
Abstract
Existing cancer therapies are often associated with drug resistance and toxicity, which results in poor prognosis and recurrence of cancer. This necessitates the identification and development of novel therapeutics against existing as well as novel cellular targets. In this study, a novel class of Benzocoumarin-Stilbene hybrid molecules were synthesized and evaluated for their antiproliferative activity against various cancer cell lines followed by in vivo antitumor activity in a mouse model of cancer. The most promising molecule among the series, i.e. compound (E)-4-(3,5-dimethoxystyryl)-2H-benzo[h]chromen-2-one (19) showed maximum antiproliferative activity in breast cancer cell lines (MDA-MB-231 and 4T1) and decreased the tumor size in the in-vivo 4T1 cell-induced orthotopic syngeneic mouse breast cancer model. The mechanistic studies of compound 19 by various biochemical, cell biology and biophysical approaches suggest that the compound binds to and inhibits the human DNA ligase I enzyme activity that might be the cause for significant reduction in tumor growth and may constitute a promising next-generation therapy against breast cancers.
Similar content being viewed by others
Introduction
Molecular hybridization is an important strategy in drug design and development based on the combination of pharmacophoric moieties of different bioactive molecules to produce a new hybrid compound with improved medicinal properties as compared to the parent molecules. The concept of hybrid molecules or molecular hybridization is now being increasingly used by pharmaceutical chemists in their quest for potent new drugs, as evidenced by the large number of recent reports on the synthesis of new bioactive hybrid molecules with the goal of creating new chemical entities that are medically more effective than their parent molecules1,2,3.
Combining the concept of molecular hybridization (MH) and biology-oriented synthesis (BIOS) we have synthesized a library of 30 stilbene (Resveratrol) and Neo-tanshinlactone based natural-product inspired hybrid molecules. The rationale behind the synthesis of natural product based hybrid molecules is the reduced general toxicity of the natural products because of their co-evolution with the targets and increased efficacy and selectivity. Out of 30 compounds one compound i.e. compound 19 showed better antiproliferative activity against the tested cancer cell lines as compared to their parent compounds (Neo-tanshinlactone and Resveratrol). The compound 19 is orally active in breast cancer syngeneic mouse model and shows good pharmacokinetic parameters to be considered as a lead molecule for future targeted therapy. Our in-silico studies showed that compound 19 may preferentially interact with human DNA ligase I (hLigI) protein, among various replication proteins that were tested. In-silico studies validated by in-vitro and ex-vivo biochemical assays showed that compound 19 specifically inhibits hLigI activity whereas FEN1 and PARP1 proteins were unaffected. Human DNA ligases are important proteins that maintain the genomic integrity of cells by joining the DNA strand breaks during DNA replication and repair processes4,5,6,7,8. An inhibition of hLigI activity leads to deficiency of DNA joining during DNA replication and repair process that leads to accumulation of DNA strand breaks leading to cell death7, 9. Since most adult cells in the human body do not replicate, or do so only sporadically (e.g., hematopoietic stem-cells), the highly proliferating cancer cells would be the primary target for hLig1 inhibitors while the side-effect on non-dividing normal cells is predicted to be minimal.
There are three major forms of human DNA ligases (human DNA ligase I, III and IV) that have been described in the literature8, 10. Among the three, hLigI is the major form in replicating cells. Elevated levels of DNA ligase I have been found in several cancer cells like breast, lung and ovarian cancer cells11. Several attempts have been made to search for specific inhibitors against DNA ligases since the year 2004 after the discovery of the crystal structure of hLigI12. Some ligase inhibitors exhibiting anticancer activity have been reported recently13,14,15,16,17,18,19,20, however none are presently used in therapy. Here, for the first time, we demonstrate an in-vivo active hLigI inhibitor which could be a lead molecule for the development of successful drug candidates.
Results
Chemistry results
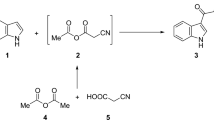
We designed and synthesized a library of E-benzocoumarin-stilbene hybrids via Horner-Wadsworth-Emmons (HWE) reaction (Fig. 1). Acid catalyzed condensation of 4-bromo ethylacetoacetate21 with 1-napthols (1–3)22 led to the formation of 4-bromomethyl-benzocoumarin derivatives 4–6 in quantitative yields. Subjecting the 4-bromomethylbenzocoumarin derivatives 4–6 to Michaelis-Arbusov reaction23, 24 with triethylphoshphite afforded the phosphonic acid diethyl esters 7–9 as crystalline compounds in 82–95% yield. Subsequently, HWE reaction25 of the phosphonic acid diethyl esters 7–9 with substituted aldehydes in dry DMF/NaOMe formed the pure compounds, (E)-4-styryl-2H-benzo[h]chromen-2-ones 10–39 in 72-96% yield (Table 1).

Synthesis of Benzocoumarin-Stilbene hybrid molecules 10–39 via Horner- Wadsworth- Emmons (HWE) reaction. Reagents and conditions: (a) H2SO4, RT, 3 h; (b) P(OEt)3, 120 °C, 2-3 h, () MeONa, DMF 0 °C −RT, 70 °C, 72–98%.
Biology results
Effect of Benzocoumarin-Stilbene hybrids on viability of different cancer cell lines
The cytotoxicity of all 30 compounds (Benzocoumarin-stilbene hybrids) were evaluated in breast cancer cell lines (MDA-MB-231) (Supplementary Table 1). At 10 μM concentration, compounds 16, 19 and 26 showed significant activity ( > 50% inhibition) in MDA-MB-231 cells (Supplementary Table 1). The activity of these compounds were further checked in different cancer cell lines such as Colorectal adenocarcinoma (DLD-1), Liver hepatoma (PLC/PRF/5), Lung carcinoma (A549), Ovarian adenocarcinoma (SK-OV-3), Brain glioblastoma (A-172), Pancreatic adenocarcinoma (PANC-1), and triple negative mouse breast cancer cell line (4T1) (Supplementary Table 2). Of all the compounds tested, compound 19 showed maximal activity in different cancer cell lines including MDA-MB-231 and 4T1 with IC50 values 12.5 ± 2.8 and 11.6 ± 1.7 μM respectively. Although the compound showed activity against MCF10A (human mammary epithelial cell line), but due to the non-toxic and cytostatic nature of the compound, we decided to test the compound in our in vivo breast cancer mouse model developed using syngeneic 4T1 cells, against which the compound showed best activity (Fig. 2B).

(A) Bright field images of MDA-MB-231 cells treated with vehicle control, Neo-tanshinlactone, Resveratrol and compound 19. The cells treated with compound 19 showed changes in cellular morphology. (B) Images of colony forming assay showed growth inhibition of colonies after the treatment with different concentrations of compound 19. Graphs C, D and E show the activities of Compound 19, Neo-tanshinlactone and Resveratrol respectively in MDA-MB-231 cells.
We studied the compound 19 induced morphological changes in MDA-MB-231 cells. The bright field images of cells shown in Fig. 2A clearly indicate that the compound induces conspicuous morphological changes in cells after 24 and 48 h, as compared to its parent compounds (Neo-tanshinlactone and Resveratrol) or vehicle control, at its IC50 concentration (12 μM). Compound 19 inhibited colony forming ability of cells (Fig. 2B) and after 24 and 48 h of treatment (at 3, 1.5 and 0.75 μM concentrations) also reduced the size of the limited colonies that formed. This indicated that the compound may have a cytostatic activity against MDA-MB-231 cells at these concentrations. We performed comparative cell viability assays for compound 19 and its parents (Neo-tanshinlactone and resveratrol) and found that compound 19 was more active as compared to parent compounds (Fig. 2C–E).
Compound 19 induced apoptosis in MDA-MB-231 cells and arrested cell cycle progression at S and G2/M phases
Quantitative analysis of the induction of apoptosis was performed in MDA-MB-231 cells using flow cytometry. MDA-MB-231 cells were treated with different concentrations (6 and 12 µM) of compound 19 for 24 and 48 h. Percentages of cells undergoing apoptosis increased with increasing concentrations of compound 19 and their treatment duration (Fig. 3A). At 24 h of treatment, 10.78 ± 1.21% and 16.37 ± 1.18% apoptosis was observed in MDA-MB-231 cells at 6 and 12 µM concentrations, whereas after 48 h of treatment a much higher 18.68 ± 4.86% and 36.17 ± 3.43% of cells were observed to undergo apoptosis at the same concentrations.

(A) Quantification of apoptosis induced by different concentrations of compound 19 in MDA-MB-231 cells at 24 and 48 h. The percentage population of early and late apoptotic cells at various concentrations of compound 19 are represented graphically. (B) The quantitative distribution of cells in different phases of the cell cycle is represented graphically. DNA histograms clearly show that compound 19 (at 12 µM for 0, 24 and 48 h) induced cell cycle arrest at the S and G2/M phase of cell cycle. The data shown are mean ± SEM of three independent experiments. Significant difference from control was observed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001.
To study the effect of compound 19 on cell cycle progression, MDA-MB-231 cells were synchronized by serum starvation for 48 h. Thereafter, cells were released into serum containing media and treated with 12 µM of compound 19 for 0, 24 and 48 h. At 0 h, both treated and untreated cells were arrested at G0/G1 phase (75.12 ± 2.23%), with only 16.18 ± 0.65% cells in the S-phase and 8.71 ± 1.57% cells in the G2/M phase (Fig. 3B). After 24 h, in control samples, 69.63 ± 0.21% cells were still present in G0/G1 phase while 18.31 ± 0.13% cells in S phase and 12.06 ± 0.08% cells were in G2/M phase. However, in the treated sample a much lower 57.61 ± 0.1% cells were in the G0/G1 phase while 20.91 ± 2.61% and 21.48 ± 2.50% cells were present in S and G2/M phases of cell cycle respectively. Figure 3B, also shows similar results after 48 h of treatment with 66.87 ± 1.23% cells in G0/G1 phase, 21.8 ± 2.72% cells in S phase, and 11.32 ± 1.48% cells in G2/M phase in control cells whereas in treated cells a significantly lower 33.78 ± 4.77% cells were in G0/G1 phase, 33.7 ± 4.2% in S phase and 32.53 ± 0.58% cells were in G2/M phase, respectively. The graph in Fig. 3B, clearly shows that at different time points, compound 19 increased the population of cells in S and G2/M phase and decreased the cell population in G0/G1 phase, indicating that it blocks the cell cycle at S and G2/M phase.
Compound 19 inhibits migration of MDA-MB-231 cells
Cancer cells possess the property of migration from their origin to distant sites which is called metastasis26. It is particularly dangerous for patients as this leads to the spread of cancer from the site of formation to other organs of the body and leads to flaring up of the condition and poor disease prognosis. We performed the scratch assay to check if the migration ability of MDA-MB-231 cells was altered in the presence of different concentrations (3 and 6 µM) of compound 19 (when treated for 12 and 24 h). We calculated the percent open area in control and treated cells at different time points with the help of TScratch software. The bright field images and graph in Fig. 4 show that the percent open area decreases more in control cells as compared to treated cells. The percent reduction in open area indicates the increase in migration ability of cells. In Fig. 4, we clearly demonstrate that compound 19 could significantly inhibit the cell migration at concentrations of 3 and 6 µM where no significant antiproliferative activity was observed.

Bright field images of MDA-MB-231 cells treated with compound 19 demonstrate the inhibition of cellular migration. The graph on the right clearly shows the decrease in the percentage of open area in control cells versus treated cells at different time points. The data shown are mean ± SEM of three independent experiments. Significant difference from control was observed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001.
Compound 19 induced DNA damage and activated the intrinsic pathway of apoptosis in MDA-MB-231 cells
We performed western blotting experiment to study the compound 19 induced apoptotic signaling events in MDA-MB-231 cells. As shown in Fig. 5, compound 19 increased DNA damage inside the cells as demonstrated by increased expression of γH2AX in cells27. We also performed comet assay to study the DNA damage caused by compound 19 and found a significant increase in the tail length of treated cells (increased tail length of DNA occurs due to DNA damage) as compared to control (Supplementary Fig. 71). Cells try to repair the damaged DNA by increasing the expression of PARP-1 protein; the level of PARP-1was found elevated cells treated with compound 19. However, during extensive DNA damage, PARP-1 is cleaved into 89 kDa and 24 kDa fragments by a caspase 3 mediated pathway that promotes the cells to undergo apoptosis instead of necrosis28. This was observed in our western blotting results in Fig. 5. Our western blotting results also showed an increase in expression of p53, decrease in expression of MDM2 (negative regulator of p53) and PCNA (cell proliferation marker). In addition, the compound promotes the cleavage of caspase 9 and caspase 3 in MDA-MB-231 cells. These results indicate that compound 19 promotes p53 mediated intrinsic pathway of apoptosis in MDA-MB-231 cells at the tested concentrations.

Western blotting images show differential expression of proteins after the treatment of compound 19 in MDA-MB-231 cells. The blot images are representative of three independent experiments.
Assessment of pharmacokinetic properties of the novel bioactive anticancer compound 19
The oral route of administration is a more preferred route than others, and the drugs must therefore be stable in SGF (simulated gastric fluid) and SIF (simulated intestinal fluid) respectively. Compound 19 showed 83.19 ± 3.22% and 96.45 ± 0.79% stability in SGF and SIF up to 180 min (Fig. 6A). Thus, compound 19 was suitable for oral administration as it was stable in SGF and SIF.

Stability studies of compound 19. (A) Simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) stability of compound 19 at 2 µg/mL concentration. (B) Metabolic stability of compound 19 at 5 µM concentration in rat liver microsomes (RLM). The data shown are mean ± SEM of three independent experiments.
In microsomal metabolic stability study, compound 19 was found to be stable in control reaction in the absence of cofactor, confirming the chemical stability as well as cofactor dependent degradation. The compound was 55.5 ± 4.77% stable up to 60 min (Fig. 6B).
The pharmacokinetic parameters followed by the oral and I.V administration were calculated using non-compartmental model listed in Table 2. After oral administration (50 mg/kg) the compound exhibited a Cmax of 13.38 ± 2.92 ng/mL at 0.5 hr (tmax). Following I.V (5 mg/kg) dosing, the plasma concentration of compound 19 was found to be 325.30 ± 101.88 ng/mL at 5 min. In-vivo t1/2 of compound 19 was found to be 19.25 ± 11.75 h and 22.77 ± 4.69 h for I.V and oral PK study, respectively. AUC0-t of compound 19 was found to be 457.15 ± 130.59 hr*ng/mL and 341.57 ± 18.64 hr*ng/mL after I.V and oral PK study, respectively. The percentage bioavailability of compound 19 was found to be 7.85 ± 0.65%. After oral dosing, compound 19 showed detectable plasma concentrations up to 72 h, and it could provide chronic onset of action at the detectable levels. This chronic onset of action could be an advantage in chronic clinical conditions like cancer.
Compound 19 arrested breast tumor progression in-vivo
Having validated the in-vitro activity of compound 19, we utilized the syngeneic 4T1 mouse orthotopic breast cancer model to evaluate the in-vivo antitumor efficacy at 50 and 100 mg/kg doses. The compound inhibited breast tumor growth in-vivo in a dose dependent manner as compared to vehicle treated animals when it was administered daily via oral gavage for 32 days (Fig. 7A,D). Pictures of harvested tumors (Fig. 7B) clearly demonstrated a marked reduction in tumor size in animals treated with the compound compared to vehicle control group. There was no significant change in the average body weight of control and treated animals for the duration of the experiment, suggesting the apparent non-toxic nature of the compound (Fig. 7C). A marked decrease in breast cancer metastasis to the lungs was also observed (Fig. 7E,F). A marked reduction in the number of 4T1 nodules on the lungs of treated mice can be seen as compared to vehicle control mice. This indicates that the compound inhibits the migration of breast cancer cells from their site of origin to the lungs as earlier indicated by our in-vitro scratch assay (Fig. 4). These experiments clearly demonstrate the antitumor and anti-metastatic activity of compound 19.

In-vivo antitumor activity of compound 19 in a syngeneic (4T1 induced) orthotopic mouse model of breast cancer. (A) Effect of the compound on breast tumor progression is represented graphically for every fourth day. (B) Representative images of control and treated tumors at 50 and 100 mg/kg body weight. (C) Effect of the compound on body weight of mice is shown graphically. (D) Graphical representation of % increase of tumor size in control and treated mice. (E) Representative image of the lungs of mice showing 4T1 nodules in control and treated mice. (F) Number of 4T1 nodules in each lung represented by a graph. The data shown are mean ± SEM of N = 6 mice of each group in two different experiments. Significant difference from control was observed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001.
Possible mode of action of compound 19
Our cell based assays showed that compound 19 arrested the cell cycle progression of MDA-MB-231 cells at S and G2/M phases and promoted the accumulation of DNA damage as seen by an increase in the level of γH2AX (Figs 3,5 and Supplementary Fig. 71). DNA replication and repair processes occurs mainly at S and G2/M phase of cell cycle29,30,31,32 suggesting that compound 19 might be targeting one or more of the DNA replication and repair proteins as its cellular target/s. This prompted us to carry out in-silico docking studies for compound 19 with various DNA replication and repair-related proteins to predict the possible cellular target/s of this compound.
On the basis of docking scores (Supplementary Table 3) obtained from our in-silico studies we predicted hLigI (lowest docking energy) as a possible cellular target for compound 19. We performed in-vitro ligation assay to confirm our in-silico prediction and found that compound 19 could inhibit hLigI activity. The ligation assays further proved compound 19 specifically inhibits the activity of hLigI (at 5, 10 and 20 µM concentrations), since the other human (hLigIIIβ and hLigIV/XRCC4) and non-human ligases (T4 DNA ligase) were unaffected by the compound at these concentrations (Fig. 8A). We also checked the functional inhibition of other proteins (PARP1 and FEN1) in the presence of compound 19 and found that the compound was not able to inhibit the function of FEN1 or PARP1 proteins even at 20 µM concentration (Supplementary Fig. 72 and 73). Compound 19, which is hybrid of Neo-tanshinlactone and Resveratrol also showed better antiligase activity when compared to parent compounds Neo-tanshinlactone and Resveratrol. We used compound 23, a known antiligase compound reported in our previous study19 as a positive control for antiligase activity (Fig. 8B).

(A) Concentration dependent inhibition of DNA ligase activity by compound 19 at 0, 5, 10 and 20 µM concentrations against different human and non-human ligases. The gel picture and graph clearly showed specific hLigI inhibition activity of compound 19. (B) A representative gel image and graph for inhibition of ligation by compound 19 and their parent molecules at 20 µM concentration. Compound 19 showed significantly better antiligase activity as compared to Neo-tanshinlactone (Neo) and Resveratrol (Res). Compound 23 was used as a positive control. The data shown are mean ± SEM of three independent experiments. Significant difference from control was observed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001.
The above in-vitro results were supported by our ex-vivo assays where the cell lysate of MDA-MB-231 cells treated with different concentrations of compound 19 showed reduced capacity for DNA ligation (performed by hLigI) whereas flap cleavage activity (performed by FEN1) remained unaffected after treatment (Supplementary Fig. 74). These experiments indicate that compound 19 can specifically inhibit hLigI activity and could be its major cellular target.
Compound 19 inhibits ligation by direct interaction with hLigI protein
Interestingly, a compound can inhibit ligation either by direct interaction with the protein where it blocks the DNA binding site (or another active or allosteric site) thereby blocking or changing its activity, or by binding with the substrate DNA, thereby occluding protein binding. In order to check both possibilities, we performed both protein and DNA interaction studies with compound 19 using various biophysical methods. Protein binding was checked by performing fluorescence quenching experiments followed by circular dichroism (CD) studies. In the presence of compound 19, fluorescence spectra of the full length hLigI protein changed as compared to control samples, indicating protein binding (Fig. 9A). The Circular Dichroism (CD) studies also showed that the compound promoted conformational changes in protein (Fig. 9B) and decreased the hLigI and DNA complex formation (Lane 2 versus Lane 3-5, Fig. 9C). Moreover, the complex formation could be rescued by addition of a higher amount of hLigI (Lane 6 and 7, Fig. 9C). This again suggested protein binding. The key amino acid residues of hLigI involved in interaction with compound 19 are shown in Supplementary Fig. 75.

Interaction studies of compound 19 with hLigI and DNA. Fluorescence spectra of (A) hLigI in the presence of compound 19, upon excitation at 280 nm. Fluorescence intensity is indicated in arbitrary units (A.U.). (B) CD spectroscopy shows conformational change in hLigI after addition of compound 19. Spectra was recorded at the wavelength of 250-195 nm. (C) EMSA shows that compound 19 breaks the interaction between DNA and hLigI protein (lanes 3-5). For complementation, increasing concentrations of hLigI, when added back to the reaction, could rescue the hLigI-DNA complex even in the presence of the inhibitor (lanes 6-7). (D) DNase I cleavage assay rules out the possibility of any kind of interaction between compound 19 and DNA. The gel images are representative of three independent experiments.
DNA binding of compound 19 was tested by the DNase I cleavage protection assay. The results from this experiment clearly suggested that unlike doxorubicin (a known DNA intercalator), methyl green (a known DNA major groove binder) or DAPI (a known DNA minor groove binder), the DNA incubated with compound 19 offered no protection from cleavage by DNase I up to 500 µM concentration (Fig. 9D, lanes 1-2 versus lanes 5-7). Thus, we may safely exclude any form of covalent interaction between compound 19 and DNA. Hence, from the above experiments, we may deduce that compound 19 exhibits its inhibitory activity through direct binding to hLigI and shows no interaction with the DNA substrate.
Discussion
In this study, a novel class of compounds belonging to the Benzocoumarin-Stilbene hybrid family have been synthesized with the help of molecular hybridization approach. The most active compound of this series (compound 19) showed significantly better antiproliferative activity as compared to their parent compounds (Neo-tanshinlactone and Resveratrol) in MDA-MB-231 (hormone receptor negative) cells (Fig. 2). Our studies showed that compound 19 could arrest cell cycle progression at S and G2/M phases of cell cycle (Fig. 3), increase DNA damage, increase PARP cleavage, and finally promote a p53 mediated intrinsic pathway of apoptotic cell death in the TNBC cell line MDA-MB-231(Fig. 5).
The compound was also tested for its pharmacokinetic properties where it demonstrated stability in SGF and SIF and was found to be metabolically stable. The compound fulfills the Lipinski’s rule of five (Supplementary Table 4) which is necessary for a compound to be considered as a drug like compound33. All these are indications that compound 19 (or new compounds derived from it) may be developed into in vivo active drug like compounds (Table 2 and Fig. 6). Since compound 19 inhibits the proliferation of MDA-MB-231 cells and is suitable for oral dosing, we tested the in-vivo efficacy of compound 19 in mouse 4T1 cell-induced orthotopic breast tumor model by oral dosing (Fig. 7). Compound 19 arrested tumor progression but could not induce tumor regression indicating the somewhat cytostatic nature of compound 19 (Figs 2B and 7B). The compound also reduced metastasis of the highly metastatic 4T1 cells into the lungs in-vivo (Fig. 7E,F) which was observed by significantly less number of nodules in the lungs of animals treated with the compound. This was indicated earlier in our in-vitro scratch assay results, where MDA-MB-231 cells were observed to migrate significantly more slowly after treatment with compound 19, as compared to control (vehicle treated) cells (Fig. 4). These results confirm the oral suitability, anti-breast cancer activity and anti-metastatic activities of compound 19.
The mode of action and target specificity of compound 19 was studied by several methods. The observations of in-vitro studies (arrest of cell cycle progression and increase in DNA damage) prompted us to explore DNA replication and repair proteins as possible targets of this compound. In-silico docking studies revealed that compound 19 preferentially interacts with hLigI as compared to other DNA replication and repair proteins that were studied (Supplementary Table 3). The top hits from in-silico studies were validated by biochemical assays in-vitro and ex-vivo and revealed the highly specific ligase-inhibition activity of compound 19. This activity of compound 19 was entirely different from that of the parent compounds which showed no ligase inhibitory activity (Fig. 8). The target specificity of compound 19 was validated by the functional activity against various proteins like hLigIIIβ, hLigIV/XRCC4, T4 DNA ligase, FEN1 and PARP1 in the presence of different concentrations of compound 19 (Fig. 8 and Supplementary Figs 72 and 73). Cell lysate from MDA-MB-231 cells treated with compound 19 demonstrated the inhibition of ligase activity whereas flap cleavage activity remained the same (Supplementary Fig. 74). This demonstrated the cellular activity of compound 19 and its specific targeting of ligase I inside the cells.
Further, we went ahead and studied the direct binding of compound 19 with hLigI by various biophysical interaction studies such as fluorescence quenching assay, CD and EMSA. Our experiments showed that compound 19 interacts specifically with hLigI but not with DNA (Fig. 9). This specific interaction of compound 19 with hligI also explains the specific antiligase activity of compound 19.
In conclusion, this is the first report of an active hLigI inhibitor with in-vivo anti-breast cancer activity. The path to discovery and activity of compound 19 is summarized in Fig. 10. The compound was found to have stable pharmacokinetic parameters and drug like properties to making it a good lead for the synthesis of even better compounds in the future. Therefore, hLigI inhibitors have the potential in the future to help in the treatment of highly metastatic breast cancers that are hormone receptor negative (e.g., TNBC) or otherwise refractory to endocrine treatments. It is likely that in the future, DNA repair proteins (including hLigI inhibitors) will be used as adjuvant therapy with existing chemotherapeutic agents and may likely demonstrate excellent synergistic activity with different DNA damaging agents used in combination therapy34, 35. This aspect is currently under active investigation in our laboratory.

The proposed mode of action of compound 19 on 4T1 induced in vivo breast cancer model.
Materials and Methods
Chemistry
Typical procedure for the synthesis of E-benzocoumarin-stilbene hybrids (10-39) through Horner-Wadsworth-Emmons (HWE) reaction
To a solution of a suitable phosphonic acid diethyl ester (10–12) (1.0 mmol) in dry DMF (3 mL) at 0 °C MeONa (1.5 mmol) was carefully added. The mixture was stirred at the same temperature for 1 h, then the suitable aldehyde (1.0 mmol) in DMF (3 mL) was added drop wise and the reaction mixture was stirred at room temperature for 1.5 h and heated at 80 °C for 1 h. After completion of the reaction, the cooled reaction mixture was poured in to ice water and left to stand for 3 h. The solid precipitate was obtained by filtration, washed with diethyl ether. Crystallization from ethyl acetate gave the desired stilbene derivative as a pure E-isomer.
(E)-4-(3,5-dimethoxystyryl)-2H-benzo[h]chromen-2-one (19)
Light yellow solid; yield: 92% mp 180-181 °C; Anal.(%) for C23H18O4: Calcd., C, 77.08; H, 5.06;, found, C, 77.14; H, 5.03; IR (KBr) 3021, 2401, 1706, 1658, 1548, 1215, 928, ν/cm−1,1H NMR (300 MHz, DMSO-d 6) δ 8.42-8.40 (m, 1 H), 8.25 (d, J = 8.8 Hz, 1 H), 8.09-8.06 (m, 1 H), 7.91 (d, J = 8.8 Hz, 1 H), 7.82 (d, J = 16 Hz, 1 H), 7.75- 7.72 (m, 2 H), 7.63 (d, J = 16 Hz, 1 H), 7.04 (d, J = 2.2 Hz, 2 H), 6.88 (s, 1 H), 6.55 (t, J = 2.1 Hz, 1 H), 3.82 (s, 6 H);13C NMR (CDCl3, 75 MHz) δ 161.2, 161.1, 151.2, 151.0, 137.8, 137.5, 134.8, 128.7, 127.6, 127.2, 124.1, 123.4, 122.7, 121.4, 120.1, 113.9, 109.9, 105.6, 101.6, 55.5; ESI- MS: (m/z); 358, found [M + H]+ 359; HRMS-ESI: C23H19O4 [M + H]+ calcd 359.1283, found 359.1284.
Biological experiments
Reagents, media and antibodies
DNA oligos were purchased from Integrated DNA Technologies (IDT, India) MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide), trypsin-EDTA, propidium iodide, DMSO and protease inhibitor cocktail, were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell growth media DMEM and RPMI were purchased from Sigma-Aldrich (St. Louis, MO, USA), FBS antibiotic and antimycotic solution were purchased from Invitrogen (Whitefield, Bangalore). Fluorescein Isothiocyanate (FITC)-labelled Annexin V (Annexin V-FITC) kit was obtained from BD Biosciences (San Diego, CA). Antibodies against human poly (ADP-ribose) polymerase (PARP), γH2AX, cleaved Caspase 3, Caspase 9, P53, and MDM2 were purchased from Cell Signalling Technology (CST, Beverly, MA). PCNA and secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO)
Cell viability and clonogenic survival assay
Cell viability assay was performed as described previously19. Briefly, cells were seeded in 96 well plates and treated with different concentrations of compound 19 for 48 h. After drug treatment, MTT reagent was added in each well of the plate and cell viability was quantified by measuring the absorbance at 590 nm.
Clonogenic survival assay was performed as described previously16 with modifications. MDA-MB-231 cells (100 cells) were seeded in 12 well plates for 24 h. Cells were then treated with different concentrations of compound 19 (0.75, 1.5 and 3 µM) for 24 and 48 h. After drug treatment, culture medium was replaced with fresh medium and cells were cultured for another 10 days. After 10 days, the cells were washed with PBS carefully and stained with a solution containing 6% glutaraldehyde and 0.5% crystal violet.
Apoptosis and cell cycle distribution analysis
Apoptotic cell death of MDA-MB-231 cells upon treatment with compound 19 was measured using a commercially available Annexin V-FITC apoptosis detection kit according to the manufacturer’s protocol (BD Biosciences, San Diego, CA). Briefly, cells were treated with different concentrations (0, 6 and 12 µM) of compound 19 for 24 and 48 h. The cells were then harvested, washed with cold PBS and stained with Annexin V-FITC and propidium iodide (PI) in binding buffer at room temperature in the dark. The stained cells were analyzed by fluorescence-activated cell sorting using a FACS-Calibur instrument (BD Biosciences, San Diego, CA) equipped with CellQuest 3.3 Software.
For the study of cell cycle distribution, cells were synchronized by serum starvation for 48 h and then grown in serum containing media in the presence or absence of 12 µM compound 19, after 0, 24 and 48 h, cells were harvested, washed twice with cold PBS and fixed with 70% ethanol. The fixed cells were washed with chilled PBS and then stained with 50 μg/mL of Propidium iodide. Cell cycle distribution was analyzed by using FACS-Calibur (BD Biosciences, San Diago, CA).
Scratch assay
The migration capacity of highly metastatic human breast cancer MDA-MB-231 cells was assessed by in-vitro scratch assay as described previously36 with some modifications. Briefly, cells were plated in 12 well plates and a scratch was created by scraping the cells with a sterile 10 μL pipet tip in the middle of the culture well. After removal of cellular debris, the cells were treated with different concentrations of the test compound. Images of the wound area were captured at 0, 12 and 24 h post-treatment. Wound area was measured by TScratch software and data were expressed as percent (%) open area of control and treated cells.
Preparation of cell lysate and Western blotting
Cells (MDA-MB-231) were seeded in 6 well plates and treated either with vehicle control (DMSO) or with different concentrations (6 and 12 µM) of compound 19 for 48 h. After the treatment cell lysate was prepared and western blotting was performed as described previously37. Briefly, the cells were lysed by very brief sonication. Sonicated samples were centrifuged and the supernatant was collected and used for ligation assay and western blotting.
In-vitro stability of compound 19 in simulated gastrointestinal fluids
Simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) were prepared according to USP specifications. The compound 19 was incubated in SGF (0.2 g NaCl, 0.32 g pepsin, 0.7 mL HCl and triple distilled water up to 100 mL, pH 1.2) or SIF (0.680 g KH2PO2, 0.616 g NaOH, 1 g pancreatin and triple distilled water up to 100 mL, pH 6.8). From the stock solution of compound 19 (1 mg/mL), equivalent concentration of 2 μg/mL of compound 19 were added to 1 mL of preincubated reaction mixture of SGF and SIF, at 37 °C in a shaking water bath in triplicate. 100 μL aliquots of sample was collected at each time point at 0, 5, 15, 30 and 60 min for SGF and 0, 5, 15, 30, 60 90, 120 and 180 min for SIF according to the human gastrointestinal transit time, followed by quenching with acetonitrile, then vortexed and centrifuged at 12000 rpm for 5 min. The supernatant was collected and analyzed by HPLC. The graph was plotted between % drug remaining and incubation time for SGF and SIF.
Metabolic stability of compound 19
Major drug clearance in the body occurs due to its metabolism in the liver. Metabolic stability in rat liver microsomes (RLM) was conducted in triplicate. Compound 19 (5 µM) was incubated in reaction milieu which contains RLM (0.5 mg/ml), MgCl2 (10 mM), NADPH (10 mM) in a 1.5 ml phosphate buffer solution (pH 7.4). Reaction was started by the addition of NADPH and kept in a shaking water bath at 37 °C. Organic content in reaction mixture was kept less than 1%. Time points at which samples were withdrawn were 0, 5, 15, 30, 45, 60 min. Testosterone was used as control to know the RLM enzyme activity. Control reaction without NADPH was performed to explore any chemical instability or non-cofactor dependent enzymatic degradation. At each time point 150 µL aliquots were withdrawn and added in 150 µL ACN, vortexed at 10,000 g for 5 minutes, 100 µL samples was pipetted out & analyzed in HPLC-PDA.
In- vivo pharmacokinetic study of compound 19
Male Sprague Dawley (SD) rats weighing 200–220 g were obtained from the Laboratory of Animal Division of CSIR-CDRI (Lucknow, India). Animals were kept in well ventilated cages in hygienic conditions at room temperature (24 ± 2 °C), 40–60% relative humidity and 12 h light-dark cycle. The animals were acclimatized for a minimum period of 3 days prior to the experiments. Pharmacokinetic study was conducted according to protocol approved by the CSIR-CDRI Institutional Animal Ethic Committee (IAEC approval no. IAEC/2012/91). Compound 19 was dissolved in DMSO:Poly Ethylene Glycol-400: 0.9%w/v saline in the ratio of 1:1:2 and was intravenously administered at the dose of 5 mg/kg to each rat (n = 6). The rats were kept on fasting overnight with water alone for oral pharmacokinetic studies. Compound 19 was dissolved in DMSO:Poly Ethylene Glycol-400: 0.5% w/v Sodium carboxymethyl cellulose in TDW in the ratio of 3:1:4 and was orally administered at the dose of 50 mg/kg to each rat (n = 6). Blood samples were collected in microcentrifuge tubes for each time point at 0.083, 0.167, 0.333, 0.5, 0.75, 1.0, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0, 24.0, 48.0 and 72 h post dosing. The blood samples were collected after light ether anesthesia from the retro-orbital plexus of rats using heparin as anticoagulant. Plasma was separated after centrifugation at 5000 rpm for 10 min and all the samples were stored at −80 °C until analysis. Rat plasma samples were processed using liquid-liquid extraction. 100 µL aliquot of plasma mixed with 150 µL of acetonitrile and 100 µL of 5 mM NaHCO3 solution was added and vortexed to mix well. In each sample, 2.5 mL of tert-Butyl methyl ether (TBME) (extraction solvent) was added and vortexed for 10 min at 2500 rpm on vibramax (Heidolph, Schwabach, Germany) followed by centrifugation (Eppendorf, Hamburg, Germany) at 5,000 rpm for 5 min. An aliquot of 2.0 mL of supernatant organic layer was separated and evaporated on vacuum concentrator (Thermo scientific, Asheville, USA) to complete dryness. The residue was reconstituted with 100 µL of mobile phase and injected into the LC-MS/MS system for analysis. The plasma samples (100 mL) were assayed along with calibration standards and QC samples and the levels of compound 19 was calculated using Analyst software. The pharmacokinetic profile and parameters were evaluated by non-compartmental model approach using Pheonix 6.3 WinNonlin (Pharsight corporation, USA) including t1/2, elimination half-life; Cmax, maximum plasma concentration; tmax, time to reach maximum plasma concentration; AUC0-t, area under concentration curve from zero to last time point.
Determination of in-vivo tumor regression
Animal studies were conducted under the protocol approved by the CSIR-CDRI Institutional Animal Ethics Committee (IAEC approval no. IAEC/2015/03). The in-vivo efficacy studies were performed using BALB/c mice with 4T1 mouse breast cancer induced syngeneic breast tumor model. Mouse tumor model was generated as described by Pulaski et al.38. Three different groups of 6 mice each were created for the experiment. Mouse breast cancer cells (4T1) were grown in tissue culture flask upto 70% confluence. Cells were then trypsinized, washed with serum containing medium to quench trypsin activity and then washed and resuspended in PBS. Then cells were injected orthotopically (1 × 106 cells in 100 μl PBS) into the mammary fat pad on the right flank of each female BALB/c mouse. Within 2 weeks, palpable tumors were formed and tumor bearing mice were divided randomly into one control and two treatment groups of six mice each. The treatment schedule began on day 16 of the injection and continued for another thirty two days. Compounds were administered by oral gavage once daily at 50 and 100 mg/kg. Tumor volumes were measured every fourth day. The group of vehicle treated animals served as control. The tumor volume, body weight and any behavioral changes in mice were monitored and recorded for the entire duration of the experiment (32 days). Tumor volumes were calculated using the formula (a × b2)/2, where a, b are two longest perpendicular diameters.
In-silico docking studies of compound 19
The crystal structures of DNA replication proteins viz., human DNA Ligase I (hLigI), Human Topoisomerase 1 (Top1), Flap Endonuclease 1 (Fen1), Proliferating cell nuclear antigen (PCNA) and Poly [ADP-ribose] polymerase 1 (PARP1) were retrieved from the PDB [PDB ID: 1 × 9 N, 1T8I, 5FV7, 3WGW and 4ZZZ]. All the crystal structures were pre-processed before docking by eliminating any co-crystallized moieties. Subsequently hydrogen atoms were added and the structures were subjected to an energy minimization step using Poling algorithm with default settings in Sybyl7.139. The structure of compounds was also drawn using Sybyl7.1 followed by an energy minimization of 1000 steps using MMFF94 force field with other default settings. For all selected targets, co-crystal structure of the protein and the inhibitor was available and the inhibitor binding site was used as active site for docking experiments except for hLig1 for which the docking site was selected as done in our previous study17.
Purification of proteins (hLigI, hLigIIIβ, hLigIV/XRCC4)
The clones for hLigI (PRSFDuet-ligI), hLigIIIβ (PQE32-hLigIIIβ) and hLigIV/XRCC4 (PRSFDuet-hLigIV/XRCC4) were kind gifts from Prof. Alan Tomkinson (University of New Mexico, Albuquerque, USA). hLigI was purified as described previously19. The clone for hLigIIIβ was transformed into Rosetta2 (DE3) pLacI cells (Novagen) and the protein was expressed at 16 °C for 18 h with 1 mM IPTG. The clone for hLigIV/XRCC4 was transformed into Rosetta2 (DE3) pLacI cells (Novagen) and the protein was expressed at 25 °C for 12 h with 1 mM IPTG. The proteins were purified following the same procedure as for hLigI.
In-vitro DNA ligation assay
DNA ligation assay was performed as described previously17. Briefly, in the presence of nicked DNA substrate, different concentrations (5, 10 and 20 µM) of compound 19 incubated with different proteins (hLigI, hLigIIIβ, hLigIV/XRCC4 and T4 DNA ligase) at 37 °C for 30 minutes. After the incubation, gel images were captured and analyzed by Image Quant LAS4010 software.
In-vitro fluorescence quenching assay
In-vitro fluorescence quenching assay was performed as described previously16 with brief modifications. At the fixed excitation wavelength 280 nm emission spectra of hLigI (100 pmol) was recorded from 300 to 400 nm in the presence (5 and 10 µM) or absence of compound 19 by spectrofluorometer. Compound 19 was used at 5 and 10 µM concentration with hLigI (100 pmol). Spectra was acquired for buffer alone (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM ATP, 10 mM MgCl2, 2 mM DTT and 5% glycerol), buffer with vehicle control, buffer with inhibitor alone and for buffer with protein and inhibitor.
Circular Dichroism (CD) study
We followed previously described protocol for CD studies40 with small modifications. Briefly, emission spectra of hLigI (100 pmol) was recorded at a wavelength of 190-250 nm in the presence (5 and 10 µM) or absence of compound 19 by spectropolarimeter. The raw values in mdeg were obtained and used for mean residual ellipticity [θ] calculation. The graph was generated by the plotting the value of mean residue ellipticity in deg cm2 dmol−1 with respect to wavelength.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described previously17. Briefly, in 20 μl reaction volume 2 pmol non-ligatable nicked DNA substrate was incubated with DMSO (vehicle control) or with different concentrations (120, 250, 500 µM) of compound 19 incubated with 10 pmol of hligI in ligation buffer. After the incubation, samples were separated by 6.5% native PAGE and bands were detected and imaged by Image Quant LAS 4010 (GE Life sciences).
DNaseI cleavage protection assay
We performed DNaseI cleavage assay as described in our previous study37. Briefly, 100 ng of plasmid DNA (pUC18) was incubated with 100 µM concentration of doxorubicin (a known DNA intercalator), DAPI (DNA minor groove binder), methyl green (DNA major groove binder) and compound 19 (at 250 and 500 µM concentrations) for 30 min at 37 °C in an incubation buffer similar to ligation buffer. After incubation, the DNA was cleaved by adding 0.0025U of DNaseI in each reaction mixture for 5 min at 37 °C and the reactions were stopped by adding stop buffer which contained 7 M Urea, 20 mM Tris-Cl (pH. 7.5), 50 mM EDTA, 20% glycerol and Bromophenol blue. The samples were resolved on 1% agarose gel and visualized under UV transilluminator and image taken with GE Image quant LAS4010.
References
Viegas-Junior, C., Danuello, A., da Silva Bolzani, V., Barreiro, E. J. & Fraga, C. A. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr Med Chem 14, 1829–1852, doi:10.2174/092986707781058805 (2007).
Meunier, B. Hybrid molecules with a dual mode of action: dream or reality? Acc Chem Res 41, 69–77, doi:10.1021/ar7000843 (2008).
Tietze, L. F., Bell, H. P. & Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew Chem Int Ed Engl 42, 3996–4028, doi:10.1002/anie.200200553 (2003).
Tomkinson, A. E. & Mackey, Z. B. Structure and function of mammalian DNA ligases. Mutat Res 407, 1–9 (1998). DOI: S0921-8777(97)00050-5.
Soderhall, S. Properties of a DNA-adenylate complex formed in the reaction between mammalian DNA ligase I and DNA containing single-strand breaks. Eur J Biochem 51, 129–136, doi:10.1111/j.1432-1033.1975.tb03913 (1975).
Soderhall, S. & Lindahl, T. Mammalian deoxyribonucleic acid ligase. Isolation of an active enzyme-adenylate complex. J Biol Chem 248, 672–675 (1973).
Prigent, C., Satoh, M. S., Daly, G., Barnes, D. E. & Lindahl, T. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol Cell Biol 14, 310–317 (1994).
Ellenberger, T. & Tomkinson, A. E. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem 77, 313–338, doi:10.1146/annurev.biochem.77.061306.123941 (2008).
Lehmann, A. R. et al. Relation between the human fibroblast strain 46BR and cell lines representative of Bloom’s syndrome. Cancer Res 48, 6343–6347 (1988).
Tomkinson, A. E. & Levin, D. S. Mammalian DNA ligases. Bioessays 19, 893–901, doi:10.1002/bies.950191009 (1997).
Sun, D. et al. Elevated expression of DNA ligase I in human cancers. Clin Cancer Res 7, 4143–4148 (2001).
Pascal, J. M., O’Brien, P. J., Tomkinson, A. E. & Ellenberger, T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature 432, 473–478, doi:10.1038/nature03082 (2004).
Zhong, S. et al. Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J Med Chem 51, 4553–4562, doi:10.1021/jm8001668 (2008).
Chen, X. et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res 68, 3169–3177, doi:10.1158/0008-5472.CAN-07-6636 (2008).
Srivastava, M. et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 151, 1474–1487, doi:10.1016/j.cell.2012.11.054 (2012).
Shameem, M. et al. Synthetic modified pyrrolo[1,4] benzodiazepine molecules demonstrate selective anticancer activity by targeting the human ligase 1 enzyme: An in silico and in vitro mechanistic study. Chem Biol Interact 237, 115–124, doi:10.1016/j.cbi.2015.05.024 (2015).
Krishna, S. et al. Pharmacophore-based screening and identification of novel human ligase I inhibitors with potential anticancer activity. J Chem Inf Model 54, 781–792, doi:10.1021/ci5000032 (2014).
Singh, D. K. et al. Human DNA ligases: a comprehensive new look for cancer therapy. Med Res Rev 34, 567–595, doi:10.1002/med.21298 (2014).
Mandalapu, D. et al. Discovery of monocarbonyl curcumin hybrids as a novel class of human DNA ligase I inhibitors: in silico design, synthesis and biology. RSC Adv 6, 26003–26018, doi:10.1039/c5ra25853g (2016).
Pandey, M. et al. Identification and characterization of novel ligase I inhibitors. Mol Carcinog 56, 550–566, doi:10.1002/mc.22516 (2016).
Choi, H. Y. & Chi, D. Y. Nonselective bromination-selective debromination strategy: selective bromination of unsymmetrical ketones on singly activated carbon against doubly activated carbon. Org Lett 5, 411–414, doi:10.1021/ol0271638 (2003).
Dong, Y. et al. Antitumor agents. 272. Structure-activity relationships and in vivo selective anti-breast cancer activity of novel neo-tanshinlactone analogues. J Med Chem 53, 2299–2308, doi:10.1021/jm1000858 (2010).
Egbe, D. A. M. B., Nowotny, C., Günther, J., Klemm, W. & Investigation, E. of the Photophysical and Electrochemical Properties of Alkoxy-Substituted Arylene-Ethynylene/Arylene-Vinylene Hybrid Polymers. Macromolecules 15, 5459–5469, doi:10.1021/ma0301395 (2003).
Solntsev, K. M., McGrier, P. L., Fahrni, C. J., Tolbert, L. M. & Bunz, U. H. Anomalous photophysics of bis(hydroxystyryl)benzenes: a twist on the para/meta dichotomy. Org Lett 10, 2429–2432, doi:10.1021/ol8006925 (2008).
Flaherty, D. P., Kiyota, T., Dong, Y., Ikezu, T. & Vennerstrom, J. L. Phenolic bis-styrylbenzenes as beta-amyloid binding ligands and free radical scavengers. J Med Chem 53, 7992–7999, doi:10.1021/jm1006929 (2010).
Gotzmann, J. et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res 566, 9–20 (2004). DOI: S1383-5742(03)00033-4.
Kuo, L. J. & Yang, L. X. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo 22, 305–309 (2008).
Los, M. et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 13, 978–988, doi:10.1091/mbc.01-05-0272 (2002).
Takata, M. et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17, 5497–5508, doi:10.1093/emboj/17.18.5497 (1998).
Branzei, D. & Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9, 297–308, doi:10.1038/nrm2351 (2008).
Takeda, D. Y. & Dutta, A. DNA replication and progression through S phase. Oncogene 24, 2827–2843, doi:10.1038/sj.onc.1208616 (2005).
Mjelle, R. et al. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst) 30, 53–67, doi:10.1016/j.dnarep.2015.03.007 (2015).
Lipinski, C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1, 337–341, doi:10.1016/j.ddtec.2004.11.007 (2004).
Yadav, B. S., Sharma, S. C., Chanana, P. & Jhamb, S. Systemic treatment strategies for triple-negative breast cancer. World J Clin Oncol 5, 125–133, doi:10.5306/wjco.v5.i2.125 (2014).
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol 13, 674–690, doi:10.1038/nrclinonc.2016.66 (2016).
Liang, C. C., Park, A. Y. & Guan, J. L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2, 329–333, doi:10.1038/nprot.2007.30 (2007).
Singh, D. K. et al. Identification of a novel human DNA ligase I inhibitor that promotes cellular apoptosis in DLD-1 cells: an in silico and in vitro mechanistic study. RSC Adv 6, 94574–94587, doi:10.1039/C6RA22364H (2016).
Pulaski, B. A. & Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr Protoc Immunol Chapter 20, Unit 20 22, doi:10.1002/0471142735.im2002s39(2001).
UNITY, Sybyl v. 7.1 (Tripos, Inc, St. Louis, MO, 2004).
Koneni, V. S. et al. Design, synthesis and anticancer activity of dihydropyrimidinone-semicarbazone hybrids as potential Human DNA Ligase1 inhibitors. Med. Chem. Commun. 7, 2349–2363, doi:10.1039/C6MD00447D (2016).
Acknowledgements
The authors acknowledge CSIR-CDRI, Govt. of India, for financial and infrastructural support. Financial support is also acknowledged from Department of Biotechnology (DBT), Govt. of India (Grant-BT/PR6421/GBD/27/436/2012), the Department of Science and Technology (DST), Govt. of India (Grant-SB/FT/LS-163/2012) and CSIR Network projects GENESIS (BSC0121) and Biopros (BSC0106). The authors are also thankful to Sophisticated Analytical Instrument Facility (SAIF) CDRI, Lucknow, India, for providing spectral analysis data of chemical compounds and for support with cell based flow cytometry experiments. MKH and DKS are thankful for their Senior Research fellowships from Council of Scientific and Industrial Research (CSIR). The manuscript bears the CDRI communication number 9558.
Author information
Authors and Affiliations
Contributions
Mohd. Kamil Hussain synthesized the compounds and Deependra Kumar Singh performed key biological experiments in the manuscript. Akhilesh Singh, Mohd. Asad, Mohd. Imran Ansari, Mohammad Shameem, Shagun Krishna, Guru R Valicherla, Vishal Makadia, Sanjeev Meena, Amit Laxmikant Deshmukh, Jiaur R Gayen, Mohammad Imran Siddiqi, performed supporting experiments in the manuscript. Dibyendu Banerjee, Kanchan Hajela, Dipak Datta, designed the study and the experiments, gave key observations and helped prepare the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hussain, M.K., Singh, D., Singh, A. et al. A Novel Benzocoumarin-Stilbene Hybrid as a DNA ligase I inhibitor with in vitro and in vivo anti-tumor activity in breast cancer models. Sci Rep 7, 10715 (2017). https://doi.org/10.1038/s41598-017-10864-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10864-3
This article is cited by
-
Synthesis and anticancer activity of novel coumarin-stilbene hybrids with different hydrocarbon chains as linkers
Medicinal Chemistry Research (2024)
-
Synthesis, cytotoxicity, Pan-HDAC inhibitory activity and docking study of new N-(2-aminophenyl)-2-methylquinoline-4-carboxamide and (E)-N-(2-aminophenyl)-2-styrylquinoline-4-carboxamide derivatives as anticancer agents
Medicinal Chemistry Research (2023)
-
Novel Curcumin Monocarbonyl Analogue-Dithiocarbamate hybrid molecules target human DNA ligase I and show improved activity against colon cancer
Medicinal Chemistry Research (2023)
-
DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy
Signal Transduction and Targeted Therapy (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
