
Abstract
Tartary buckwheat seeds are rich in various nutrients, such as storage proteins, starch, and flavonoids. To get a good knowledge of the transcriptome dynamics and gene regulatory mechanism during the process of seed development and nutrients accumulation, we performed a comprehensive global transcriptome analysis using rice tartary buckwheat seeds at different development stages, namely pre-filling stage, filling stage, and mature stage. 24 819 expressed genes, including 108 specifically expressed genes, and 11 676 differentially expressed genes (DEGs) were identified. qRT-PCR analysis was performed on 34 DEGs to validate the transcriptome data, and a good consistence was obtained. Based on their expression patterns, the identified DEGs were classified to eight clusters, and the enriched GO items in each cluster were analyzed. In addition, 633 DEGs related to plant hormones were identified. Furthermore, genes in the biosynthesis pathway of nutrients accumulation were analyzed, including 10, 20, and 23 DEGs corresponding to the biosynthesis of seed storage proteins, flavonoids, and starch, respectively. This is the first transcriptome analysis during seed development of tartary buckwheat. It would provide us a comprehensive understanding of the complex transcriptome dynamics during seed development and gene regulatory mechanism of nutrients accumulation.
Similar content being viewed by others

Introduction
Seed is the primary storage organ in plants for storing nutrients such as starch, lipids, and proteins1. Therefore, it is of great importance for unraveling the mechanism and regulatory networks during seed development. Seed development in higher plants is a highly complex process. It begins from the double fertilization in ovules, goes through the differentiation and development of various tissues and organs, and finally forms a mature seed2.
The processes of seed development and nutrients accumulation are dependent on the expression and regulation of massive expressed genes. Studying the expression patterns of these genes will greatly help us to understand the molecular mechanism of the accumulation of various nutrients during seed development. Transcriptome analyses in seed development of Arabidopsis 2, rice1, wheat3, soybean4, Brassica napus 5, and barley6 have identified large amounts of related regulatory and functional genes. These genes include transcription factors (TFs)1,2,4,5,6, hormone related genes1, genes encoding seed storage proteins (SSPs)4,5, genes involved in lipid metabolism5, and genes involved in carbohydrate metabolism5.
Tartary buckwheat (Fagopyrum tataricum) is an annual dicotyledonous crop of Fagopyrum Mill. As its origin center, China has the largest planting area and highest yield of tartary buckwheat7. Especially in the poor mountain areas of the west of China, tartary buckwheat has gradually become the primary food and economical crop8. Tartary buckwheat seed is rich in the primary nutrients, such as starch, fatty acid (linoleic acid), and seed proteins8,9,10. Besides, compared to common buckwheat and cereal crops, tartary buckwheat is richer in the flavonoids (such as rutin and quercetin), dietary fiber, vitamins, chiro-inositol, minerals, and is more balanced in the amino acid composition11,12,13. Therefore, tartary buckwheat is recognized as a particular medicinal and edible plant. Its pharmacological efficiency has been studied massively, including protecting hepatic cells against high glucose-induced oxidative stress and insulin resistance14, antioxidant activity15, protective effects on high TMAO diet-induced vascular dysfunction and liver injury16, inducing G2/M cell cycle arrest and apoptosis17.
Massive researches are focused on the contents of various nutrients in tartary buckwheat seeds and their pharmacological efficiency. However, little is known on the transcriptome dynamics and gene regulatory network during seed development of tartary buckwheat. Rice tartary buckwheat is a special type of tartary buckwheat that is with thin shell and easily dehulled. Here we performed a comprehensive global transcriptome analysis using rice tartary buckwheat seeds at different development stages, namely pre-filling stage (PS), filling stage (FS), and mature stage (MS). The expression patterns of genes related to various aspects during seed development were identified. This study would provide a comprehensive understanding of the molecular mechanism of the accumulation of nutrients during seed development at the transcriptional level.
Results
Seed development stages and sampling
Tartary buckwheat seeds at 12 developmental stages from their emergence to maturation were selected, and the seed coats were detached. The morphology of the seeds and their longitudinal sections were shown on Fig. 1. At the first three stages, no obvious change was found on their surface, but the embryo gradually developed, from just appearance at the tip of seed (stage 1), to half-length of seed (stage 2), finally growing to full length of seed (stage 3) (embryo was marked by arrow at each stage). Thus, these three stages were named as embryo developing stage or pre-filling stage. From stage 4 to stage 8, seeds plumped up little by little, and the contents of seeds increased as well. Most of the nutrients accumulated at these stages, and they were named as filling stage. From the stage 9 to 12, there seemed no big difference on the longitudinal sections, however, seeds become stronger. At the stage 12, the color of seed coat turned into yellow (figure not shown), which means seeds were maturated completely. The stages from 9 to 12 were named as maturation stage. Next, we chose seeds at these stages for RNA-Seq. Seeds at stage 1 to stage 3 were taken as samples for pre-filling stage (PS). Considering gene expression was usually prior to the nutrients actually accumulation, we just took seeds at stage 4 to stage 6 as samples for filling stage (FS). As seeds have been completely mature and the RNA was degraded at stage 12 (figure not shown), we took seeds at stage 9 to stage 11 as samples for maturation stage (MS). Thus, samples for RNA-Seq were determined.

The morphology and their longitudinal sections of tartary buckwheat seeds at different developmental stages. Arrow indicated the embryo in seed.
High-throughput RNA-Seq and global analysis of gene expression
To have a global view of the seed development at transcriptional level, high-throughput RNA-Seq was performed on RNAs from three seed developmental stages (PS, FS, MS). Three biological replicates at each stage were used for the analysis. Pearson’s rank correlation analysis was performed to evaluate the reproducibility among biological replicates. As shown in Table S1, the correlations between samples among the same biological replicates were good, with a value ranging from 0.983 to 1.000. However, the correlations between samples out of the biological replicates were various, with a value ranging from 0.136 to 0.905. These illustrate that our results were with high reliability and reproducibility of the biological replicates and the sequence method.
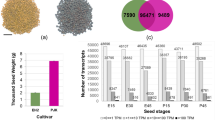
After high-throughput RNA-Seq, we obtained 66 107 888 to 81 404 646 raw reads and 62 033 722 to 75 373 688 clean reads for each library. The clean reads were subsequently mapped to the reference genome data of tartary buckwheat. 73.78% to 81.52% clean reads were mapped to the genome and 67.70% to 71.17% clean reads were mapped to the predicted coding sequences of the genome. 24 819 genes were identified in total, with 23 122, 23 067, 23 077, 22 513, 22 550, 22 327, 21 884, 21 772, and 21 692 genes in the libraries of PS1, PS2, PS3, FS1, FS2, FS3, MS1, MS2, and MS3, respectively. Novel transcripts that not included in the reference genes were also identified, ranging from 2 085 to 2 696 for each library (Table 1).
We performed a principle component analysis (PCA) of all samples during seed development. Consistent with their distinct developmental stages, samples from different biological replicates were clustered separately (Fig. 2a). Samples at FS showed closer to PS, rather than MS. This was likely due to only seeds at the early filling stages were selected, and those at later filling stages were ignored. The cluster dendrogram by hclust showed similar results as the PCA analysis (Fig. S1).

Global analysis of gene expression. (a) Principle component analysis (PCA) of all samples during seed development. Samples 1 to 9 represent PS1, PS2, PS3, FS1, FS2, FS3, MS1, MS2, and MS3, respectively. (b) Biological process of the stage specifically expressed genes based on GO annotation.
In addition, we compared the identified genes at different seed developmental stages. An interesting finding was that 108 genes were specifically expressed at MS (Table S2). Of these, only 26 genes (24%) were successfully annotated to at least one item of GO functions. Based on the sub-categories in biological process, these genes were classified to 9 sub-categories, including response to stimulus (7), metabolic process (6), cellular process (3), multicellular organismal process (2), developmental process (2), localization (2), establishment of localization (2), reproduction (1), and reproductive process (1) (Fig. 2b).
Differentially expressed genes (DEGs) during seed development
Using NOISeq, a total of 11 676 DEGs were identified at three stages of seed development (Table S3), among which 2 745, 5 876, and 11 163 DEGs were identified in PS-vs-FS, FS-vs-MS, and PS-vs-MS, respectively (Fig. 3a). Of these DEGs, 1 105 were differentially regulated in all the comparisons of stages with each other, whereas 112, 1 398, and 4 388 were differentially regulated in the comparisons of PS-vs-FS and FS-vs-MS, PS-vs-FS and PS-vs-MS, and FS-vs-MS and PS-vs-MS, respectively. Remaining DEGs were differentially regulated in the comparisons of two stages (Fig. 3b). Besides, qRT-PCR was performed on 34 DEGs that annotated to SSPs biosynthesis (five DEGs), flavonoids biosynthesis (18 DEGs), and hormone biosynthesis and signal pathway (11 DEGs) to validate the transcriptome data. The expression patterns obtained by qRT-PCR were well consistent with those obtained by transcriptome analysis, with a Pearson correlation coefficient of 0.83 (P < 0.0001) (Fig. 3c). In addition, we performed an additional analysis to identify DEGs during seed development of tartary buckwheat, using edgeR. We made a comparison of the results obtained by NOISeq and that obtained by edgeR. We get a good consistency of two methods, with the Pearson correlation coefficient of 0.95, 0.95, and 0.97 in PS-vs-FS, FS-vs-MS, and PS-vs-MS, respectively (Fig. S2).

Differentially expressed genes (DEGs) during seed development. (a) Statistics of DEGs in PS-vs-FS, FS-vs-MS, and PS-vs-MS, respectively. (b) Venn diagram of DEGs in PS-vs-FS, FS-vs-MS, and PS-vs-MS, respectively. (c) Validation of the transcriptome data by qRT-PCR.
Hierarchical clustering of all 11 676 DEGs were performed, using the Pearson correlation method associated with average linkage clustering. As a result, eight clusters were identified, namely C1–C8 (Fig. 4a,b). Similarly, they could be divided into two major groups based on the expression patterns. The first group contained C1 to C5, genes in which showed up-regulation patterns. The second group contained C6 to C8, genes in which showed down-regulation patterns. In detail, C1, C2 and C3 were up-regulated at FS, followed by a down-regulation at MS. C4 were up-regulated at MS compared to PS and FS. C5 showed a gradually up-regulation with the seed developmental stage going. C6 were down-regulated at FS and MS compared to PS. C7 and C8 were gradually down-regulated along with seed development. Based on GO annotation, the biological processes at each cluster were analyzed, and 59 significantly enriched biological processes were identified in total, with the FDR ≤ 0.05 (Table S4; Fig. 4c). Among all clusters, C7 (40) and C2 (15) contained most number of enriched biological processes, whereas C1 did not contain any enriched biological process. Generally, numbers of sugar and carbohydrate metabolic and catabolic processes were enriched in C2 and C3, including the metabolic and catabolic processes of hexose, monosaccharide, glucose, and carbohydrate. Fatty acid metabolic process was enriched in C2 as well. Genes in C2 and C3 were higher expressed at FS, suggesting nutrients were massively accumulated at this stage. Genes in C7 and C8 were mostly linked to the various biological processes of biosynthesis and organization, including RNA, ATP, purine nucleotide, and nucleotide biosynthetic processes, ribonucleoprotein complex biogenesis, and cellular component, chromosome, macromolecular complex subunit, cellular macromolecular complex, subunit, organelle, and nucleosome organization. All of these are essential for cell maintenance and growth, but were down regulated during seed development, suggesting maturation was the last step of a seed’s life.

Hierarchical clustering analysis of differentially expressed genes (DEGs) during seed development. (a) Hierarchical cluster of the identified DEGs. Eight main clusters were presented as C1 – C8. (b) Expression profile of eight clusters correspondance to the Hierarchical cluster result. (c) Biological processes of the DEGs significantly enriched in eight clusters, based on GO annotation. The significance was presented as −Log2 transformed P-value. Missing GO-slim was represented by grey color.
To further understand the metabolic pathways in which the DEGs involved, KEGG pathways were identified (Table S5). The significantly enriched pathways for PS-vs-FS and FS-vs-MS were shown on Table 2. For PS-vs-FS, 1 668 DEGs were successfully annotated to 126 pathways, among which 5 typical nutrients related pathways were significantly enriched (Qvalue < 0.05), including photosynthesis - antenna proteins, photosynthesis, carbon fixation in photosynthetic organisms, cysteine and methionine metabolism, and flavone and flavonol biosynthesis. For FS-vs-MS, 4 013 DEGs were successfully annotated to 133 pathways, among which 10 pathways were significantly enriched (Qvalue < 0.05), including ribosome, proteasome, spliceosome, RNA transport, DNA replication, ribosome biogenesis in eukaryotes, aminoacyl-tRNA biosynthesis, protein processing in endoplasmic reticulum, RNA degradation, and citrate cycle (TCA cycle). Genes in these pathways played important roles on definite processes at stages of seed development.
Phytohormones that regulated during seed development
Phytohormones are reported to be important signals in controlling seed development, maturation, and nutrients accumulation1. Therefore, we identified the hormone related DEGs. As a result, 633 DEGs were annotated to genes related to eight major hormones, including abscisic acid (ABA, 149), auxin (AUX, 132), ethylene (ET, 130), salicylic acid (SA, 67), brassinosteroid (BR, 59), cytokinin (CK, 33), jasmonic acid (JA, 33), and gibberellin (GA, 30). These genes were linked to various aspects of plant hormone homeostasis, including biosynthesis (60), metabolism (25), receptor (95), signal transduction (421) and transportion (32) (Table S6, Table 3). Numbers of them that have been found in the complex regulation network of seed development were presented on Fig. 5. Genes involving the pathways of ABA, AUX, ET, SA, BR, CK, JA, and GA could be clustered to two groups dependent on their presented expression patterns. One group was linked to genes that showed up-regulation trends during seed development, whereas the other group was linked to genes that showed down-regulation trends during seed development. In addition, the expression patterns of 11 important hormone related DEGs were verified by qRT-PCR (Fig. 6). These included 7, 1, 1, 1, and 1 DEGs in the biosynthesis and signal pathway of ABA, ET, CK, JA, and GA, respectively. Their expression patterns obtained by qRT-PCR were similar to that obtained by RNA-Seq during seed development, with the exception of sample1_00026062-RA. These differentially expressed hormone related genes suggest that the specific process of seed development of tartary buckwheat might be governed by the regulation of complex phytohormone signal pathways.

Hierarchical cluster of the hormone related differentially expressed genes (DEGs). ABA, abscisic acid; AUX, auxin; ET, ethylene; SA, salicylic acid; BR, brassinosteroid; CK, cytokinin; JA, jasmonic acid; GA, gibberellin.

qRT-PCR confirmation of 11 hormone related differentially expressed genes (DEGs).
Expression patterns of the SSP biosynthesis genes during seed development
Tartary buckwheat is famous for its abundant nutrients, including protein, starch, and flavonoids8,10,18. Therefore, characterization and analysis of their biosynthesis genes is of great importance. Among the four types of SSPs (globulin, albumin, glutelin, and prolamin), we identified 10 globulin encoding transcripts that were differentially expressed during seed development, including seven 13 S globulin encoding DEGs and three 7 S globulin-like protein encoding DEGs (Fig. 7). Interestingly, all of the seven 13 S globulin encoding DEGs exhibited up-regulation patterns with seed development, whereas three 7 S globulin-like encoding DEGs exhibited down-regulation patterns with seed development. In addition, of the seven 13 S globulin encoding genes, four were annotated to the existing 13 S globulin sequences in common buckwheat (sample1_00022718-RA, sample1_00021677-RA, sample1_00021674-RA, and sample1_00021668-RA), and two were annotated to the existing 13 S globulin sequences in tartary buckwheat (sample1_00013128-RA and sample1_00013130-RA). This indicates that most of the important genes for the accumulation of 13 S globulin during seed development have been found. Except for globulin, genes encoding albumin, glutelin, and prolamin were not significantly changed during seed development in our data.

Hierarchical cluster showing the expression patterns of seed storage protein encoding genes that were differentially expressed during seed development.
DEGs involved in the biosynthesis of flavonoid during seed development
Tartary buckwheat is rich in flavonoids, including rutin and other flavonoids12,13,18. We characterized 20 DEGs that related to flavonoid biosynthesis pathway19, including five chalcone synthase (CHS), one chalcone isomerase (CHI), two flavones 3-hydroxylase (F3H), one flavonoid 3′-hydroxylase (F3′H), two flavonol synthase (FLS), four dihydroflavonol-4-reductase (DFR), two UDP-glycose: glycosyltransferase (UGT), and three leucoanthocyantin reductase (LAR) (Fig. 8). Five genes encoding CHS were differentially expressed during seed development. Among them, the expression of sample1_00002939-RA and sample1_00002940-RA were increased at FS, followed by a decrease at MS; sample1_00006854-RA was up-regulated along with seed development; whereas sample1_00021115-RA and sample1_00016770-RA were gradually down-regulated along with seed development. Sample1_00012486-RA, orthologous to CHI, was up-regulated at FS and followed by a down-regulation at MS. Two genes encoding F3H exhibited two different expression patterns: sample1_00003908-RA was up-regulated at FS but later down-regulated at MS, whereas sample1_00021254-RA was down-regulated with seed development. One F3′H, sample1_00012969-RA, whose expression level was firstly increased at FS but later decreased at MS, was also identified. Two genes encoding FLS showed two different expression patterns: sample1_00013849-RA was up-regulated at FS but later down-regulated at MS, whereas sample1_00013850-RA was down-regulated with seed development. Four genes encoding DFR were identified (sample1_00019968-RA, sample1_00009801-RA, sample1_00009055-RA, and sample1_00019963-RA), with similar expression trend that down-regulated with seed development. Two UGT encoding genes were also identified (sample1_00011838-RA and sample1_00011837-RA), with up-regulation pattern during seed development. In addition, we identified three LAR genes and they showed two expression patterns: sample1_00005844-RA was up-regulated at FS but down-regulated at MS; sample1_00017445-RA and sample1_00013708-RA had higher expression level at PS and FS, but were down-regulated at MS. Overall, most of DEGs in flavonoid biosynthesis pathway showed a higher expression at PS and MS, suggesting the biosynthesis of tartary buckwheat flavonoids occurred before seed maturation.

Expression patterns of flavonoid biosynthesis genes that were differentially expressed during seed development.
Identification of starch biosynthesis genes differentially regulated during seed development
Starch accumulation is an essential process for seed development. We identified 23 DEGs involved in the starch biosynthesis pathway, including sucrose synthase (SUS), UDP glucose pyrophosphorylase (UGPase), ADP glucose pyrophosphorylase (AGPase), granule bound starch synthase (GBSS), starch synthase (SS), starch-branching enzyme (BE), and debranching enzyme (DBE) (Fig. 9). Three SUS genes were differentially expressed, of which, sample1_00016979-RA was up-regulated at FS comparing to PS, and followed by a down-regulation at MS; sample1_00019718-RA was up-regulated with seed development stages; sample1_00015597-RA was down-regulated with seed development stages. Two UGPase genes, sample1_00027519-RA and sample1_00011476-RA, were up-regulated at FS but followed by a down-regulation at MS. Five AGPase encoding genes showed three different expression patterns: sample1_00019415-RA and sample1_00012299-RA were down-regulated with seed development stage going; sample1_00024766-RA was up-regulated with seed development stage going; whereas sample1_00020580-RA and sample1_00008018-RA were firstly up-regulated at FS but down-regulated at MS. Six GBSS genes also showed three different expression patterns: sample1_00010052-RA and sample1_00015883-RA were down-regulated with seed development stage going; sample1_00007331-RA and sample1_00014869-RA were up-regulated with seed development stage going; whereas sample1_00021989-RA and sample1_00025030-RA were firstly up-regulated at FS but down-regulated at MS. Two SS genes, sample1_00002556-RA and sample1_00009846-RA, were down-regulated with seed development. Two BE genes, sample1_00025798-RA and sample1_00010572-RA were up-regulated at FS but down-regulated at MS. At last, three DBEs showed definitely opposite expression patterns: sample1_00017386-RA and sample1_00000397-RA were down-regulated with seed development whereas sample1_00001532-RA was up-regulated with seed development.

Expression patterns of starch biosynthesis genes that were differentially expressed during seed development.
Discussion
Identification of stage specifically expressed genes
Based on our analysis, 108 specifically expressed genes involved in 9 sub-categories of GO biological process were identified at MS (Table S2; Fig. 2b). Some of them were annotated to the well-defined genes whose functions have been characterized. For example, fagopyritols are accumulated in the embryos of mature seeds in F. esculentum 20, and three genes for their biosynthesis have been identified, namely fagopyritol synthase 1 (FeGolS-1), FeGolS-2, and FeGolS-3 21. In our result, sample1_00011242-RA, annotated to FeGolS-2 of F. esculentum, was specifically expressed at MS. This suggests fagopyritols in tartary buckwheat are also accumulated in the embryos of mature seeds, similar to that in common buckwheat. One SSP encoding gene, sample1_00013130-RA, was specifically expressed at MS. It encodes a 13 S globulin in tartary buckwheat, suggesting an accumulation of 13 S globulin at MS of seeds. Function of MADS-box TFs in seed development have been found in several species, including Arabidopsis 22, rice23, and tomato24. We identified a MADS-box gene (sample1_00005419-RA) that specifically expressed at MS, suggesting it has also a role in seed maturation of tartary buckwheat. In addition, two specifically expressed genes were related to plant hormone biosynthesis (sample1_00006058-RA and sample1_00009789-RA). Sample1_00006058-RA was homologous to gibberellin 20-oxidase. Gibberellin 20-oxidase catalyze C20-GA substrates to C19-GA products, and is necessary for the formation of bioactive GA25. Allene oxide synthase (AOS) is important in the JA biosynthesis pathway26. One homolog of AOS, sample1_00009789-RA, was also specifically expressed at MS. Furthermore, some specifically expressed genes were related to plant defense responses, including three genes encoding cytochrome P45027 (sample1_00019807-RA, sample1_00017522-RA, and sample1_00026585-RA), three genes encoding heat shock protein28 (sample1_00015507-RA, sample1_00019961-RA, and sample1_00021423-RA), and one gene belonging to HSF TF family29 (sample1_00018030-RA).
Cluster of DEGs during seed development
Base on the expression patterns of DEGs, we clustered them to eight clusters, namely C1 – C8 (Fig. 4a,b). They could be roughly divided to two groups: the up-regulation group (C1 to C5) and the down-regulation group (C6 to C8). The number of DEGs in the down-regulation group was several times more than that in the up-regulation group, suggesting more inactive events are occurred in the progress of seed development than the active events. This is further elucidated by the enriched biological processes based on GO annotation (Table S4; Fig. 4c). Massive genes linked to cell maintenance and organization were included in the down-regulation group. All of these were essential for cell stabilization and growth, but were down regulated during seed development, suggesting although the last step morphologically of a seed’s life is maturation and desiccation, their corresponding molecular events occur far earlier, which begin at the completion of embryo development (PS). The decrease of genes related to cell maintenance and energy as the cotyledons approached the mature was also found in soybean4. The active events occurs during seed development are more biologically particular and meaningful for seeds. This is demonstrated by the biological processes involving genes in the up-regulation group, including numbers of carbohydrate and fatty acid metabolic processes which are correlated to the nutrients accumulation and storage in seeds, leading seeds to maturation30.
Hormone signals involved in seed development
We identified 633 DEGs that in the hormone biosynthesis and signal pathway, including 149 ABA, 132 AUX, 130 ET, 67 SA, 59 BR, 33 CK, 33 JA, and 30 GA related genes during tartary buckwheat seed development (Table 3; Figs 5 and 6). Most hormone related DEGs identified were linked to ABA. ABA is a key hormone required for seed development31,32,33. Previous study has revealed that CYP707A in ABA catabolism has high expression level throughout seed development in bean31. Three homologs of the CYP707A were differentially regulated in our data (sample1_00009748-RA, sample1_00009778-RA, sample1_00009427-RA), suggesting that ABA 8’-hydroxylases during ABA catabolism may also occur in seed development of tartary buckwheat. PP2C has a major role in the ABA response in seeds, and its expression is increased during late seed maturation32. Its homolog in tartary buckwheat was also up-regulated as seed development (sample1_00026417-RA). LEC1 and ABI3 are two other genes that connect to seed maturation in ABA signaling33, and their homologs were differentially expressed during seed development in tartary buckwheat (sample1_00021697-RA and sample1_00015232-RA). In addition, a homolog of the essential gene in ABA biosynthesis, AAO3, was identified and exhibited an up-regulation during tartary buckwheat seed development (sample1_00009713-RA).
AUX is also necessary for seed development. Most of the studies focus on its role in determining seed size, with the influence of the expression of gene in AUX biosynthesis (ZmTar3, ZmTar1, and ZmYuc1) and signaling (auxin efflux carriers, PIN; ARF2)34,35. Homologs of ZmYuc1 (sample1_00021785-RA and sample1_00000287-RA), PIN (sample1_00006555-RA, sample1_00026719-RA, sample1_00026829-RA, sample1_00019675-RA), ARF2 (sample1_00015872-RA, sample1_00015871-RA, and sample1_00026266-RA) were significantly differentially expressed during tartary buckwheat seed development. Most of these DEGs had higher expression level at PS and FS, this is correspondent to that cell division and expansion is faster to form larger size seeds at these stages.
The molecular evidence illustrate ethylene is in the complex regulation of seed size and seed shape, in which genes in ethylene biosynthesis (EIN2, ERS1, ETR1), signaling (CTR1, ETO1, ETR1, EIN2), and catabolism (ACC deaminase) are involved36,37. Among the 130 ET related DEGs we identified, six were homologous to the characterized genes regulating seed development. Two homologs of ETO1 (sample1_00001473-RA and sample1_00023983-RA) were down-regulated as seed development. One homologs of ERS1 (sample1_00007359-RA) was down-regulated as seed development as well. Three homologs of ETR1 (sample1_00002821-RA, sample1_00009139-RA and sample1_00009140-RA) exhibited two opposite expression patterns: sample1_00002821-RA was up-regulated with seed development; whereas sample1_00009139-RA and sample1_00009140-RA were down-regulated with seed development.
The effects of SA on seed development exhibit two distinct opinions. On one hand, exogenous application of SA would increase seed’s dry mass, and pod and seed number38. On the other hand, however, SA delays fruit ripening, and SA deficiency mutants increase seed yield and antioxidant vitamin concentration39. Although no SA biosynthesis DEGs was identified in our data, three homologs in SA catabolism, and 8 genes in SA signal were identified. These included two DEGs homologous to UGT74F2 (sample1_00013064-RA and sample1_00009861-RA), one DEGs homologous to BSMT1 (sample1_00006811-RA), one DEGs homologous to PAD4 (sample1_00006909-RA), two DEGs homologous to EDS5 (sample1_00016328-RA and sample1_00006574-RA), two DEGs homologous to NPR1 (sample1_00000985-RA and sample1_00010619-RA), two DEGs homologous to TGA6 (sample1_00019877-RA and sample1_00022827-RA), and one DEGs homologous to SNI1 (sample1_00024341-RA). They showed two opposite expression patterns: parts of them were up expressed during seed development, whereas the rest were down expressed during seed development.
BR regulates embryo development, seed size, seed shape, seed length, and seed yield40. The characterized genes include BZR1, BES1, DWF4, and DWAFR11. Five of their homologs were identified in our data, including one in BR biosynthesis(sample1_00014237-RA, homolog of DWF4) and four in BR signal pathway (sample1_00015915-RA, sample1_00006053-RA, sample1_00006896-RA, and sample1_00009571-RA, homologs of BZR1). All of their expression levels were gradually falling down with seed development, suggesting BR might regulate seed development at the early stage.
CK is also reported to function on seed development, such as seed size, seed yield, embryonic growth, with the involvement of genes encoding isopentenyl transferase (IPT), cytokinin oxidase/dehydrogenase (CKX), and histidine kinase (AHK)41. Four DEGs of the homologous genes were identified, including one homolog of IPT8 (sample1_00002766-RA), two homologs of CKX1 (sample1_00006930-RA and sample1_00020506-RA), one homolog of CKX3 (sample1_00000991-RA). Sample1_00002766-RA and sample1_00020506-RA were up-regulated during seed development, whereas sample1_00006930-RA and sample1_00000991-RA were down-regulated during seed development. This suggests they may have different functions in regulating seed development of tartary buckwheat.
JA related processes in seed development contain embryo development, fruit ripeness, nutrients compositions (proximates, amino acids, fatty acids, isoflavones, and antinutrients), and seed production. These are accompanied with the expression of numbers of JA-inducible genes, including OPDA, AOS1 and JAZs, JMT 42. Nine homologs of them were identified in our data, of which three DEGs were homologous to AOS (sample1_00009789-RA, sample1_00020469-RA, sample1_00001703-RA), two were homologous to JMT (sample1_00003907-RA, sample1_00011765-RA), four were homologous to JAZ3 (sample1_00024846-RA, sample1_00000715-RA, sample1_00025602-RA, sample1_00022744-RA). All of them but one showed down-regulation with seed development, suggesting that JA might also be at the early regulatory network of seed development stages in tartary buckwheat.
Previous studies have revealed that genes encoding GA 2-oxidase and GA 3-oxidase in GA biosynthesis pathway can affect seed development, starch biosynthesis, embryo and seed coat development43,44. Four homologs of these genes, sample1_00019238-RA, sample1_00026062-RA, sample1_00024004-RA, and sample1_00016169-RA, were identified, all of which showed down-regulation expression as seed development.
Accumulation of the nutrients related genes during seed development of tartary buckwheat
Tartary buckwheat is famous for its abundant nutrients, including proteins, starch, and flavonoids8,10,18. Therefore, characterization and analysis of their biosynthesis genes is of great importance. SSPs are the main buckwheat allergen, and tens of their encoding sequences have been reported, mostly in common buckwheat45,46. In tartary buckwheat, three SSPs have been identified, including a 16-kDa major allergen belonged to 2S albumin47, a 24 kDa allergenic protein homology with a legumin-like protein48, and an allergen with high similarity to legume-like 13S globulin storage protein49. In our transcriptome, we identified 10 DEGs encoding 13S globulins (7) and 7S globulin-like proteins (3) (Fig. 7). All of the seven 13S globulin encoding DEGs exhibited up-regulation patterns with seed development. And six of them were matched to the existing buckwheat 13S globulin sequences. Sample1_00021677-RA, sample1_00021674-RA and sample1_00021668-RA were matched to a 13S globulin of common buckwheat (GenBank: BAO50862.1). Sample1_00022718-RA was also matched to a 13S globulin of common buckwheat (GenBank: BAO50870.1). Sample1_00013128-RA and sample1_00013130-RA were matched to a 13S globulin of tartary buckwheat (GenBank: ABI32184.1). Our transcriptome data is in accordance with previous reports that 13S globulin is one of the most dominant allergens, and accounts for 43% of total seed proteins in buckwheat seeds46.
Flavonoids, especially rutin, are the most widely studied nutrients in buckwheat13,50. Comparing with common buckwheat, the content of rutin in tartary buckwheat is higher11. At the transcriptome level, genes in the flavonoid biosynthesis have been thoroughly characterized and analyzed in buckwheat19,50,51,52. As shown on Fig. 8, 20 DEGs involved in flavonoid biosynthesis were identified. Most of them in the upstream of flavonoid biosynthesis pathway exhibited higher expression at PS and MS. However, two UGT encoding genes that catalase quercetin substrate to rutin, sample1_00011838-RA and sample1_00011837-RA, were up-regulated during seed development, consistence with rutin is the main accumulated flavonoid at matured buckwheat seeds50. In addition, the flavonoid biosynthesis genes were mostly reported in flower19,51,52. This is largely due to the inflorescence stage has the highest rutin content in all of the buckwheat species50. In tartary buckwheat and rice tartary buckwheat, matured seeds have the second highest rutin content50. Our results will provide molecular evidence for previous study, as well as candidate genes that primarily participate in the seed flavonoid biosynthesis of tartary buckwheat for further research.
Starch is the major form of carbohydrates accumulated at mature seeds and the main nutrient that make the seeds and other storage organs expand and enlarge. Genes in the starch biosynthesis pathway have been identified entirely, involving SUS, UGPase, AGPase, GBSS, SS, BE, and DBE 53. In buckwheat, starch account for over 70% of the seeds’ dry weight, thus lead its quality to be the main determinant of the quality of buckwheat seeds production8. However, the buckwheat starch biosynthesis genes remain largely unknown, except for two GBSS genes54,55. In our data, we identified 23 genes in starch biosynthesis which were differentially regulated during seed development of tartary buckwheat, including 3, 2, 5, 6, 2, 2, and 3 corresponding to the genes encoding SUS, UGPase, AGPase, GBSS, SS, BE, and DBE, respectively (Fig. 9). Of these, sample1_00010052-RA is matched to the GBSSI gene in tartary buckwheat (AHA36967.1). GBSS, also known as waxy protein, is the most direct enzyme that influences amylose content and starch quality53. The identified starch biosynthesis DEGs provide expression patterns for starch biosynthesis genes in seed development of tartary buckwheat, and would accelerate function analysis of them as well.
Conclusions
We performed a comprehensive global transcriptome analysis at seed developmental stages (PS, FS, and MS) of rice tartary buckwheat. Totally, 24 819 expressed genes were identified, including 108 genes that were specifically expressed at MS; and 11 676 DEGs were identified. The transcriptome data was confirmed by a qRT-PCR analysis on 34 DEGs, and a good consistence was obtained. All DEGs were classified into eight clusters based on their expression patterns. The GO annotation and KEGG pathways were also enriched for these DEGs. In addition, 633 hormone-related DEGs were also identified, which would be the key regulators at different stages of seed development of tartary buckwheat. Furthermore, 10 DEGs encoding SSPs, 20 DEGs involving in flavonoid biosynthesis, and 23 DEGs in starch biosynthesis were identified, suggesting the expression patterns of the nutrients accumulation related genes during seed development of tartary buckwheat. To our knowledge, this is the first study that revealed a global transcriptomic dynamics during seed development in tartary buckwheat. It would help us to understand the complex transcriptome dynamics and gene regulatory mechanism during the process of seed development and nutrients accumulation.
Materials and Methods
Plant materials and paraffin-embedded sections
A thin seed shell, high yield new line, Gui Miku 1503–11 (simply Mi 11) was used as our material, which is derived from the cross progenies of tartary buckwheat (thick shell) Jinqiaomai 2 native to Shanxi and local rice tartary buckwheat (thin shell) Xiaomiqiao native to Yunnan. It was planted in our testing field in spring, and with normal field management during the growth periods. Seeds were taken at 12 developmental stages from their emergence to maturation after pollination. Then seed coats were stripped and the remaining seeds were fixed in formalin-aceto-alcohol solution immediately for paraffin-embedded sections. Making and observation of the paraffin-embedded sections was performed as previous described procedure56. Based on the morphology of the seeds and their longitudinal sections, samples for RNA-Seq were collected at three stages, namely PS (corresponding to stage 1 to stage 3), FS (corresponding to stage 4 to stage 6), and MS (corresponding to stage 9 to stage 12) (see Results for details). For RNA extraction, the materials were frozen in liquid nitrogen immediately. Seed coats were stripped on a mixture of ice and liquid nitrogen, and the remaining seeds were stored at −80 °C.
RNA extraction, librayry construction, and sequencing
RNA was extracted using plant RNA purification kit that containing Dnase I (TianGen, Beijing, China), according to the manufacturer’s instruction. RNA samples with A260/280 ≥ 1.8, A260/230 ≥ 1.8, RIN ≥ 6.5, and 28 S/18 S ≥ 1.0 in quality, and concentration ≥20 ng/μL and total RNA amount ≥2 μg in quantity was acceptable for next step experiment, including library construction and qRT-PCR analysis. Samples from each stage consisted of three biological replicates, thus coming to nine RNA samples. All of the RNA samples were used to construct cDNA libraries for transcriptome sequencing.
For library construction, 2 μg RNA was used for each sample. mRNA was purified using Dynabeads mRNA purification kit in accordance to the manufacturer’s instruction(Invitrogen). Then the mRNA was cleaved to 200–250 bp small fragments in Fragment buffer. First strand cDNA was then generated using these small fragments by N6 primers, and second strand cDNA was generated by Second Strand Master Mix. Then cDNA was purified using QIAquick PCR Purification Kit (QIAGEN), combined with End Repair Mix, and add with A-tailing using A-Tailing Mix. To select cDNA with 300–350bp size, whole cDNA was used to perform a 2% agarose gel electrophoresis, and then purified the right size cDNA using QIAquick Gel Extraction kit (QIAGEN). PCR amplification was subsequently carried out using PCR Primer Cocktail to enrich the cDNA fragments, and the PCR products were purified using Ampure XP Beads (AGENCOURT). The quality of the libraries were determined by Agilent 2100 bioanalyzer instrument (Agilent DNA 1000 Reagents) and qRT-PCR (TaqMan Probe).
At last, the qualified libraries were amplified to generate cluster using TruSeq PE Cluster Kit V3-cBot-Hs (Illumina). Then they were sequenced on the Illumina HiSeq. 4000 System with read length of 101 bp and paired-end method. All of the raw data generated by sequencing have been deposited in NCBI SRA under Accession: PRJNA352888.
Mapping and analysis of DEGs
Trimmomatic was used to obtain clean reads. Raw data were filtered by removing adapter containing reads, reads with N (unknown base) percentage more than 10%, and low quality reads. Then clean reads were mapped to the reference genome data of tartary buckwheat (SRA accession number: PRJNA395279), using TopHat2. All of the gene sequences analyzed in this manuscript were presented in Table S7. These include 108 stage specifically expressed genes, 633 hormone related genes, 10 SSP encoding genes, 20 flavonoid biosynthesis involving genes, and 23 starch biosynthesis involving genes. The expressed genes were normalized and quantified as FPKM value, using RSEM57. The cluster of samples was analyzed using Euclidean distance by ward method in R language. All of the detected genes were annotated based on NR database, GO database, and KEGG database, with the e value of 1e-5.
DEGs were identified using two packages: 1) NOISeq package, with the threshold of absolute value of log2Ratio ≥ 1 and Probability ≥ 0.858; 2) edgeR package, for an additional analysis to confirm the DEGs obtained by NOISeq. Hierarchical clustering of DEGs was performed using the Pearson correlation method associated with average linkage clustering by MeV. GO enrichment was performed using agriGO. KOBAS 2.0 was then used to identify the significantly enriched metabolic pathways. And a local blastp was performed with the AHD2.0 to identify plant hormone related genes. The threshold for blast was set as 1e-5.
qRT-PCR analysis
qRT-PCR analysis was performed to verify transcriptome results. RNA samples used for qRT-PCR were identical to those used for the RNA-Seq. 34 DEGs were selected, and gene-specific primers were designed using Primer Premier 6.0 (Table S8). Actin was used as the inner reference gene. qRT-PCR was carried out using SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (RR820b, TAKARA, DaLian, China) on an ABI ViiA™ 7 real-time PCR system (Applied Biosystems, USA) with three technical replicates. Amplification reactions were initiated with a denaturing step (95 °C for 10 min), followed by 40 cycles of denaturing (95 °C for 10 s), annealing (60 °C for 30 s) and extension (72 °C for 40 s). Data were analyzed by 2−(∆∆Ct) method to obtain relative mRNA expression data.
References
Xue, L. J., Zhang, J. J. & Xue, H. W. Genome-wide analysis of the complex transcriptional networks of rice developing seeds. PloS one 7, e31081, https://doi.org/10.1371/journal.pone.0031081 (2012).
Le, B. H. et al. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proceedings of the National Academy of Sciences of the United States of America 107, 8063–8070, https://doi.org/10.1073/pnas.1003530107 (2010).
Cantu, D. et al. Effect of the down-regulation of the high Grain Protein Content (GPC) genes on the wheat transcriptome during monocarpic senescence. BMC genomics 12, 492, https://doi.org/10.1186/1471-2164-12-492 (2011).
Jones, S. I., Gonzalez, D. O. & Vodkin, L. O. Flux of transcript patterns during soybean seed development. BMC genomics 11, 136, https://doi.org/10.1186/1471-2164-11-136 (2010).
Basnet, R. K. et al. Genome-wide analysis of coordinated transcript abundance during seed development in different Brassica rapa morphotypes. BMC genomics 14, 840, https://doi.org/10.1186/1471-2164-14-840 (2013).
Kohl, S. et al. Metabolic and transcriptional transitions in barley glumes reveal a role as transitory resource buffers during endosperm filling. Journal of experimental botany 66, 1397–1411, https://doi.org/10.1093/jxb/eru492 (2015).
Ohnishi, O. Search for the wild ancestor of buckwheat III. The wild ancestor of cultivated common buckwheat, and of tatary buckwheat. Economic Botany 52, 123–133, https://doi.org/10.1007/BF02861199 (1998).
Zhu, F. Buckwheat starch: Structures, properties, and applications. Trends in Food Science & Technology 49, 121–135, https://doi.org/10.1016/j.tifs.2015.12.002 (2016).
Costantini, L. et al. Development of gluten-free bread using tartary buckwheat and chia flour rich in flavonoids and omega-3 fatty acids as ingredients. Food chemistry 165, 232–240, https://doi.org/10.1016/j.foodchem.2014.05.095 (2014).
Guo, X. & Yao, H. Fractionation and characterization of tartary buckwheat flour proteins. Food chemistry 98, 90–94, https://doi.org/10.1016/j.foodchem.2005.05.055 (2006).
Qin, P., Qiang, W., Fang, S., Hou, Z. & Ren, G. Nutritional composition and flavonoids content of flour from different buckwheat cultivars. International Journal of Food Science & Technology 45, 951–958, https://doi.org/10.1111/j.1365-2621.2010.02231.x (2010).
Jiang, S. et al. Separation of five flavonoids from tartary buckwheat (Fagopyrum tataricum (L.) Gaertn) grains via off-line two dimensional high-speed counter-current chromatography. Food chemistry 186, 153–159, https://doi.org/10.1016/j.foodchem.2014.08.120 (2015).
Zielinska, D., Turemko, M., Kwiatkowski, J. & Zielinski, H. Evaluation of flavonoid contents and antioxidant capacity of the aerial parts of common and tartary buckwheat plants. Molecules 17, 9668–9682, https://doi.org/10.3390/molecules17089668 (2012).
Hu, Y., Hou, Z., Liu, D. & Yang, X. Tartary buckwheat flavonoids protect hepatic cells against high glucose-induced oxidative stress and insulin resistance via MAPK signaling pathways. Food & function 7, 1523–1536, https://doi.org/10.1039/c5fo01467k (2016).
Peng, L. X. et al. Flavonoids, Antioxidant Activity and Aroma Compounds Analysis from Different Kinds of Tartary Buckwheat Tea. . Indian journal of pharmaceutical sciences 77, 661–667 (2015).
Hu, Y., Zhao, Y., Yuan, L. & Yang, X. Protective effects of tartary buckwheat flavonoids on high TMAO diet-induced vascular dysfunction and liver injury in mice. Food & function 6, 3359–3372, https://doi.org/10.1039/c5fo00581g (2015).
Li, Y. et al. Flavonoids from tartary buckwheat induce G2/M cell cycle arrest and apoptosis in human hepatoma HepG2 cells. Acta biochimica et biophysica Sinica 46, 460–470, https://doi.org/10.1093/abbs/gmu023 (2014).
Liu, B. & Zhu, Y. Extraction of flavonoids from flavonoid-rich parts in tartary buckwheat and identification of the main flavonoids. Journal of Food Engineering 78, 584–587, https://doi.org/10.1016/j.jfoodeng.2005.11.001 (2007).
Li, X. et al. Differential Expression of Flavonoid Biosynthesis Genes and Accumulation of Phenolic Compounds in Common Buckwheat (Fagopyrum esculentum). Journal of Agricultural & Food Chemistry 58, 10911–10917, https://doi.org/10.1021/jf103310g (2010).
Horbowicz, M., Brenac, P. & Obendorf, R. L. Fagopyritol B1, O-alpha-D-galactopyranosyl-(1– > 2)-D-chiro-inositol, a galactosyl cyclitol in maturing buckwheat seeds associated with desiccation tolerance. Planta 205, 1–11, https://doi.org/10.1007/s004250050290 (1998).
Ueda, T., Coseo, M. P., Harrell, T. J. & Obendorf, R. L. A multifunctional galactinol synthase catalyzes the synthesis of fagopyritol A1 and fagopyritol B1 in buckwheat seed. Plant Science 168, 681–690, https://doi.org/10.1016/j.plantsci.2004.09.029 (2005).
Ehlers, K. et al. The MADS Box Genes ABS, SHP1, and SHP2 Are Essential for the Coordination of Cell Divisions in Ovule and Seed Coat Development and for Endosperm Formation in Arabidopsis thaliana. PloS one 11, e0165075, https://doi.org/10.1371/journal.pone.0165075 (2016).
Yang, X. et al. Live and let die - the B(sister) MADS-box gene OsMADS29 controls the degeneration of cells in maternal tissues during seed development of rice (Oryza sativa). PloS one 7, e51435, https://doi.org/10.1371/journal.pone.0051435 (2012).
Busi, M. V. et al. MADS-box genes expressed during tomato seed and fruit development. Plant molecular biology 52, 801–815, https://doi.org/10.1023/A:1025001402838 (2003).
Huang, S. & Brown, S. M. Overexpression of 20-oxidase confers a gibberellin-overproduction phenotype in Arabidopsis. Plant physiology 118, 773–781, https://doi.org/10.1104/pp.118.3.773 (1998).
Laudert, D., Pfannschmidt, U. & Lottspeich, F. Cloning, molecular and functional characterization of Arabidopsis thaliana allene oxide synthase (CYP 74), the first enzyme of the octadecanoid pathway to jasmonates. Plant molecular biology 31, 323–335, https://doi.org/10.1007/BF00021793 (1996).
Hwang, I. S. & Hwang, B. K. Role of the pepper cytochrome P450 gene CaCYP450A in defense responses against microbial pathogens. Planta 232, 1409–1421, https://doi.org/10.1007/s00425-010-1266-y (2010).
Maimbo, M., Ohnishi, K., Hikichi, Y., Yoshioka, H. & Kiba, A. Induction of a small heat shock protein and its functional roles in Nicotiana plants in the defense response against Ralstonia solanacearum. Plant physiology 145, 1588–1599, https://doi.org/10.1104/pp.107.105353 (2007).
Pajerowska-Mukhtar, K. M. et al. The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Current biology: CB 22, 103–112, https://doi.org/10.1016/j.cub.2011.12.015 (2012).
Gutierrez, L., Van Wuytswinkel, O., Castelain, M. & Bellini, C. Combined networks regulating seed maturation. Trends in plant science 12, 294–300, https://doi.org/10.1016/j.tplants.2007.06.003 (2007).
Yang, S. H. & Zeevaart, J. A. Expression of ABA 8′-hydroxylases in relation to leaf water relations and seed development in bean. The Plant journal: for cell and molecular biology 47, 675–686, https://doi.org/10.1111/j.1365-313X.2006.02815.x (2006).
Nishimura, N. et al. ABA-Hypersensitive Germination1 encodes a protein phosphatase 2C, an essential component of abscisic acid signaling in Arabidopsis seed. The Plant journal: for cell and molecular biology 50, 935–949, https://doi.org/10.1111/j.1365-313X.2007.03107.x (2007).
Finkelstein, R. R., Gampala, S. S. & Rock, C. D. Abscisic acid signaling in seeds and seedlings. The Plant cell 14(Suppl), S15–45, https://doi.org/10.1105/tpc.010441 (2002).
Bernardi, J. et al. The Auxin-Deficient Defective Kernel18 (dek18) Mutation Alters the Expression of Seed-Specific Biosynthetic Genes in Maize. Journal of Plant Growth Regulation 35, 770–777, https://doi.org/10.1007/s00344-016-9581-6 (2016).
Schruff, M. C. et al. The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development 133, 251–261, https://doi.org/10.1242/dev.02194 (2006).
Walton, L. J. et al. Ethylene involvement in silique and seed development of canola, Brassica napus L. Plant physiology and biochemistry: PPB 58, 142–150, https://doi.org/10.1016/j.plaphy.2012.06.016 (2012).
Robert, C., Noriega, A., Tocino, A. & Cervantes, E. Morphological analysis of seed shape in Arabidopsis thaliana reveals altered polarity in mutants of the ethylene signaling pathway. Journal of plant physiology 165, 911–919, https://doi.org/10.1016/j.jplph.2007.10.005 (2008).
Fariduddin, Q., Hayat, S. & Ahmad, A. Salicylic Acid Influences Net Photosynthetic Rate, Carboxylation Efficiency, Nitrate Reductase Activity, and Seed Yield in Brassica juncea. Photosynthetica 41, 281–284, https://doi.org/10.1023/B:PHOT.0000011962.05991.6c (2003).
Abreu, M. E. & Munne-Bosch, S. Salicylic acid deficiency in NahG transgenic lines and sid2 mutants increases seed yield in the annual plant Arabidopsis thaliana. Journal of experimental botany 60, 1261–1271, https://doi.org/10.1093/jxb/ern363 (2009).
Jiang, W. B. et al. Brassinosteroid regulates seed size and shape in Arabidopsis. Plant physiology 162, 1965–1977, https://doi.org/10.1104/pp.113.217703 (2013).
Bartrina, I., Otto, E., Strnad, M., Werner, T. & Schmulling, T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. The Plant cell 23, 69–80, https://doi.org/10.1105/tpc.110.079079 (2011).
Nam, K. H. et al. Comparative analysis of chemical compositions between non-transgenic soybean seeds and those from plants over-expressing AtJMT, the gene for jasmonic acid carboxyl methyltransferase. Food chemistry 196, 236–241, https://doi.org/10.1016/j.foodchem.2015.09.046 (2016).
Singh, D. P., Jermakow, A. M. & Swain, S. M. Gibberellins are required for seed development and pollen tube growth in Arabidopsis. The Plant cell 14, 3133–3147, https://doi.org/10.1105/tpc.003046 (2002).
Nakayama, A., Park, S., Zheng-Jun, X., Nakajima, M. & Yamaguchi, I. Immunohistochemistry of active gibberellins and gibberellin-inducible alpha-amylase in developing seeds of morning glory. Plant physiology 129, 1045–1053, https://doi.org/10.1104/pp.010921 (2002).
Sano, M., Nakagawa, M., Oishi, A., Yasui, Y. & Katsube-Tanaka, T. Diversification of 13S globulins, allergenic seed storage proteins, of common buckwheat. Food chemistry 155, 192–198, https://doi.org/10.1016/j.foodchem.2014.01.047 (2014).
Park, J. W. et al. Identification and characterization of the major allergens of buckwheat. Allergy 55, 1035–1041, https://doi.org/10.1034/j.1398-9995.2000.00763.x (2000).
Chen, P., Guo, Y. F., Yan, Q. & Li, Y. H. Molecular cloning and characterization of Fag t 2: a 16-kDa major allergen from Tartary buckwheat seeds. Allergy 66, 1393–1395, https://doi.org/10.1111/j.1398-9995.2011.02657.x (2011).
Wang, Z. et al. Cloning, expression, and identification of immunological activity of an allergenic protein in tartary buckwheat. Bioscience, biotechnology, and biochemistry 70, 1195–1199, https://doi.org/10.1271/bbb.70.1195 (2006).
Zhang, X., Yuan, J. M., Cui, X. D. & Wang, Z. H. Molecular cloning, recombinant expression, and immunological characterization of a novel allergen from tartary buckwheat. Journal of agricultural and food chemistry 56, 10947–10953, https://doi.org/10.1021/jf801855a (2008).
Gupta, N., Sharma, S. K., Rana, J. C. & Chauhan, R. S. Expression of flavonoid biosynthesis genes vis-a-vis rutin content variation in different growth stages of Fagopyrum species. Journal of plant physiology 168, 2117–2123, https://doi.org/10.1016/j.jplph.2011.06.018 (2011).
Li, C. et al. Cloning, characterization, and activity analysis of a flavonol synthase gene FtFLS1 and its association with flavonoid content in tartary buckwheat. Journal of agricultural and food chemistry 60, 5161–5168, https://doi.org/10.1021/jf205192q (2012).
Yao, H. et al. Deep sequencing of the transcriptome reveals distinct flavonoid metabolism features of black tartary buckwheat (Fagopyrum tataricum Garetn.). Progress in biophysics and molecular biology. https://doi.org/10.1016/j.pbiomolbio.2016.11.003 (2016).
Nougue, O., Corbi, J., Ball, S. G., Manicacci, D. & Tenaillon, M. I. Molecular evolution accompanying functional divergence of duplicated genes along the plant starch biosynthesis pathway. BMC evolutionary biology 14, 103, https://doi.org/10.1186/1471-2148-14-103 (2014).
Wang, X. et al. Identification and characterization of granule bound starch synthase I (GBSSI) gene of tartary buckwheat (Fagopyrum tataricum Gaertn.). Gene 534, 229–235, https://doi.org/10.1016/j.gene.2013.10.053 (2014).
Chrungoo, N. K., Devadasan, N., Kreft, I. & Gregori, M. Identification and characterization of granule bound starch synthase (GBSS-I) from common buckwheat (Fagopyrum esculentum Moench). Journal of Plant Biochemistry and Biotechnology 22, 269–276, https://doi.org/10.1007/s13562-012-0153-y (2013).
Liu, J. et al. iTRAQ-facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). Journal of proteomics 126, 68–81, https://doi.org/10.1016/j.jprot.2015.05.031 (2015).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323, https://doi.org/10.1186/1471-2105-12-323 (2011).
Tarazona, S., Garcia-Alcalde, F., Dopazo, J., Ferrer, A. & Conesa, A. Differential expression in RNA-seq: a matter of depth. Genome research 21, 2213–2223, https://doi.org/10.1101/gr.124321.111 (2011).
Acknowledgements
The authors give thanks to the Natural Science Foundation of China (31471562), Initial Fund for Doctor Research in Guizhou Normal University (11904/0516031, 11904/0516026), Science and Technology Foundation of Guizhou Province (QianKeHeJiChu[2016]1107), and Foundation of Guizhou Natural Science (QiankeheJzi [2014]2011) for financial support. We give thanks to Jiehong Zhao and Jing Yu (Guizhou Academy of Tobacco Science, Guiyang 550081, Guizhou, China) to give us help for this manuscript.
Author information
Authors and Affiliations
Contributions
J.H. analyzed the data, created the figures, and drafted the manuscript. J.D. and T.X.S sampled the seeds’ materials and prepared total RNA for sequencing. Q.J.C. and C.G.L. planted and managed the tartary buckwheat materials in the field. Z.Y.M. and L.W.Z. performed laboratorial experiments. Y.W. revised the manuscript. F.L.Z. and S.Z.Y. analyzed the data and revised the manuscript. Q.F.C. conceived and designed this study. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, J., Deng, J., Shi, T. et al. Global transcriptome analysis and identification of genes involved in nutrients accumulation during seed development of rice tartary buckwheat (Fagopyrum Tararicum). Sci Rep 7, 11792 (2017). https://doi.org/10.1038/s41598-017-11929-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11929-z
This article is cited by
-
Comparative proteomic analyses of Tartary buckwheat (Fagopyrum tataricum) seeds at three stages of development
Functional & Integrative Genomics (2022)
-
Integrated microRNA and transcriptome profiling reveal key miRNA-mRNA interaction pairs associated with seed development in Tartary buckwheat (Fagopyrum tataricum)
BMC Plant Biology (2021)
-
Mapping QTLs for 1000-grain weight and genes controlling hull type using SNP marker in Tartary buckwheat (Fagopyrum tataricum)
BMC Genomics (2021)
-
Buckwheat (Fagopyrum sp.) genetic resources: What can they contribute towards nutritional security of changing world?
Genetic Resources and Crop Evolution (2020)
-
Charting oat (Avena sativa) embryo and endosperm transcription factor expression reveals differential expression of potential importance for seed development
Molecular Genetics and Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
