4,8,11,13,14,15,16,17,18,20,22,24,26,28,29,32, while others include combined pathogenic CNVs and likely pathogenic CNVs10,23,25,30 or combined pathogenic CNVs and VOUS12,19,21,27. In addition, previous studies have had different inclusion criteria for participating patients and used various microarray platforms with different resolutions. A summary of microarray studies in patients with ASD is given in Supplementary Table S3. Of note, because the diagnostic yield of karyotype abnormalities in ASD patients has been shown to be lower than 3%7,23, it is not surprising that there were no statistically significant differences in the detected frequencies of pathogenic CNVs and VOUS between group 1 patients with normal karyotype and group 2 patients with the use of CMA as the first-tier test. Therefore, our findings support the recommendation of using CMA as the first-tier diagnostic test in patients with ASD of unknown cause.
Among pathogenic CNVs, the16p13.11 microduplication was initially considered as a rare benign CNV38, however, we interpreted this duplication as pathogenic because several previous studies have supported the positive association between this duplication and a variety of neuropsychiatric disorders including ASD, unexplained ID, schizophrenia, epilepsy, and attention-deficit hyperactivity disorder (ADHD). This duplication region contains two strong candidate genes, NTAN1 and NDE1, which are both expressed in the brain and have been proposed as candidate genes for ASD and other neuropsychiatric disorders39,40,41,42. Most of the pathogenic, likely pathogenic CNVs and VOUS identified in this study were transmitted from a healthy parent (18 cases from 23 families, from which parental DNA samples were available). These inherited CNVs may suggest incomplete penetrance or some other factors interacting with the CNVs. Likewise, pathogenic CNVs in 1q21.1-1q21.2 (duplication)43,44,45, 15q13.2-15q13.3 (deletion)46,47, and 16p13.11 (duplication)40,41 were also found to be inherited in our study. The other possible theory would be the two-hit model, i.e. a second mutation not detected by CMA48. Our findings further emphasize that parental array study alone may not be conclusive enough for CNV interpretation, the VOUS found in microarray studies need to be further analyzed before making a definite conclusion as to whether they are pathogenic or benign CNVs.
In the five de novo CNVs, one was a large deletion associated with the known 9q21.13 microdeletion syndrome, while the other four were duplications ranging from 127–220 kb in size. These duplications contained recently identified ASD-associated genes, DPP10 49 and CHL1 50,51, and two genes that are highly expressed in the brain, but have not yet been linked to ASD, namely carnosine dipeptidase 1 (CNDP1) and serine incorporator 2 (SERINC2). Duplications of neither SERINC2 nor CNDP1 have been reported in the Thai CNV database34. The 1p35.2 duplication containing SERINC2 in particular is rare, and is not seen in the DGV database. Loss-of-function (LoF) variants (nonsense, frameshift and splice site variants) of these two genes were not observed in the 195 unrelated Thais from our in-house exome sequencing database (data not shown). In addition, 16 rare LoF variants of SERINC2 (MAF < 0.001) and 27 rare LoF variants of CNDP1 (MAF < 0.001) were found in the Exome Aggregation Consortium (ExAC) database. However, clinical phenotype data was not available for these individuals.
A de novo duplication of chromosome 18q22.3 included the ZNF407, CNDP2 and CNDP1 genes. Balanced translocation and point mutations in the ZNF407 gene were identified in one study of ID patients with autism52, but deletions/duplications of ZNF407 and CNDP2 genes have not been previously reported in patients with autism. The Carnosine dipeptidase 1 gene (CNDP1, OMIM 609064), also known as serum carnosinase (CN1), is involved in neuroprotective actions and neurotransmitter functions in the brain. It is mainly expressed in the cytosol of pyramidal neurons in the hippocampus and in large and small neurons of the temporal cortex in the human brain53. Alterations of serum carnosinase expression and activity have been shown to be associated with neurological diseases including multiple sclerosis and Parkinson’s disease54. Although carnosinase deficiency has been reported in patients with ID and DD55,56,57, the overexpression of CNDP1 from copy number gain as seen in our patient has never, to our knowledge, been previously reported. Additionally, carnosinase is an enzyme whose alterations is likely to be recessively inherited, so CNDP1 gene duplication may not affect the carnosinase function. A search of the DGV database found no CNVs which encompassed the exact same duplicated region, but we did find several smaller duplications encompassing CNDP1 in the general population. A search of the DECIPHER database did not reveal cases with deletion and duplication relatively similar in size to this duplication region. In addition, two DD cases (nssv581777 and nssv707115) with 178.39 and 279.02 kb duplications of the CNDP1 region were submitted to the ISCA/ClinGen database and interpreted as benign and uncertain-likely benign (Supplementary Figure 1a). Thus, these findings do not support CNDP1 as an ASD candidate gene.
A de novo 220 kb duplication of chromosome 1p35.2 encompassed the FABP3, SNRNP40, NKAIN1 and SERINC2 genes. Although FABP3 knockout mice have shown decreased social memory and novelty seeking58 and one study found the NKAIN1 protein strongly expressed in the hippocampus and cerebellar granular cell layer59, no evidence for association between duplication of FABP3, SNRNP40, and NKAIN1 genes and neurodevelopmental disorders has been reported. Of particular interest is the serine incorporator 2 gene (SERINC2, OMIM 614549), also known as tumor differentially expressed 2-like (TDE2), which encodes a transmembrane protein that facilitates incorporation of serine into phosphatidylserine and sphingolipids60. The concentration of sphingolipids is highest in the brain, they play important roles in neural plasticity, signaling and axonal guidance61. Expression of Serinc2 mRNA was upregulated in the dentate gyrus of the hippocampus and the cerebellar Purkinje cell layer following kainate-induced seizures in rat60. In addition, overexpressed SERINC2 protein was reported in patients with developmental delay carrying deletions or mutations of the UPF2 or UPF3B genes, both of which are implicated in nonsense-mediated mRNA decay pathway (NMD)62. Furthermore, rare SERINC2 variants were significantly associated with alcohol dependence in subjects of European descent63. Transcript expressions of genes in the NKAIN1-SERINC2 genomic region were significantly associated with expressions of genes in the dopaminergic, serotoninergic, cholinergic, GABAergic, glutamatergic, histaminergic, endocannabinoid, metabolic, neuropeptide and opioid pathways64. Several studies have reported that alcohol dependence was associated with increased risk of neuropsychiatric disorders, including bipolar disorder and autism65,66,67. Autism and alcohol dependence have been shown to share some genetic factors according to earlier studies, where a higher incidence of alcoholism in the family members of ASD patients compared with the general population was observed. Also, the links between the autism susceptibility candidate 2 gene (AUTS2), a known ASD risk gene, in the regulation of alcohol consumption was reported67,68. These findings support the hypothesis that SERINC2 may be one of the dosage-sensitive genes and an increased SERINC2 protein may predispose to neurodevelopmental and neuropsychiatric disorders partly via abnormal NMD pathways and neurotransmitter or metabolic systems. In the DECIPHER database, we identified two CNVs of slightly larger size encompassing SERINC2; a 377.2-kb microdeletion that has been reported in a patient with seizure (patient 289527 in Supplementary Figure 1b), and a 434.6-kb microduplication reported in a patient with ID and short attention span who also has Potocki-Shaffer syndrome. While the microdeletion was deemed possibly pathogenic, the microduplication was inherited from healthy parent, raising the possibility of incomplete penetrance. In addition, two larger duplications containing SERINC2 were seen in the ISCA/ClinGen database. These are a de novo 1.27-Mb duplication identified in a patient with global DD that was interpreted as pathogenic (nssv578518), and a 1.15-Mb VOUS identified in a patient with dysmorphic facial features (nssv1602204). Although the effects of other genes in these large duplication regions in the ISCA database could not be excluded, SERINC2 is likely one of the susceptibility genes that may contribute to neuropsychiatric disorders as noted in the supportive evidence discussed above. Because this gene have not been previously linked to autism, we proposed the SERINC2 gene as a novel ASD candidate gene. The supportive evidences of SERINC2 as a potential ASD candidate gene are summarized in Supplementary Figure 4.
Alpha-thalassemia (α-thalassemia) is the most common incidental finding of CMA in our Thai population. In Northern Thailand and Laos, the prevalence of α-thalassemia has been estimated as between 30–40% including 3.6–10% of α-thalassemia 1 and 16.4–20% of α-thalassemia 269. Since the majority of alterations in α-globin genes are deletions, a CMA with enough resolution should be able to detect these mutations. In this study, CMA testing could detect α-thalassemia in 9 of 114 ASD patients (7.89%); 4.39% α-thalassemia 1 (5/114), 1.75% α-thalassemia 2 (2/114), and 0.88% Hb H disease (1/114). A comparison of our CMA, which was a high resolution SNP array, and traditional single-tube multiplex PCR results indicated that CMA could accurately detect HBA gene deletion. In previous studies, oligonucleotide microarrays with custom design probes were successfully used to detect both α-thalassemia and β-thalassemia70,71,72. Thus, we suggest that a tentative diagnosis of α-thalassemia should be considered in patients who underwent CMA testing because this is important for genetic counseling and prevention of α-thalassemia, particularly in populations where the disease is endemic. Of note, distal deletion of chromosome 16p (16pter-16p13.3) causes alpha-thalassemia-intellectual deficit syndrome linked to chromosome 16 (ATR-16 syndrome, MIM 141750). Patients with ATR-16 syndrome present with either alpha-thalassemia trait or Hb H disease associated with mild to moderate ID, and with dysmorphic features in some cases. Deletion at 16p13.3 includes the loss of multiple genes including HBA1 and HBA2 resulting in alpha thalassemia phenotype, however the genes responsible for ID and other development abnormalities are not yet clearly identified. Some studies have suggested that deletion of SOX8 at chromosome 16p13.3 could contribute to ID in patients with ATR-16 syndrome because of its high expression in the brain73,74. However, our ASD patients, who have alpha thalassemia trait due to 16p13.3 deletions ranging from 3,924–149,072 bp, were unlikely to have ATR-16 syndrome because the 16p13.3 deletions in previously reported patients were much larger (>800 kb)74,75,76 and included many other genes including SOX8.
We also reported an atypical PWS case with ASD caused by a UPD of chromosome 15. Although, the results of the microarray and MS-PCR analyses were consistent with Prader-Willi syndrome (PWS), the clinical features of this patient were milder than in typical PWS patients. Various studies have found that PWS patients with UPDs of 15q11-q13 had higher IQs, milder behavior problems77,78, and more pronounced clinical features of ASD than PWS patients with deletion of 15q11-q1379. Our patient carried two AOH regions at 15q11.1–15q22.2 and 15q26.1-15q26.3. The B-allele frequency (BAF) plot indicated both uniparental isodisomy and heterodisomy regions on chromosome 15, demonstrating results of the recombination process in meiosis (Supplementary Figure 1). From the BAF plot, we can also deduce that the non-disjunction occurred in maternal meiosis II, followed by post-zygotic loss of the paternal chromosome 15 by trisomy rescue80. Due to the many genes located within these two large AOH regions, there is a possibility that in addition to the maternal UPD15 there exists the interplay between multiple genes that may contribute to the atypical phenotype of this patient. However, more investigations are required to further elucidate the underlying pathogenesis.
In conclusion, our study is the first CMA testing in a large cohort of 114 patients with ASD from Thailand, a country which has to date been underrepresented in ASD studies. Our findings support the usefulness of CMA as a routine diagnostic test in patients with ASD of unknown cause. The CMA information can help not only medical management, but also in providing appropriate genetic counseling and evaluation of recurrence risk in families. In addition, we propose that the SERINC2 gene is a novel ASD candidate gene. However, because of the limitation of the number of studies related to SERINC2, further investigations are required to confirm the effect of SERINC2 in patients with ASD.
Methods
Patients
A total of 114 Thai patients with ASD of unknown cause were recruited in the study, 91 males and 23 females, with ages ranging from 1 to 18 years at the time of recruitment. They were divided into two cohorts. The first cohort (Group 1) was first evaluated for the common genetic causes of ASD and all patients, selected from a previous report7, had normal karyotype and negative DNA tests for Fragile X syndrome and MECP2 mutations (defined as “non-syndromic ASD”). Therefore, they represented non-syndromic ASD patients with the use of CMA as a second line test. Group 1 patients comprised 68 retrospective cases with non-syndromic ASD, who met the DSM-IV criteria for ASD including 61 non-syndromic ASD with ID patients (nonverbal IQ < 70) and 7 non-syndromic ASD without ID patients (nonverbal IQ ≥ 70). The second cohort (Group 2) included 46 prospective ASD and DD cases with a clinical diagnosis of ASD based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria. DD is defined as performance at least two standard deviations below the age-appropriate mean and is not quantifiable with IQ testing. Group 2 patients were referred for CMA testing at Ramathibodi Hospital, Bangkok according to the American College of Medical Genetics and Genomics (ACMG) recommendations for the use of CMA as the first-tier diagnostic test in patients with unexplained ASD and DD.
The study was approved by the Institutional Ethics Committee of Faculty of Medicine, Prince of Songkla University (REC 48/364-006-3) and Faculty of Medicine Ramathibodi Hospital, Mahidol University (ID 12-57-03). The methods were carried out in accordance with the approved guidelines and relevant regulations. Written informed consent was obtained from the parents of each ASD individual.
Chromosomal Microarray Analysis
CMA was performed with the Infinium CytoSNP-850K v1.1 Beadchip (Illumina, San Diego, California, USA), according to the manufacturer’s instructions. The array contains approximately 850,000 single nucleotide polymorphisms (SNPs) markers spanning the entire genome with an average probe spacing of 1.8 kb. The data were analyzed using BlueFuse Multi software v4.3 and GenomeStudio Data Analysis Software v. 2011.1 based on the reference human genome (hg19/GRCh37). Microarrays were carried out in available parents of the patients with pathogenic CNVs and VOUS to investigate the inheritance pattern in the families using either the Infinium CytoSNP-850K Beadchip (850,000 SNP markers) or HumanCytoSNP-12 DNA Analysis BeadChip v2.1 kit (~300,000 SNP markers) depending on the size of the interested CNVs.
CNVs interpretation
In this study, only detected CNV with at least 10-probe coverage were reported. The detected CNVs were classified into pathogenic, benign, or variant of uncertain clinical significance (VOUS) according to established guidelines from the International Standard Cytogenomic Array (ISCA) and the ACMG5,81. The classification was based on the size of CNV, gene content, the inheritance pattern, and information in the medical literature and public databases. The Database of Genomic Variants (DGV) and the Thai CNV database (http://www.thaicnv.icbs.mahidol.ac.th/thaicnv/index.php)34, which contains CNVs from 3,017 Thai individuals, were used to exclude common structural variations in the Thai population, while the Online Mendelian Inheritance in Man (OMIM) database was used to identify disease-causing genes. The ISCA and Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) were used as reference for known microdeletion and microduplication syndromes. In addition, three ASD-related databases including the Simons Foundation for Autism Research Initiative (SFARI) (https://gene.sfari.org/autdb), the Autism DataBase (AUTDB) (http://autism.mindspec.org/autdb), and Autism Chromosome Rearrangement Database (ACRD; http://projects.tcag.ca/autism/) were utilized for interpretation of clinical significance.
CNVs overlapping with a region of known microdeletion or microduplication syndromes and/or disease-causing genes were classified as pathogenic. Even though a CNV was inherited from healthy parent, it could still be classified as pathogenic if incomplete penetrance was commonly observed. CNVs with insufficient information to determine whether they were pathogenic or benign were classified as VOUS. VOUS were further divided into likely pathogenic, likely benign and VOUS (no subclassification). CNVs were considered to be possibly pathogenic when they were de novo or inherited from an affected parent, and included genes associated with ID, ASD or neurological development or function. CNVs were considered to be possibly benign when they were inherited from a healthy parent and did not include genes known to be associated with a specific disease. CNVs were still considered to be VOUS with no subclassification when they contained genes with no or less known functions and had, to date, been considered as of unknown clinical significance in multiple publications and/or databases. CNVs that had been reported as common polymorphisms occurring in more than 1% of the population, and previously reported in multiple publications and normal population databases, were classified as benign and excluded from further analysis. Additionally, AOH regions larger than 10 Mb in size were also reported.
Methylation-specific PCR (MS-PCR) for uniparental disomy detection
DNA samples were treated with sodium bisulfite, followed by SNRPN methylation specific PCR (MS-PCR). MS-PCR was performed with original primer sets designed by Kubota et al.37 and alternative primer sets designed by Hussain Askree et al.36. Untreated DNA was used as a control to ensure complete sodium bisulfate conversion of DNA, and amplified using primer set as previously described36. Subsequently, the PCR products were run on 2.5% agarose gel and visualized through ethidium bromide staining under a UV transilluminator.
Statistical analysis
All statistical comparisons were made by Chi-square test using the online GraphPad QuickCalcs software (https://graphpad.com/quickcalcs/contingency1.cfm). An alpha level of 0.05 was used for all statistical analyses.
References
Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summa. 65, 1–23 (2016).
Poolsuppasit, S., Panyayong, B., Liknapichitkul, D., Serisathien, P. & Chutha, W. Holistic Care for Thai Autism. J Ment Health Thai. 13, 10–16 (2005).
Huguet, G., Ey, E. & Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 14, 191–213 (2013).
Battaglia, A. et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 17, 589–599 (2013).
Miller, D. T. et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 86, 749–764 (2010).
Rauch, A. et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A. 140, 2063–2074 (2006).
Charalsawadi, C. et al. A case with a ring chromosome 13 in a cohort of 203 children with non-syndromic autism and review of the cytogenetic literature. Cytogenet Genome Res. 144, 1–8 (2014).
Baldwin, E. L. et al. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med. 10, 415–429 (2008).
Bremer, A. et al. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. 156, 115–124 (2011).
Coulter, M. E. et al. Chromosomal microarray testing influences medical management. Genet Med. 13, 770–776 (2011).
Ellison, J. W. et al. Clinical Utility of Chromosomal Microarray Analysis. Pediatrics. 130, e1085–e1095 (2012).
Eriksson, M. A. et al. Rare copy number variants are common in young children with autism spectrum disorder. Acta Paediatr. 104, 610–618 (2015).
Filges, I. et al. High resolution array in the clinical approach to chromosomal phenotypes. Gene. 495, 163–169 (2012).
Henderson, L. B. et al. The impact of chromosomal microarray on clinical management: a retrospective analysis. Genet Med. 16, 657–664 (2014).
Iourov, I. Y. et al. Molecular karyotyping by array CGH in a Russian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogenet. 5, 46 (2012).
Moreiraa, E. S. et al. Detection of small copy number variations (CNVs) in autism spectrum disorder (ASD) by custom array comparative genomic hybridization (aCGH). Research in Autism Spectrum Disorders. 23, 145–151 (2016).
Nava, C. et al. Prospective diagnostic analysis of copy number variants using SNP microarrays in individuals with autism spectrum disorders. Eur J Hum Genet. 22, 71–78 (2014).
Nicholl, J. et al. Cognitive deficit and autism spectrum disorders: prospective diagnosis by array CGH. Pathology. 46, 41–45 (2014).
Oikonomakis, V. et al. Recurrent copy number variations as risk factors for autism spectrum disorders: analysis of the clinical implications. Clin Genet. 89, 708–718 (2016).
Riggs, E. R. et al. Chromosomal microarray impacts clinical management. Clin Genet. 85, 147–153 (2014).
Roberts, J. L., Hovanes, K., Dasouki, M., Manzardo, A. M. & Butler, M. G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene. 535, 70–78 (2014).
Rosenfeld, J. A. et al. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med. 12, 694–702 (2010).
Shen, Y. et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 125, e727–735 (2010).
Sorte, H. S., Gjevik, E., Sponheim, E., Eiklid, K. L. & Rodningen, O. K. Copy number variation findings among 50 children and adolescents with autism spectrum disorder. Psychiatr Genet. 23, 61–69 (2013).
Stobbe, G. et al. Diagnostic yield of array comparative genomic hybridization in adults with autism spectrum disorders. Genet Med. 16, 70–77 (2014).
Xu, Q. et al. Chromosomal microarray analysis in clinical evaluation of neurodevelopmental disorders-reporting a novel deletion of SETDB1 and illustration of counseling challenge. Pediatr Res. 80, 371–381 (2016).
Al-Mamari, W. et al. Diagnostic Yield of Chromosomal Microarray Analysis in a Cohort of Patients with Autism Spectrum Disorders from a Highly Consanguineous Population. J Autism Dev Disord. 45, 2323–2328 (2015).
Chong, W. W. et al. Performance of chromosomal microarray for patients with intellectual disabilities/developmental delay, autism, and multiple congenital anomalies in a Chinese cohort. Mol Cytogenet. 7, 34 (2014).
Shin, S., Yu, N., Choi, J. R., Jeong, S. & Lee, K. A. Routine chromosomal microarray analysis is necessary in Korean patients with unexplained developmental delay/mental retardation/autism spectrum disorder. Ann Lab Med. 35, 510–518 (2015).
Siu, W. K. et al. Diagnostic yield of array CGH in patients with autism spectrum disorder in Hong Kong. Clin Transl Med. 5, 18 (2016).
Soueid, J. et al. RYR2, PTDSS1 and AREG genes are implicated in a Lebanese population-based study of copy number variation in autism. Sci Rep. 6, 19088 (2016).
Tao, V. Q. et al. The clinical impact of chromosomal microarray on paediatric care in Hong Kong. PLoS One. 9, e109629 (2014).
Gazzellone, M. J. et al. Copy number variation in Han Chinese individuals with autism spectrum disorder. J Neurodev Disord. 6, 34 (2014).
Suktitipat, B. et al. Copy number variation in Thai population. PLoS One. 9, e104355 (2014).
Chong, S. S., Boehm, C. D., Higgs, D. R. & Cutting, G. R. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 95, 360–362 (2000).
Hussain Askree, S. et al. Allelic dropout can cause false-positive results for Prader-Willi and Angelman syndrome testing. J Mol Diagn. 13, 108–112 (2011).
Kubota, T. et al. Methylation-specific PCR simplifies imprinting analysis. Nat Genet. 16, 16–17 (1997).
Hannes, F. D. et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 46, 223–232 (2009).
Brownstein, C. A. et al. Overlapping 16p13.11 deletion and gain of copies variations associated with childhood onset psychosis include genes with mechanistic implications for autism associated pathways: Two case reports. Am J Med Genet A. 170a, 1165–1173 (2016).
Ramalingam, A. et al. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 56, 541–544 (2011).
Ullmann, R. et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 28, 674–682 (2007).
Ingason, A. et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry. 16, 17–25 (2011).
Brunetti-Pierri, N. et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 40, 1466–1471 (2008).
Mefford, H. C. et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 359, 1685–1699 (2008).
Bernier, R. et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med. 18, 341–349 (2016).
Ben-Shachar, S. et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 46, 382–388 (2009).
van Bon, B. W. et al. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 46, 511–523 (2009).
Girirajan, S. et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 42, 203–209 (2010).
Marshall, C. R. et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 82, 477–488 (2008).
Li, C., Liu, C., Zhou, B., Hu, C. & Xu, X. Novel microduplication of CHL1 gene in a patient with autism spectrum disorder: a case report and a brief literature review. Mol Cytogenet. 9, 51 (2016).
Shoukier, M. et al. Microduplication of 3p26.3 in nonsyndromic intellectual disability indicates an important role of CHL1 for normal cognitive function. Neuropediatrics. 44, 268–271 (2013).
Ren, C. M. et al. Balanced translocation t(3;18)(p13;q22.3) and points mutation in the ZNF407 gene detected in patients with both moderate non-syndromic intellectual disability and autism. Biochim Biophys Acta. 1832, 431–438 (2013).
Teufel, M. et al. Sequence identification and characterization of human carnosinase and a closely related non-specific dipeptidase. J Biol Chem. 278, 6521–6531 (2003).
Wassif, W. S. et al. Serum carnosinase activities in central nervous system disorders. Clin Chim Acta. 225, 57–64 (1994).
Cohen, M. et al. Serum carnosinase deficiency: a non-disabling phenotype? J Ment Defic Res. 29(Pt 4), 383–389 (1985).
Murphey, W. H. et al. Serum carnosinase deficiency concomitant with mental retardation. Pediatr Res. 7, 601–606 (1973).
van Heeswijk, P. J., Trijbels, J. M., Schretlen, E. D., van Munster, P. J. & Monnens, L. A. A patient with a deficiency of serum-carnosinase activity. Acta Paediatr Scand. 58, 584–592 (1969).
Shimamoto, C. et al. Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies. Hum Mol Genet. 23, 6495–6511 (2014).
Gorokhova, S., Bibert, S., Geering, K. & Heintz, N. A novel family of transmembrane proteins interacting with beta subunits of the Na,K-ATPase. Hum Mol Genet. 16, 2394–2410 (2007).
Inuzuka, M., Hayakawa, M. & Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem. 280, 35776–35783 (2005).
Guirland, C., Suzuki, S., Kojima, M., Lu, B. & Zheng, J. Q. Lipid rafts mediate chemotropic guidance of nerve growth cones. Neuron. 42, 51–62 (2004).
Nguyen, L. S. et al. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet. 22, 1816–1825 (2013).
Zuo, L. et al. Rare SERINC2 variants are specific for alcohol dependence in individuals of European descent. Pharmacogenet Genomics. 23, 395–402 (2013).
Zuo, L. et al. NKAIN1-SERINC2 is a functional, replicable and genome-wide significant risk gene region specific for alcohol dependence in subjects of European descent. Drug Alcohol Depend. 129, 254–264 (2013).
Klimkiewicz, A., Klimkiewicz, J., Jakubczyk, A., Kieres-Salomonski, I. & Wojnar, M. Comorbidity of alcohol dependence with other psychiatric disorders. Part I. Epidemiology of dual diagnosis. Psychiatr Pol. 49, 265–275 (2015).
Regier, D. A. et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 264, 2511–2518 (1990).
Miles, J. H., Takahashi, T. N., Haber, A. & Hadden, L. Autism families with a high incidence of alcoholism. J Autism Dev Disord. 33, 403–415 (2003).
Schumann, G. et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci USA 108, 7119–7124 (2011).
Fucharoen, S. & Winichagoon, P. Hemoglobinopathies in Southeast Asia. Hemoglobin. 11, 65–88 (1987).
Bang-Ce, Y., Hongqiong, L., Zhuanfong, Z., Zhengsong, L. & Jianling, G. Simultaneous detection of alpha-thalassemia and beta-thalassemia by oligonucleotide microarray. Haematologica. 89, 1010–1012 (2004).
Chan, K., Wong, M. S., Chan, T. K. & Chan, V. A thalassaemia array for Southeast Asia. Br J Haematol. 124, 232–239 (2004).
Zesong, L., Ruijun, G. & Wen, Z. Rapid detection of deletional alpha-thalassemia by an oligonucleotide microarray. Am J Hematol. 80, 306–308 (2005).
Gibson, W. T. et al. Phenotype-genotype characterization of alpha-thalassemia mental retardation syndrome due to isolated monosomy of 16p13.3. Am J Med Genet A. 146a, 225–232 (2008).
Pfeifer, D., Poulat, F., Holinski-Feder, E., Kooy, F. & Scherer, G. The SOX8 gene is located within 700 kb of the tip of chromosome 16p and is deleted in a patient with ATR-16 syndrome. Genomics. 63, 108–116 (2000).
Gibbons, R. J. alpha-Thalassemia, mental retardation, and myelodysplastic syndrome. Cold Spring Harb Perspect Med. 2, a011759 (2012).
Harteveld, C. L. et al. Refinement of the genetic cause of ATR-16. Hum Genet. 122, 283–292 (2007).
Roof, E. et al. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. 44(Pt 1), 25–30 (2000).
Torrado, M. et al. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A. 143a, 460–468 (2007).
Veltman, M. W. et al. Prader-Willi syndrome–a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry. 13, 42–50 (2004).
Jinawath, N. et al. Mosaic trisomy 13: understanding origin using SNP array. J Med Genet. 48, 323–326 (2011).
Kearney, H. M., Thorland, E. C., Brown, K. K., Quintero-Rivera, F. & South, S. T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 13, 680–685 (2011).
Boudry-Labis, E. et al. A novel microdeletion syndrome at 9q21.13 characterised by mental retardation, speech delay, epilepsy and characteristic facial features. Eur J Med Genet. 56, 163–170 (2013).
Achsel, T., Ahrens, K., Brahms, H., Teigelkamp, S. & Luhrmann, R. The human U5-220kD protein (hPrp8) forms a stable RNA-free complex with several U5-specific proteins, including an RNA unwindase, a homologue of ribosomal elongation factor EF-2, and a novel WD-40 protein. Mol Cell Biol. 18, 6756–6766 (1998).
Jerng, H. H., Qian, Y. & Pfaffinger, P. J. Modulation of Kv4.2 channel expression and gating by dipeptidyl peptidase 10 (DPP10). Biophys J. 87, 2380–2396 (2004).
Frints, S. G. et al. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet. 12, 1463–1474 (2003).
Leshchyns’ka, I. et al. The adhesion molecule CHL1 regulates uncoating of clathrin-coated synaptic vesicles. Neuron. 52, 1011–1025 (2006).
Acknowledgements
We would like to thank the patients and their families who participated in the study. We also thank Prof. Dr. Wasun Chantratita for providing the in-house Thai WES database. This work was supported by grants from the Faculty of Medicine, Prince of Songkla University (48/364-006-3) and the National Center for Genetic Engineering and Biotechnology (BT-B-01-MG-18-4814) (P.L.), the Development Potentials of Thai people Project, Faculty of Medicine Ramathibodi Hospital, Mahidol University (ID 12-57-03) and the Crown Property Bureau Foundation (N.J.). T.T., D.W., and N.J. are recipients of the Research Career Development Awards from the Faculty of Medicine at Ramathibodi Hospital.
Author information
Authors and Affiliations
Contributions
A.H. performed the experiments, analyzed data and co-wrote the manuscript. W.T. performed the experiments. T.T., K.R., T.H., T.S., R.R., J.W., J.C., D.W. and N.R. performed the clinical diagnoses for samples recruitment. S.F. performed experiments for confirmation of α-thalassemia. P.L. and N.J. supervised the project, designed the study, obtained funding, analyzed data and co-wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hnoonual, A., Thammachote, W., Tim-Aroon, T. et al. Chromosomal microarray analysis in a cohort of underrepresented population identifies SERINC2 as a novel candidate gene for autism spectrum disorder. Sci Rep 7, 12096 (2017). https://doi.org/10.1038/s41598-017-12317-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12317-3
This article is cited by
-
Parental Perspectives on Early Life Screening and Genetic Testing for ASD: A Systematic Review
Journal of Autism and Developmental Disorders (2024)
-
Effects of stressful life-events on DNA methylation in panic disorder and major depressive disorder
Clinical Epigenetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
