
Abstract
In the melon exotic accession PI 161375, the gene cmv1, confers recessive resistance to Cucumber mosaic virus (CMV) strains of subgroup II. cmv1 prevents the systemic infection by restricting the virus to the bundle sheath cells and impeding viral loading to the phloem. Here we report the fine mapping and cloning of cmv1. Screening of an F2 population reduced the cmv1 region to a 132 Kb interval that includes a Vacuolar Protein Sorting 41 gene. CmVPS41 is conserved among plants, animals and yeast and is required for post-Golgi vesicle trafficking towards the vacuole. We have validated CmVPS41 as the gene responsible for the resistance, both by generating CMV susceptible transgenic melon plants, expressing the susceptible allele in the resistant cultivar and by characterizing CmVPS41 TILLING mutants with reduced susceptibility to CMV. Finally, a core collection of 52 melon accessions allowed us to identify a single amino acid substitution (L348R) as the only polymorphism associated with the resistant phenotype. CmVPS41 is the first natural recessive resistance gene found to be involved in viral transport and its cellular function suggests that CMV might use CmVPS41 for its own transport towards the phloem.
Similar content being viewed by others


Introduction
Plant viruses are considered to be responsible for half of the emerging infectious diseases in plants1. Moreover, they can cause complete harvest lost and substantial economic losses, when they affect crops (reviewed by ref. 2). The use of naturally resistant cultivars is one of the most successful control measures against viral infections. While introgression, selection and breeding have been used for many years to develop resistant cultivars, nowadays the identification of the host factors governing resistance is providing potential targets for improving viral disease management.
About half of the approximately 200 known virus resistance genes in plants are recessively inherited3. Considering that recessive resistance seems to be more durable than dominantly inherited resistance4, focusing research on the genes involved provides a key tool to control plant diseases. The vast majority of the recessive resistance genes in plants identified to now encode eukaryotic translation initiation factors (eIFs), implicated in viral translation, but also in viral replication and in some cases, in cell-to-cell movement (for a review see refs 5 and 6). Significant advances have been recently achieved in the identification of host factors different than eIFs as naturally occurring recessive resistance genes in different pathosystems. For example, in barley, a protein disulfite isomerase-like (HvPDIL5-1), involved in protein folding, is encoded by the gene rym11 and confers resistance to Bymoviruses7. In Arabidopsis thaliana, rwm-1 encodes a phosphoglyceratekinase implicated in Watermelon mosaic virus accumulation8. As eIFs have been used in several plant species to identify or develop new resistant sources6, 9,10,11,12, identifying host factors other than eIFs involved in other steps of the viral cycle will provide new insights to understand plant-pathogen interactions and to identify new targets to develop resistant cultivars.
Cucumber mosaic virus (CMV), the type member of the Cucumovirus genus, has the broadest host range among viruses, being able to infect more than 1,200 species, including important crop plants within the Solanaceae, Cruciferae and Cucurbitaceae families, where they can cause severe damage worldwide13. Such extended host range involves an important genomic variability in CMV, which results in a high number of strains, usually classified into two subgroups, I and II, on the basis of their sequence14. Genetic control of resistance to CMV is heterogeneous, including dominant, recessive, monogenic and polygenic controls. The most common is recessive and polygenic and results in an impairment of long-distance movement of the virus15,16,17,18. In melon (Cucumis melo L.), studies evaluating a high number of accessions from different geographical origins reported three main strain-specific sources of resistance against CMV: “Freeman´s Cucumber” melon, the Japanese accession C-189, and the Korean accession PI 161375 cultivar “Songwhan Charmi”(SC)19,20,21,22. The CMV resistance present in SC was described as recessive, oligogenic21 and quantitative, with several QTLs involved and a major QTL mapping in linkage group XII (LGXII)23. Further studies demonstrated that cmv1, a single gene mapping in LG XII was able to confer by itself total recessive resistance to CMV subgroup II strains24, 25. Besides, at least two additional QTLs (cmvqw3.1 and cmvqw10.1) must act together and cooperatively with cmv1 to confer resistance to the more aggressive subgroup I strains26.
Reassortants and chimaeras between CMV-FNY (subgroup I) and CMV-LS (subgroup II) revealed that the determinant of the virulence against cmv1 was the movement protein (MP). Moreover, a combination of four residues or group of residues in the MP of CMV-FNY was identified as the only requirement of the MP to overcome cmv1-mediated resistance25. CMV-LS, was able to replicate and move cell-to-cell in the resistant plant. However, it was never detected in the phloem. Immunogold labeling experiments established that the virus was present in the bundle sheath (BS) cells of the resistant plant, but never reached the vascular parenchyma (VP) cells or the Intermediary cells (IC)27. Therefore, Cmv1/cmv1allows or prevents systemic infection by acting as a molecular gate, only in the bundle sheath (BS) cells, and determining the entrance of the virus in the phloem in a manner dependent on the viral MP present. Thus, cmv1 represents a new host factor implicated on a poorly studied step of viral infections, the transport from mesophyll to phloem to establish a systemic infection.
In the present study, we have first narrowed the interval carrying cmv1 from 2.2cM24 to a region of 132 Kb and demonstrate that cmv1 encodes a Vacuolar Protein Sorting 41 (VPS41), a protein involved in vesicle trafficking from late Golgi to the vacuole. We show that the transgenic expression of the susceptible Piel de Sapo (PS) allele of CmVPS41 in the SC resistant background is able to restore the infection by CMV and that a TILLING mutant in the CmVPS41gene impairs CMV-LS infection. Besides, a collection of 52 melon accessions was used to identify the causal polymorphism in the CmVPS41 gene. This is the first cloning of a natural recessive resistance gene involved in a new mechanism of resistance based on blocking long distance viral transport by preventing virus entry into the phloem.
Results
Fine mapping of cmv1
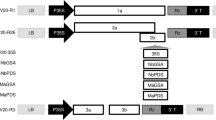
The melon cmv1 gene, which confers total resistance to CMV strains from subgroup II25, was located in LG XII within a 5.78 Mb interval flanked by markers CMN61_44 and CMN21_5524. Fine mapping of cmv1 was addressed using an F2 population from a cross between the susceptible accession PS and the NIL SC12-1, which carries the resistant allele24. A first set of 780 F2 individuals, screened with the above flanking markers, allowed identifying 55 recombinant individuals. Genotyping of those individuals with eight SSR markers distributed in the 5.78 Mb interval (Fig. 1a, Supplementary Table S1) identified five different recombination regions. Inoculation of F3 plants with CMV-LS, mapped cmv1 within an interval of 1.37 Mb flanked by SSR markers Sca02308.2 and Sca07080.7. The 14 recombinants lying within this 1.37 Mb interval were then genotyped with internal markers, defining a 373 Kb interval, from marker M2R2 to M1. Finally, three out of 14 recombinants allowed reducing the cmv1 interval to 132Kb between markers Sca4_345 and Sca4_358. Since no further recombinants in this interval were available, we carried out a screening of a new set of 3455 F2 individuals that finally rendered only three additional recombinants between those markers. The genotyping of these individuals with internal markers showed that their recombination breakpoints were located between markers Sca4_358 and Internal 4. Subsequent phenotyping of the corresponding F3 progeny did not allow us to further reduce the cmv1 interval (Fig. 1a).

Fine mapping of cmv1. (a) Sequential intervals screened during fine mapping. Each horizontal bar represents a group of recombinants inside the region, colors indicate the origin of each region. Red, SC; green, PS; yellow, heterozygous and white, recombination interval. The phenotype of the recombinants is noted at the right. R, S and R/S for resistant, susceptible and segregating F3 plants. (b) Annotated features inside the smallest cmv1 interval.
Functional analysis of cmv1 interval and candidate gene identification
According to the available in silico annotations of the melon genome, the 132 Kb cmv1interval contains eight predicted genes and part of the MELO3C004827 gene (Fig. 1b, Supplementary Table S2). Five of them correspond to sequences smaller than 500 bp, not validated by ESTs. Thus, they are unlikely functional genes. The four remaining genes are MELO3C004835, encoding a possible Lysosomal Pro-X carboxypeptidase (PCP), MELO3C004833, encoding a Crooked neck-like protein 1 (CNL) and two annotated genes, MELO3C004831 and MELO3C004827, that correlate with the C-terminal and N-terminal part, respectively, of a Vacuolar protein sorting-associated protein 41 (VPS41). Analysis of the polymorphisms between both parental lines PS and SC in the 132 Kb cmv1 interval28 revealed no variable transposon insertions or structural variations. However, they detected 1,318 polymorphisms including SNPs and INDELs. From those polymorphisms, 283 lay inside or close (1 kb up/downstream) to the PCP, CNL or VPS41 genes, and could modify their function. We detected 10 SNPs inside the exons of the three genes, although only five of them were non-synonymous: one in PCP, one in CNL and three in MELO3C004827 VPS41. When the predicted VPS41 ORFs were manually curated, the results strongly suggested that both predicted genes corresponded to a single ORF whose genomic sequence would span 28.9Kb and would have 19 exons (Fig. 2 and Supplementary Table S3). Cloning of the complete VPS41 cDNA from both PS and SC lines confirmed that, indeed, it was a unique ORF of 2,883 bp. Thus the cmv1 interval includes 3 likely genes and five non-synonymous polymorphisms between PS and SC that might be responsible for the resistance to CMV. Considering that cmv1 blocks the movement of the virus from the BS cells to the phloem cells27, our main candidate gene was CmVPS41, since it is involved in the transport of vesicles and cargo proteins towards the vacuole29, 30 and this function could be recruited by the virus for its own transport. In fact, VPS41 has been implicated in animal virus release from the infected cell31.

Structure of CmVPS41 gene. (a) CmVPS41 structure according to annotation on the melon genome. Both genes MELO3C004831 and MELO3C004827 correspond to a single open reading frame. The genomic DNA expands 28.9Kb. (b) CmVPS41 gene structure based on the complete coding sequence. The 19 exons produce a 2,883 bp cDNA. SNPs between PS and SC are detailed below in blue. In red, the amino-acid changes in exons 4, 5 and 13. The red line demarks one of the ends of the cmv1interval.
cmv1 encodes a Vacuolar Protein Sorting 41
Validation of CmVPS41 as the cmv1 gene was carried out by two complementary approaches. First, we generated transgenic susceptible melon plants expressing the dominant PS susceptible allele in the resistant SC background. As a second approach, we searched for CmVPS41 mutants with enhanced resistance to CMV-LS in a susceptible background. The susceptible PS allele was cloned under its own promoter and transformed into the SC resistant accession to generate three independent lines expressing the gene (Supplementary Figure S1). Given that cmv1is sufficient to confer resistance to CMV-LS, but also necessary for resistance to CMV-FNY25, 26, expression of the PS allele of CmVPS41 should allow both CMV-LS and CMV-FNY to spread in the transgenic plants. The three transgenic T0 lines were tetraploid, a frequent event in melon transformation32, 33. Thus, as we could not obtain diploid lines after self-pollination, we directly used the T0 generation for phenotyping. In vitro replicated T0 clones from the three independent transgenic lines were newly rooted, acclimated and then inoculated with either CMV-LS or CMV-FNY. CMV-LS infection was detected as a mosaic only in one line (SC-T0-17), two leaves above the inoculated leaves and was subsequently confirmed by RT-PCR using specific primers from CMV-LS (Fig. 3a). However, inoculation with CMV-FNY produced mosaic symptoms in all plants from the three transgenic lines, whereas the untransformed in vitro regenerated SC control plants remained symptomless and untransformed in vitro regenerated PS control plants were systemically infected (Fig. 3c). Infection was confirmed by detection of CMV-FNY by RT-PCR using specific primers from CMV-FNY (Fig. 3b). Thus, the results show that the expression of the susceptible CmVPS41allele in the resistant SC accession allows CMV systemic infection and, therefore, that CmVPS41 protein is indeed encoded by Cmv1. The limited infection by CMV-LS suggests that as the transgenic lines are tetraploid, and they may carry only one copy of the susceptible allele, the limited amount of PS CmVPS41 protein could be a limiting factor for CMV-LS infection.

CMV infection of T0 transgenic plants expressing PS CmVPS41. Representative results 21 days after the inoculation with either CMV-LS or CMV-FNY are shown (a). RT-PCR detection of CMV-LS-infected plants. (b) RT-PCR results after inoculation with CMV-FNY. (c) In vitro acclimatized T0 transgenic plants inoculated with CMV-FNY. PS: non-transgenic, in vitro regenerated susceptible line; SC: non-transgenic, in vitro regenerated resistant line; SC-T0: transgenic SC lines (16, 17 and 38) expressing CmVPS41 from PS under its own promoter. C- negative RT-PCR control. Arrows indicate the 1200 bp band of the size marker.
Screening of 6,200 M2 families of a TILLING population developed on the CharMono background34, a melon accession susceptible to CMV, identified 14 mutants, with six of the mutations lying within the coding sequence (Fig. 4a and Supplementary Table S4). One of the mutations was synonymous, two of them produced amino acid changes predicted as neutral by PROVEAN software and three were predicted as deleterious. The TILLING mutants were subjected to three rounds of self-pollination selecting for the five non-synonymous mutations in CmVPS41 to get homozygous plants and to prevent the presence of contaminating mutations in other parts of the genome related to the phenotype. After challenging plants of the five families carrying non-synonymous substitutions with CMV-LS, all of them showed infected plants, indicating that none of the mutations conferred total resistance to the virus. However, mutant plants carrying the mutation D360N showed milder symptoms than CharMono, the susceptible control parental line of the TILLING population. Moreover, after 21 dpi, the plant upper leaves of the mutant D360N seemed to recover, unlike PS and CharMono susceptible control plants. CMV detection in the fourth leaf of each plant at 21 dpi showed that, while in some of the mutant plants the virus was still present, the viral load was significantly lower when compared with PS (p-value = 9.306e-06) and CharMono (p-value = 0.0379) plants (Fig. 4b). The SC 12-1-99 subNIL resistant control carrying cmv1 remained uninfected. The experiment with the mutant D360N was repeated three times, inoculating 10 plants in each experiment, with similar results. Interestingly, the mutation D360N was predicted as the most deleterious for the CmVPS41 protein of all TILLING mutations found (Supplementary Table S4). Thus, these results indicate that mutations in the CmVPS41 gene lead to lower susceptibility to CMV-LS, confirming that CmVPS41 is encoded by cmv1.

(a) Amino acid sequence of the CmVPS41 fragment screened in the TILLING population (exons 5 and 6). The sequence was aligned against the corresponding sequence from representative plant species. Detailed below are the nonsynonymous TILLING mutants detected. (b) CMV-LS accumulation in the inoculated D360N TILLING family. Samples were collected after 21dpi. ELISA results from the fourth true leaf from a representative experiment are shown. PS, Piel de Sapo susceptible line; SC12-1-99, introgression line carrying cmv1; CM, CharMono susceptible TILLING parental line; D360N, TILLING family carrying the mutation D360N in CmVPS41. *p-value for the t-test comparing against M3425 lower than 0.05, **lower than 0.01, ***lower than 0,001.
Identification of a mutation associated with the resistant phenotype by the analysis of a diverse collection of melon accessions
Out of the three non-synonymous changes between PS and SC found in exons 4, 5 and 13 of CmVPS41 (Fig. 2), only the substitution L348R at exon 5 is predicted as deleterious for the protein by PROVEAN and MutPred software (Supplementary Table S5), whereas the substitutions P262A and S620P should be neutral. This suggests that the change L348R could be the causal polymorphism of the resistant phenotype given by the SC allele of CmVPS41. To confirm this hypothesis, we genotyped CmVPS41 in a collection of 52 diverse melon accessions35, 36 with molecular markers developed to detect the three SNPs (Supplementary Table S1) and studied the correlation of each mutation with the susceptibility or resistance to CMV-LS. The collection included 41 accessions of C. melo ssp.melo and 11 C. melo ssp. agrestis (Supplementary Table S6). Most of the accessions carried the SC allele at exons 4 and 13, and the PS allele at exon 5.The PS haplotype was shared by only four accessions from the cantalupensis group, all French varieties. Can-PMRUSA a reticulatus type, and Am-Korsa-Rus an ameri type, carried PS alleles at exons 5 and 13, and SC allele at exon 4. Con-GMJa, an Asiatic melon belonging to the conomon type, was the only accession that shared SC haplotype and carried the SC allele at exon 5 (Supplementary Table S6). A subset of nine melon accessions carrying all observed haplotypes were selected for phenotyping: five with the most frequent haplotype (In-Kirk-Turk, In-Ps-PipaSp, Mom-PI124Ind, Ag-Tri Ind and Mom-MR1Ind) two with PS haplotype (Can-Ved Fran and Can-NC Fran), the one with SC haplotype (Con-GM Ja) and one with SC allele only at exon 4 (Can-PMR USA) (Table 1). After challenge with CMV-LS, all of them were systemically infected except Con-GM Ja, the only one carrying the SC allele at the SNP in exon 5 (Table 1). Thus, the only SNP associated with the resistance was located in CmVPS41 exon 5, suggesting that the mutation L348R is the causal polymorphism for the resistance given by cmv1. Accordingly, real-time experiments to monitor gene expression between PS and SC showed no significant variations in leaf, vein and petiole tissues of both parental lines (Supplementary Figure S2), confirming that the causal mutation was located in the coding sequence and not in regulatory regions.
VPS41 gene is present as a single gene in most plant species
VPS41 gene is part of the “Homotypic Fusion and Vacuole Protein Sorting” (HOPS) complex, which is present in a wide range of species from animals to yeast37. A search in the PLAZA 3.0 database (http://bioinformatics.psb.ugent.be/plaza/), which contains data of plant genomes that have been fully sequenced, shows that CmVPS41 belongs to the orthologue family ORTHO03D004047. This gene is present in 29 species out of 31 included in PLAZA 3.0 and in 22 of these species the gene is present as a single copy (Supplementary Table S7), as it is the case in C. melo. In these species, VPS41 might also have a role in CMV transport in the susceptible species. The coded protein, contains a Clathrine domain, characteristic of many proteins involved in vacuolar maintenance and protein sorting38, and a WD-40 domain, presumably involved in protein-protein interaction. The causal polymorphism L348R is outside both of them and is located in a highly conserved region of the gene (Fig. 4a, Supplementary Figure S3). All species, including C. melo, have a Leucine at position 348 except the resistant melon accession SC, which has an Arginine. The location and identity of the mutation L348R in this conserved region, where only one TILLING mutant (D360N) showed a mild effect on CMV resistance, suggests that this could be the key amino-acid for the interaction with CMV and suggests the possibility of testing the effect of this mutation in other species susceptible to CMV.
Discussion
In melon, cmv1 confers recessive resistance to CMV strains of subgroup II. It is also necessary to confer resistance to subgroup I strains. However, in this case, the cooperation of other two QTLs with cmv1is also required25. Here we have identified CmVPS41, a protein involved in membrane trafficking to the vacuole as part of the “Homotypic Fusion and Vacuole Protein Sorting” (HOPS) complex, as the product of cmv1. We have validated CmVPS41 as cmv1 both by generating transgenic susceptible lines expressing the susceptible allele in the resistant parental line, and by characterizing a TILLING mutant with reduced susceptibility to CMV-LS.
The HOPS complex is involved in regulating the vesicle transport from the late endosome, but also directly from the Golgi complex, through the AP-3 pathway, to the vacuole37. It is a multiprotein complex composed of six subunits. Four of them constitute the core proteins and are shared with CORVET, another complex guiding endosome life cycle. The remaining two subunits, VPS41 and VPS39, are specific for HOPS and confer the tethering function that enables HOPS to promote vacuolar fusion, being VPS41 the effector subunit38,39,40,41.
Our previous studies have determined that Cmv1/cmv1 is a checkpoint for CMV phloem entry, acting only in the BS cells. The susceptible allele Cmv1would permit virus transport from BS cells to VP cells or ICs to develop a systemic infection, whereas the resistant allele cmv1 would prevent this movement27. According to our data, a single amino acid change (L348R) in the CmVPS41 protein would modify the putative interaction with the CMV movement protein (MP), the viral interacting factor that determines the onset of the systemic infection25. Therefore, in the resistant plant, this putative interaction would not happen. However, a set of four simultaneous mutations in the MP of the virus are able to restore the systemic infection in the resistant plant25, suggesting that these changes are able to restore the putative interaction with CmVPS41encoded by the resistant plant as well. Altogether, this suggests that, in the susceptible plant, CMV could be recruiting some CmVPS41 activity either to mediate its own transport towards the PDs or alternatively, it could be needed already in the PDs to gate them for CMV movement to the phloem. In this scenario, the main activity of CmVPS41 in vesicle fusion would remain unchanged, since both resistant and uninfected susceptible plants have the same phenotype. Likewise, zip-2, an Arabidopsis missense mutant in AtVPS41 in a highly conserved amino acid has no physiological or morphological phenotype but is able to suppress zig-1, a mutant impaired in membrane trafficking towards the vacuole. It has been suggested that the zip-2 mutation alters part of AtVPS41 function, but the essential function remains intact42. The inability to obtain null mutants from a T-DNA insertion collection strongly suggests lethality in homozygosis of AtVPS41 mutants. In a similar way, in our TILLING screening in the CmVPS41gene, we did not identify any stop codon mutant, supporting again an essential function of CmVPS41 for the cell. In fact, out of the five mutants identified presenting amino acid change, only one showed an enhanced (but not fully) resistant phenotype. The rest did not show any altered phenotype. This is expected for a gene with an essential function that cannot be easily modified without strongly compromising plant viability.
Recessive resistance genes, like cmv1, are host factors recruited by the virus to complete their cycle, and thus they can be exploited to protect plants from viral infections. Most of the natural recessive resistance genes identified and cloned to date encode translation initiation factors (eIFs)5, 43 and are involved in virus replication. Since first identified in pepper44, their key role in viral infections has also been demonstrated in more than 20 plant species and 30 viruses and have been exploited as targets to produce plants resistant to a broader spectrum of viruses in several crop species (reviewed by Sanfaçon, 2015). Only a few examples of natural recessive resistance involve genes other than eIFs, although they also restrict virus multiplication7, 8. cmv1 is the first natural recessive resistance gene whose function has been located in a particular cell type, where it is involved in the transport of the virus to the phloem to produce a systemic infection27. Since cmv1 is able to block CMV-LS in the BS cells and VPS41 is involved in intracellular membrane trafficking, this suggests that CmVPS41 protein is involved in CMV transport in the boundary BS cells/phloem, rather than in viral replication. Our findings suggest a new mechanism for a natural recessive resistance whose molecular characterization could provide a whole new range of plant virus control strategies targeting either CmVPS41 or other participants in the same step of CMV systemic transport. Furthermore, the identification of the causal mutation in CmVPS41 enables the directed gene editing by CRISPR/Cas9 to generate melon resistant plants and this possibility could also be explored in the VPS41 gene from other crops.
To date, while no other VPS has been demonstrated to be involved in a natural virus resistance, VPSs and other components of the vesicle trafficking machinery have been implicated in virus life cycle. Most of both animal and plant viruses associate to the host secretory pathways components for different purposes: to accomplish replication, but also for movement and budding45, 46. In animals, enveloped viruses exploit the endomembrane system to enter their hosts47. The VPS pathway is necessary for the release of Marburg virus through VPS431, whereas hVPS41 is increasingly associated with human pathologies and together with other HOPS components, was shown to be essential for Ebolavirus infections48. Several plant viruses are known to interact with components of membrane trafficking complexes on its own benefit, therefore facilitating the transport and replication of the viral material. For example, the Tombusvirus Tomato bushy stunt virus (TBSV) interacts through its p33 protein with VPS23 and Vps4p, two members of ESCRT, another complex involved in endosome life cycle mediating vesicle formation49. VPS23 is also the interacting protein for Carnation Italian ringspot virus (CIRV) p36, involving the ESCRT complex with viral replication50. COP II, the complex that regulates vesicle trafficking between ER and GA, is also involved in replication of Turnip mosaic virus (TuMV) through its 6K2 protein51 and of MNSV replication, through its p/B protein52. All the above examples involve interactions between viral and host proteins found by different methods, but not natural resistances against the viruses involved, suggesting again that mutations in genes encoding components of vesicle trafficking machinery can often be deleterious for the plant. Only in cucumber, a VPS4 was recently proposed, although not validated, as a candidate for the zym1gene that confers resistance to Zucchini Yellow Mosaic Virus (ZYMV)53. As VPS4 is often recruited as a permanent component of the viral replication complexes, (VRCs), it could be likely affecting ZYMV replication.
Due to the nature of CmVPS41, our results support the idea that vesicular endocytosis and plasmodesmata may share some regulatory mechanisms regarding virus movement54. Further effort should be done to determine the molecular mechanism of the resistance conferred by this gene and its relation with the viral MP. The identification and cloning of CmVPS41allow the possibility to address new questions related to the precise mechanism of resistance such as how and where does the interaction between CmVPS41 and viral MP take place or how this interaction allows the virus to go through the PDs that communicate BS and phloem cells to produce a systemic infection. Moreover, since the participation of the CmVPS41 seems to be necessary only to go through these PDs, they could be different than those communicating BS cells with other mesophyll cells. Additionally, it remains to be seen if VPS41 is a general checkpoint for CMV phloem invasion in other species. Identification of cmv1also opens up possibilities as a first step to start pyramiding in melon elite cultivars all the QTLs involved in resistance to CMV once they are identified.
Materials and Methods
Plant and virus material
For fine mapping of the cmv1 gene, a melon F2 population from the cross between the susceptible line T111 of the Spanish cultivar Piel de Sapo (PS) type and the resistant Near Isogenic Line (NIL) SC12-1 was used. The NIL SC12-1 carries a single introgression from the resistant Korean accession PI 161375, cultivar ‘Songwhan Charmi’ (SC) in LG XII containing the cmv1 gene in the genetic background of PS24. F2 recombinant plants were selected using flanking molecular markers (see below) and self-pollinated to obtain the F3 plants used for phenotyping. The C. melo accessions representing a wide range of melon diversity35, 36 were provided by the Cucurbits group of the COMAV-UPV (Instituto de Conservación y Mejora de la Agrodiversidad Valenciana, Universidad Politécnica de Valencia, Spain). For the CMV resistance assays, PS was used as susceptible control and SC and the sub NIL SC12-1-99, which carries a shorter introgression in LG XII containing cmv1 24, were used as resistant controls. Negative controls were prepared from leaf tissue extracts of mock-inoculated plants. Virus strains used in this study were CMV-LS, belonging to subgroup II, and CMV-FNY, belonging to subgroup I, kindly provided by Dr Peter Palukaitis as infectious clones55, 56.
Virus inoculation
For virus inoculation experiments, seeds were pre-germinated and grown as previously described25. Inoculation and virus detection were carried out as previously described by Essafi and collaborators24. Briefly, viral inocula were prepared from freshly symptomatic leaves of zucchini squash ‘Chapin F1’ (Semillas Fito SA, Barcelona, Spain) and rub-inoculated onto the cotyledons of young 7-10-day old melon plants. Symptoms were scored visually at 14, 21 and 28 days post-inoculation (dpi). For inoculation of in vitro acclimated transgenic plants, six newly rooted in vitro clones from each of the T0-SC independent lines, four PS and four SC control in vitro regenerated plants were transferred to soil pots inside individual plastic bags. Plants were kept in growth chambers (Sanyo MLR-350H, Sanyo Electric Biomedical Co, Osaka, Japan) under long-day conditions of 22 °C for 16 h with 5000 lx of light and 18 °C for 8 h in the dark during 3 days, inside plastic bags to keep high humidity, and then one day with the plastic bag open. Then, the plastic bag was removed and the plants were kept for four days longer in the chamber until two newly formed leaves were inoculated with CMV, as already described25.
Virus detection
Virus detection by DAS-ELISA was carried out at 21 dpi from young developed leaves using CMV coat protein-specific polyclonal antiserum (Lowe Biochemica GmbH, Otterfing, Germany) following manufacturer´s protocol. ELISA reactions were measured spectrophotometrically after 60 minutes reaction at 405 nm in a VICTOR3 V multilabel plate reader (Perkin Elmer Inc., Waltham, MA, USA). Viral detection by RT-PCR was performed as described25. For CMV-FNY detection, specific primers F109-3′R (TGGTCTCCTTTTAGAGACCC) and F109-2200F (CGGGACCATTAGTCAAGTTG) were used to amplify a 1,147 bp fragment. For CMV-LS, specific primers LS1-1400R (GAAGCATTCCACATATCGTAC) and LS1-1F (GTTTTATTTACAAGAGCGTACG) were used to amplify a 1,400 bp fragment.
Development of molecular markers
Molecular markers used for fine mapping of cmv1 (SSRs and SNPs) and for cmv1 analysis in diverse melon accessions are described in Supplementary Table S1. The flanking markers of the cmv1 interval, SSRs CMN61_44 and CMN21_5524 had previously been developed57. The remaining markers were developed from polymorphisms detected by re-sequencing PS and SC lines58. The polymorphisms were selected based on their physical position in the melon genome and the sequence flanking the polymorphisms was used to develop the molecular markers (sequence available from http://melonomics.net). For SSRs markers, primers flanking the repetitive sequence were designed using Primer359 (http://bioinfo.ut.ee/primer3-0.4.0/). One of the primers was labelled with IRD-800 (MWG Biotech AG, Ebersteg, Germany). After PCR amplification, fragments were visualized with LICOR IR2 sequencer (Li-corInc, Lincon, New England, USA) as described previously60. For SNPs genotyping, three methods were used: Sanger sequencing, Taqman probes and CAPS assay (SNPs/sanger, SNPs/Taqman and SNPs/CAPS). For SNPs/Sanger and Indel/Sanger sequencing markers, primers flanking the polymorphism were designed also with Primer3. PCR amplified fragments were sequenced by capillary electrophoresis at Macrogen (Macrogen Europe, Amsterdam, The Netherlands). Sequences were analyzed with Sequencher® version 5.0 sequence analysis software (Gene Codes Corporation, Ann Arbor, MI USA http://www.genecodes.com) and aligned with PS and SC sequences in order to determine the SNPs allele. Probes for SNPs/Taqman markers were ordered as Custom TaqMan® SNP Genotyping Assays (Applied Biosystems, Foster City, CA, USA) by submitting the SNP and flanking sequence. Genotyping was performed using a LightCycler 480 instrument (Roche Applied Science, IN, USA), mixing Taqman probes with LightCycler 480 Probe Master (Roche Applied Science, IN, USA) following manufacturer’s instructions. Genotype calling was done with the endpoint genotyping analysis procedure available from the LightCycler 480 software version 1.5. (Roche Applied Science, IN, USA). For SNPs/CAPS markers, primers flanking the polymorphism were designed also with Primer3. PCR amplified fragments were digested with the restriction enzyme, either BsiEI or TseI (New England BioLabs Inc., MA, USA) following manufacturer’s protocol. Restriction fragments were resolved on a 2% agarose gel.
cmv1 fine mapping
Search for recombinants was conducted for successive small intervals on an F2 population from the cross between PS and the NIL SC12-1. DNA was extracted from 100 mg of a young leaf from F2 seedlings using the CTAB method61. 780 plants were genotyped with the two flanking markers of the cmv1 interval (CMN61_44 and CMN21_55 (Supplementary Table S1)) and recombinant plants were selected and self-pollinated. 55 F2 recombinant plants were genotyped with a set of internal markers (Supplementary Table S1) in the first interval to determine the exact point of recombination. To position the cmv1 gene, 20 F3 plants from each F2 recombinant were inoculated as described above and symptoms were visually scored. A recombinant was considered resistant only when none of the F3 plants showed symptoms. Recombination breakpoints and phenotypic information were combined to narrow the cmv1 interval. Once the interval was reduced, if several recombinants inside the new interval were available a new set of markers inside the interval (Supplementary Table S1) was used to genotype the recombinants. If no recombinants were available, new F2 plants were screened until less than 0.1% of F2 plants screened were recombinant.
Functional analysis of the cmv1 region
The smallest cmv1 interval was analyzed using the annotation of the C. melo genome version 3.5.162 which is available from the melon genome website (http://melonomics.net/files/Genome/Melon_genome_v3.5.1/). The available gff3 file was used to identify genes and genetic features present in the cmv1 region. Manual curation of the predicted genes was performed by Blastp of the predicted proteins against the non-redundant protein sequences database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Polymorphisms between PS and SC lines had been previously detected from re-sequencing data28, 58. The Plaza database, version 3.0 dicots (http://bioinformatics.psb.ugent.be/plaza/)63, was used to identify orthologues and structure of the candidate genes in other plant species whose genome was available.
Cloning of cmv1 candidate gene
The complete coding sequence (CDS) of the cmv1 candidate from PS and SC was cloned using GATEWAY system (Gateway®Gene Cloning, Thermo Fisher Scientific, MA, USA). Briefly, total RNA was extracted from 100 mg of young leaves using TRI Reagent (Sigma–Aldrich, MO, USA) following manufacturer’s instructions. cDNA was synthesised using SuperScript III Reverse Transcriptase Kit (Invitrogen, CA, USA) and oligo(dT)20 as reverse primer. Complete cmv1 CDS was obtained by two PCR reactions, using primers pAG06F (ATGGCTCCCATTCTATCGGTA) and pAG23R (CTAATTGATGCAATCTTGAC) for the first 767 nucleotides, of the gene, and pAG22F (GTCAAGATTGCATCAATTAG) with pAG11R (TCAAGTCTTGGAAGCAGCAG) for the remaining 2,136 nucleotides, corresponding to the 3′ part of the coding sequence. These PCR products overlapped 20 base pairs and were assembled in a chimeric PCR to obtain the full-length CDS using primers pAG61F (ACGTCGACTATGGCTCCCATTCTATCGGTA) (incorporating SalI restriction enzyme site, in bold) and pAG61R (TCGATATCAGTCTTGGAAGCAGCAG) (incorporating EcoRV restriction enzyme site, in bold). The chimeric PCR product was inserted between the SalI and EcoRV sites in pENTR3C (Thermo Fisher Scientific, MA, USA) generating the pENVPS41-PS and the pENVPS41-SC constructs. Constructs were sequenced at Macrogen (Macrogen Europe, Amsterdam, The Netherlands) using primers listed in Supplementary Table S8. Effects of the polymorphisms in the cmv1 candidate protein were predicted with PROVEAN (http://provean.jcvi.org/index.php)64 and MutPred65. The binary construct used for melon transformation was made transferring the CmVPS41PS allele from pENVPS41-PS to the pEARLY301 binary vector66. First, a 1597 bp fragment containing cmv1 putative UTR and promoter was PCR-amplified from PS genomic DNA using primers AGGTCGACGTTCACTGAGACATTCG and TGGTCGACTGTGATGAACGGCCGATT and cloned into the SalI site of pENVPS41-PS, upstream of the starting codon to generate pENProVPS41-PS. Then, pENProVPS41-PS was recombined into the pEARLY301 expression vector66, using LR clonase mix enzyme II (Gateway®Gene Cloning, Thermo Fisher Scientific, MA, USA), obtaining the pEX301VPS41-PS, which carries the HA tag sequence at the 3′ end of the gene.
Determination of the expression levels of CmVPS41mRNA by Real Time-PCR
Quantitative Real Time-PCR (qRT-PCR) amplification was performed on three experiments, with three biological replicates each, from SC and PS. 100 mg from three different parts of the leaves (limbo, vein and petiole) were collected from each plant. RNA extraction was carried out using the RNeasy PLANT MINI KIT (Qiagen, Germany) and samples were treated with RNAse free TURBO-DNase I (Applied Biosystem, Ambion, CA, USA) following manufacturer’s instructions. Total RNA was reverse transcribed with SuperScript III Reverse Transcriptase Kit (Invitrogen, CA, USA) and oligo (dT)20 as reverse primer. Quantitative PCR was performed on a LightCycler 480 Real-Time PCR System (Roche Applied Science, IN, USA), using SYBR Green I Mix (Roche Applied Science, IN, USA). Primers TCAACTTCCCGTATTAGTTCCATACA and ACAATGAGTTTGAAGCAAGAGCAAC were designed for CmVPS41mRNA amplification using PRIMER EXPRESS® SOFTWARE v 2.0. (Applied Biosystems, CA, USA).Melon cyclophilin (CmCYP7)67 was used as reference gene. The experiment was done by triplicate. Melting curve analysis at the reaction end-point and no-template controls were used to ensure product-specific amplification and avoid primer-dimer quantification. A five-point standard curve from 10-fold dilutions was built for the cyclophilin reference and CmVPS41 genes. The efficiency of primers was calculated from the slope of the linear correlation between Cp (Crossing points) and each dilution (E = 10(−1/slope)). As the efficiencies between genes did not differ significantly from 1, we used the 2-∆∆Cp method68 to calculate the relative expression of CmVPS41. Differences were considered significant when fold change was larger than 2.
Identification and analysis of CmVPS41 TILLING mutants
A CharMono mutant TILLING collection34 was screened for mutants inside the CmVPS41 coding region. Briefly, pooled DNA from 6,200 M2 families was first PCR amplified with specific primers TAGACTAAAGCTCAAAAGGCTTGCA and GAGTTGTTAGCATACTACATAGGAC, flanking a 732 bp amplicon including exons 5 and 6. This PCR was then used as template in a nested PCR reaction using internal primers carrying a M13 tail (CGACGTTGTAAAACGACGCTATTTCCCATTTCCTC and GATAACAATTTCACACAGGGTCCATGTCATACTCC), plus M13 universal primers, M13F700 (5′-CACGACGTTGTAAAACGAC-3′) and M13R800 (5′-GGATAACAATTTCACACAGG-3′), labeled at the 5′end with infra-red dyes IRD700 and IRD800 (LI-COR®, Lincoln, NE, USA), respectively. The 574 bp amplicon obtained was used for mutant detection as previously reported34, 69. The mutants obtained were subsequently sequenced to identify the nucleotide change. The effect of the observed mutations in the protein was analysedwith PROVEAN (http://provean.jcvi.org/index.php, ref. 64). Families carrying a non-synonymous mutation were selected for phenotyping. Mutant homozygous plants were obtained for each selected family by self-pollinating M2 plants.
Melon transformation
The plasmid pEX301VPS41-PS was introduced into the AGL0 strain of Agrobacterium tumefaciens. Melon transgenic lines were developed by co-cultivation of the Agrobacterium culture with three-days-old cotyledons of SC as previously described33. T0 plants that had integrated the transgene were confirmed by PCR. Briefly, after a DNA extraction performed as explained above, the transgene, as well as, the endogenous CmVPS41 were PCR-amplified with primers ATGGCTCCCATTCTATCG and CACATATTCACCTTCTGTATCA. The endogenous gene produced a 757 bp band, whereas in the transgene, as no introns are present, a 324pb band was amplified. Among the T0 plants that amplified a band of 324 pb, the expression of CmVPS41PS allele in leaves was confirmed by RT-PCR. Briefly, total RNA was extracted from 100 mg of young leaves, treated with DNAse and reverse transcribed to cDNA as detailed above, using oligo (dT)20 as reverse primer. Subsequent PCR amplification was carried out with primer ATGTTATTGGAGCACACAGT that anneals with the CmVPS41sequence and AGCGTAATCTGGAACATCGT that anneals with the pEX301VPS41-PS, HA tag sequence at the 3′ end of the gene, amplifying a fragment of 642pb (Supplementary Figure S1). The response to CMV-LS and FNY strains was tested in T0 plants expressing the CmVPS41PS allele as indicated above. Plants were considered susceptible when symptoms appeared in non-inoculated new leaves.
Data availability statement
The C. melo VPS41 sequence is available at http://melonomics.net.
References
Anderson, P. K. et al. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends in Ecology & Evolution 19, 535–544, doi:10.1016/j.tree.2004.07.021 (2004).
Nicaise, V. Crop immunity against viruses: outcomes and future challenges. Frontiers in Plant Science 5, 660, doi:10.3389/fpls.2014.00660 (2014).
Kang, B.-C., Yeam, I. & Jahn, M. Genetics of plant virus resistance. Annual Review of Phytopathology 43, 581–621, doi:10.1146/annurev.phyto.43.011205.141140 (2005).
Fraser, R. S. S. The genetics of plant-virus interactions: implications for plant breeding. Euphytica 63, 175–185, doi:10.1007/bf00023922 (1992).
Truniger, V. & Aranda, M. Recessive resistance to plant viruses. Advances in virus research 119–159 (2009).
Sanfaçon, H. Plant translationfactors and virus resistance. Viruses 7, 3392–3419, doi:10.3390/v7072778 (2015).
Yang, P. et al. PROTEIN DISULFIDE ISOMERASE LIKE 5-1 is a susceptibility factor to plant viruses. Proceedings of the National Academy of Sciences of the United States of America 111, 2104–2109, doi:10.1073/pnas.1320362111 (2014).
Ouibrahim, L. et al. Cloning of the Arabidopsis rwm1 gene for resistance to Watermelon mosaic virus points to a new function for natural virus resistance genes. The Plant Journal 79, 705–716, doi:10.1111/tpj.12586 (2014).
Rubio, M., Nicolaï, M., Caranta, C. & Palloix, A. Allele mining in the pepper gene pool provided new complementation effects between pvr2-eIF4E and pvr6-eIF(iso)4E alleles for resistance to pepper veinal mottle virus. Journal of General Virology 90, 2808–2814, doi:10.1099/vir.0.013151-0 (2009).
Ibiza, V. P., Cañizares, J. & Nuez, F. EcoTILLING in Capsicum species: searching for new virus resistances. BMC Genomics 11, 631–631, doi:10.1186/1471-2164-11-631 (2010).
Chandrasekaran, J. et al. Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Molecular Plant Pathology. doi:10.1111/mpp.12375 (2016).
Rodríguez-Hernández, A. et al. Melon RNA interference (RNAi) lines silenced for Cm-eIF4E show broad virus resistance. Molecular plant pathology 13, 755–763, doi:10.1111/j.1364-3703.2012.00785.x (2012).
Edwardson, J. R. & Christie, R. G. In CRC Handbook of Viruses Infecting Legumes (eds J.R. Edwardson & R.G. Christie) 293–319 (CRC Press, 1991).
Roossinck, M. J. Cucumber mosaic virus, a model for RNA virus evolution. Molecular Plant Pathology 2, 59–63 (2001).
Caranta, C. et al. QTLs involved in the restriction of Cucumber mosaic virus (CMV) long-distance movement in pepper. Theor Appl Genet 104, 586–591 (2002).
Valkonen, J. P. & Watanabe, K. N. Autonomous cell death, temperature sensitivity and the genetic control associated with resistance to Cucumber mosaic virus (CMV) in diploid potatoes (Solanum spp). Theor Appl Genet 99, 996–1005 (1999).
Ohnishi, S. et al. Multigenic system controlling viral systemic infection determined by the interactions between Cucumber mosaic virus genes and quantitative trait loci of soybean cultivars. Phytopathology 101, 575–582, doi:10.1094/phyto-06-10-0154 (2011).
Kobori, T., Satoshi, T. & Osaki, T. Movement of Cucumber mosaic virus is restricted at the interface between mesophyll and phloem pathway in Cucumis figarei. Journal of General Plant Pathology 66, 159–166, doi:10.1007/pl00012939 (2000).
Argyris, J. M., Pujol, M., Martín-Hernández, A. M. & Garcia-Mas, J. Combined use of genetic and genomics resources to understand virus resistance and fruit quality traits in melon. Physiologia Plantarum 155, 4–11, doi:10.1111/ppl.12323 (2015).
Diaz, J. A. et al. Potential sources of resistance for melon to nonpersistently aphid-borne viruses. Plant Disease 87, 960–964 (2003).
Karchi, Z., Cohen, S. & Govers, A. Inheritance of resistance to Cucumber Mosaic virus in melons. Phytopathology 65, 479–481 (1975).
Risser, G., Pitrat, M. & Rode, J. C. Etude de la résistance du melon (Cucumis melo L.) au virus de la mosaïque du concombre. Ann Amél Plant 27, 509–522 (1977).
Dogimont, C. et al. Identification of QTLs contributing to resistance to different strains of Cucumber mosaic cucumovirus in melon. Acta Hortic 510, 391–398 (2000).
Essafi, A. et al. Dissection of the oligogenic resistance to Cucumber mosaic virus in the melon accession PI 161375. Theoretical and Applied Genetics 118, 275–284 (2009).
Guiu-Aragonés, C., Díaz-Pendón, J. A. & Martín-Hernández, A. M. Four sequence positions of the movement protein of Cucumber mosaic virus determine the virulence against cmv1-mediated resistance in melon. Molecular Plant Pathology 16, 675–684, doi:10.1111/mpp.12225 (2015).
Guiu-Aragonés, C. et al. The complex resistance to Cucumber mosaic cucumovirus (CMV) in the melon accession PI 161375 is governed by one gene and at least two quantitative trait loci. Molecular Breeding 34, 351–362, doi:10.1007/s11032-014-0038-y (2014).
Guiu-Aragonés, C. et al. cmv1 is a gate for Cucumber mosaic virus transport from bundle sheath cells to phloem in melon. Mol. Plant Pathology 17, 973–984 (2016).
Sanseverino, W. et al. Transposon Insertions, Structural Variations, and SNPs Contribute to the Evolution of the Melon Genome. Molecular Biology and Evolution 32, 2760–2774, doi:10.1093/molbev/msv152 (2015).
Pols, M. S., ten Brink, C., Gosavi, P., Oorschot, V. & Klumperman, J. The HOPS Proteins hVps41 and hVps39 Are Required for Homotypic and Heterotypic Late Endosome Fusion. Traffic 14, 219–232, doi:10.1111/tra.12027 (2013).
Asensio, C. S. et al. Self-assembly of VPS41 promotes sorting required for biogenesis of the regulated secretory pathway. Developmental cell 27, 425–437, doi:10.1016/j.devcel.2013.10.007 (2013).
Kolesnikova, L. et al. Vacuolar protein sorting pathway contributes to the release of Marburg virus. Journal of Virology8 3, 2327–2337, doi:10.1128/JVI.02184-08 (2009).
Nuñez-Palenius, H. G. et al. Melon Fruits: Genetic Diversity, Physiology, and Biotechnology Features. Critical Reviews in Biotechnology 28, 13–55, doi:10.1080/07388550801891111 (2008).
Castelblanque, L. et al. Improving the genetic transformation efficiency of Cucumis melo subsp. melo “Piel de Sapo” via Agrobacterium. Procedings of the IXth UECARPIA Meeting on Genetics and Breeding of Cucurbitaceae, 21-24th May, Avignon,, 627–631 (2008).
Dahmani-Mardas, F. et al. Engineering melon plants with improved fruit shelf life using the TILLING approach. PLoS ONE 5, doi:10.1371/journal.pone.0015776 (2010).
Esteras, C. et al. SNP genotyping in melons: genetic variation, population structure, and linkage disequilibrium. Theoretical and Applied Genetics 126, 1285–1303, doi:10.1007/s00122-013-2053-5 (2013).
Leida, C. et al. Variability of candidate genes, genetic structure and association with sugar accumulation and climacteric behavior in a broad germplasm collection of melon (Cucumis melo L.). BMC Genetics 16, 28, doi:10.1186/s12863-015-0183-2 (2015).
Balderhaar, H. Jk & Ungermann, C. CORVET and HOPS tethering complexes – coordinators of endosome and lysosome fusion. Journal of Cell Science 126, 1307–1316, doi:10.1242/jcs.107805 (2013).
Darsow, T., Katzmann, D. J., Cowles, C. R. & Emr, S. D. Vps41p Function in the Alkaline Phosphatase Pathway Requires Homo-oligomerization and Interaction with AP-3 through Two Distinct Domains. Molecular Biology of the Cell 12, 37–51 (2001).
Ostrowicz, C. W. et al. Defined subunit arrangement and Rab interactions are required for functionality of the HOPS tethering complex. Traffic 11, 1334–1346, doi:10.1111/j.1600-0854.2010.01097.x (2010).
Solinger, J. A. & Spang, A. Tethering complexes in the endocytic pathway: CORVET and HOPS. FEBS Journal 280, 2743–2757, doi:10.1111/febs.12151 (2013).
Rehling, P., Dawson, T., Katzmann, D. J. & Emr, S. D. Formation of AP-3 transport intermediates requires Vps41 function. Nature Cell Biology 1, 346–353 (1999).
Niihama, M., Takemoto, N., Hashiguchi, Y., Tasaka, M. & Morita, M. T. ZIP genes encode proteininvolved inmembrane trafficking of the TGN–PVC/Vacuoles. Plant and Cell Physiology 50, 2057–2068, doi:10.1093/pcp/pcp137 (2009).
Wang, A. & Krishnaswamy, S. Eukaryotic translation initiation factor 4E-mediated recessive resistance to plant viruses and its utility in crop improvement. Molecular Plant Pathology 13, 795–803, doi:10.1111/j.1364-3703.2012.00791.x (2012).
Ruffel, S. et al. A natural recessive resistance gene against potato virus Y in pepper corresponds to the eukaryotic initiation factor 4E (eIF4E). The Plant Journal 32, 1067–1075, doi:10.1046/j.1365-313X.2002.01499.x (2002).
Morosky, S., Lennemann, N. J. & Coyne, C. B. BPIFB6 regulatessecretory pathway trafficking and Enterovirus replication. Journal of Virology 90, 5098–5107, doi:10.1128/jvi.00170-16 (2016).
Serra-Soriano, M., Pallás, V. & Navarro, J. A. A model for transport of a viral membrane protein through the early secretory pathway: minimal sequence and endoplasmic reticulum lateral mobility requirements. The Plant Journal 77, 863–879, doi:10.1111/tpj.12435 (2014).
Vale-Costa, S. & Amorim, M. J. Recycling Endosomes and Viral Infection. Viruses 8, 64, doi:10.3390/v8030064 (2016).
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343, doi:10.1038/nature10348 (2011).
Barajas, D., Martín, I. Fd. C., Pogany, J., Risco, C. & Nagy, P. D. Noncanonical role for the host Vps4 AAA + ATPase ESCRT protein in the formation of Tomato bushy stunt virus replicase. PLoS Pathog 10, e1004087, doi:10.1371/journal.ppat.1004087 (2014).
Richardson, L. G. L. et al. A unique N-Tterminal sequence in the Carnation Italian ringspot virus p36 replicase-associated protein interacts with the host cell ESCRT-I component Vps23. Journal of Virology 88, 6329–6344, doi:10.1128/JVI.03840-13 (2014).
Jiang, J., Patarroyo, C., Garcia Cabanillas, D., Zheng, H. & Laliberté, J.-F. The vesicle-forming 6K2 protein of Turnip mosaic virus interacts with the COPII coatomer Sec. 24a for viral systemic infection. Journal of Virology 89, 6695–6710, doi:10.1128/jvi.00503-15 (2015).
Genovés, A., Navarro, J. A. & Pallás, V. Theintra- and intercellular movement of Melon necrotic spot virus (MNSV) depends on an active secretory pathway. Molecular Plant-Microbe Interactions 23, 263–272, doi:10.1094/MPMI-23-3-0263 (2010).
Amano, M. et al. High-resolution mapping of zym, a recessive gene for Zucchini yellow mosaic virus resistance in cucumber. Theoretical and Applied Genetics 126, 2983–2993, doi:10.1007/s00122-013-2187-5 (2013).
Lewis, J. D. & Lazarowitz, S. G. Arabidopsis synaptotagmin SYTA regulates endocytosis and virus movement protein cell-to-cell transport. Proceedings of the National Academy of Sciences of the United States of America 107, 2491–2496, doi:10.1073/pnas.0909080107 (2010).
Rizzo, T. & Palukaitis, P. Construction of full-length cDNA clones of Cucumber mosaic virus RNAs 1, 2 and 3: Generation of infectious RNA transcripts. Molecular and General Genetics 122, 249–256 (1990).
Zhang, L., Handa, K. & Palukaitis, P. Mapping local and systemic symptom determinants of Cucumber mosaic cucumovirus in tobacco. The Journal of General Virology 75, 3185–3191 (1994).
Fukino, N., Sakata, Y., Kunihisa, M. & S., M. Characterisation of novel simple sequence repeat (SSR) markers for melon (Cucumis melo L.) and their use for genotype identification. Journal of Horticultural Science & Biotechnology 82, 330–334 (2007).
Garcia-Mas, J. et al. The genome of melon (Cucumis melo L.). Proceedings of the National Academy of Sciences 109, 11872–11877, doi:10.1073/pnas.1205415109 (2012).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Research 40, e115–e115, doi:10.1093/nar/gks596 (2012).
Gonzalo, M. J. et al. Simple-sequence repeat markers used in merging linkage maps of melon (Cucumis melo L.). Theor Appl Genet 110, 802–811 (2005).
Doyle, J. J. & Doyle, J. L. Isolation of plant DNA from fresh tissue. Focus1 2, 13–15 (1990).
Argyris, J. M. et al. Use of targeted SNP selection for an improved anchoring of the melon (Cucumis melo L.) scaffold genome assembly. BMC Genomics 16, 4, doi:10.1186/s12864-014-1196-3 (2015).
Proost, S. et al. PLAZA 3.0: an access point for plant comparative genomics. Nucleic Acids Research 43, D974–D981, doi:10.1093/nar/gku986 (2015).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 7, e46688, doi:10.1371/journal.pone.0046688 (2012).
Li, B. et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750, doi:10.1093/bioinformatics/btp528 (2009).
Earley, K. W. et al. Gateway-compatible vectors for plant functional genomics and proteomics. The Plant Journal 45, 616–629, doi:10.1111/j.1365-313X.2005.02617.x (2006).
Gonzalez-Ibeas, D. et al. MELOGEN: an EST database for melon functional genomics. BMC Genomics 8, 306 (2007).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Research 29, 2002–2007 (2001).
Dalmais, M. et al. UTILLdb, a Pisum sativum in silico forward and reverse genetics tool. Genome Biology 9, R43–R43, doi:10.1186/gb-2008-9-2-r43 (2008).
Saladié, M. et al. Comparative transcriptional profiling analysis of developing melon (Cucumis melo L.) fruit from climacteric and non-climacteric varieties. BMC Genomics 16, 440, doi:10.1186/s12864-015-1649-3 (2015).
Acknowledgements
We thank Fuensanta Garcia for technical support and Miguel Aranda for critical reading. The TILLING platform is supported by the Program Saclay Plant Sciences (SPS, ANR-10-LABX-40) and the European Research Council (ERC-SEXYPARTH). This work was supported by grants AGL2009-12698-C02-01 and AGL2012-40130-C02-01 from the Spanish Ministry of Science and Innovation, the Spanish Ministry of Econom and Competitiveness, through the “Severo Ochoa Programme for Centres of Excellence in R&D” 2016-2019 (SEV‐2015‐0533)” and the CERCA Programme/Generalitat de Catalunya.
Author information
Authors and Affiliations
Contributions
A.G. and L.P. performed equally most of the experiments and wrote the first draft. M.B. initiated map-based cloning. G.G. participated in map-based cloning. P.R. performed the TILLING screening. B.P. contributed with exotic DNA material and critical reading of the manuscript. C.T. supervised the TILLING screening. A.B. supervised the TILLING and did critical reading of the manuscript. J.G.-M. designed some experiments and complemented the writing. A.M.M.-H. designed the experiments and wrote the manuscript. All authors read and agreed on the content of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Giner, A., Pascual, L., Bourgeois, M. et al. A mutation in the melon Vacuolar Protein Sorting 41prevents systemic infection of Cucumber mosaic virus . Sci Rep 7, 10471 (2017). https://doi.org/10.1038/s41598-017-10783-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10783-3
This article is cited by
-
Insights into the early transcriptomic response against watermelon mosaic virus in melon
BMC Plant Biology (2024)
-
Vegetable biology and breeding in the genomics era
Science China Life Sciences (2023)
-
Fine-mapping and identification of a candidate gene controlling seed coat color in melon (Cucumis melo L. var. chinensis Pangalo)
Theoretical and Applied Genetics (2022)
-
A novel introgression line collection to unravel the genetics of climacteric ripening and fruit quality in melon
Scientific Reports (2021)
-
A single resistance factor to solve vineyard degeneration due to grapevine fanleaf virus
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
