
Abstract
Myopia (short-sightedness) and hyperopia (long-sightedness) occur when the eye grows too long or short, respectively, for its refractive power. There are currently approximately 1.45 billion myopes worldwide and prevalence is rising dramatically. Although high myopia significantly increases the risk of developing a range of sight-threatening disorders, the molecular mechanisms underlying ocular growth regulation and its relationship to these secondary complications remain poorly understood. Thus, this study meta-analyzed transcriptome datasets collected in the commonly used chick model of optically-induced refractive error. Fifteen datasets (collected across five previous studies) were obtained from GEO, preprocessed in Bioconductor, and divided into 4 conditions representing early (≤1 day) and late (>1 day) myopia and hyperopia induction. Differentially expressed genes in each condition were then identified using Rank Product meta-analysis. The results provide novel evidence for transcriptional activation of the complement system during both myopia and hyperopia induction, and confirm existing literature implicating cell signaling, mitochondrial, and structural processes in refractive error. Further comparisons demonstrated that the meta-analysis results also significantly improve concordance with broader omics data types (i.e., human genetic association and animal proteomics studies) relative to previous transcriptome studies, and show extensive similarities with the genes linked to age-related macular degeneration, choroidal neovascularization, and cataract.
Similar content being viewed by others


Introduction
Recent world-wide increases in the prevalence of myopia (short-sightedness) suggest that environmental and/or lifestyle factors are extraordinarily important to the underlying pathogenesis of this complex disorder1. Myopia typically results when excessive axial growth causes a mismatch between the eyes refractive power and its length2. By comparison, hyperopic (long-sighted) eyes are usually small3. Environmentally-driven changes to ocular axial growth and consequent refractive state have been investigated using animal models, in which rearing with negatively powered defocusing lenses or form deprivation occluders increases eye growth resulting in myopia, while rearing with positive lenses induces growth arrest and hyperopia (long-sightedness)4. Although early studies in these models were primarily limited to hypothesis-driven approaches (for example refs 5,6,7,8) the recent availability of microarray and RNA-sequencing technologies has enabled a number of discovery-driven investigations of gene expression in the posterior eye. To date, eleven such transcriptome studies have been published encompassing a range of species (primate, mouse, and chick), platforms (commercial microarrays, custom microarrays, and RNA-sequencing), optical manipulations (lens-induced myopia; LIM, form deprivation occlusion myopia; FDM, lens-induced hyperopia; LIH, and FDM recovery) and tissue samples (retina, RPE, choroid, sclera)9,10,11,12,13,14,15,16,17,18,19. These studies have offered many insights into the biology of optically-induced ocular growth changes, however, the key expression responses underlying refractive error induction remain unclear as >1500 genes have been implicated with poor cross-study replication20, 21.
Thus, the present study aimed to use meta-analysis techniques to generate a more reliable understanding of the generalized transcriptome responses underlying refractive error induction in animal models. Studies were considered for inclusion in the meta-analysis if they used commercially available microarray chips or RNA-sequencing to profile mRNA expression in the posterior eye of animal models of optically-induced myopia or hyperopia and matched no lens controls. Based on these criteria, five of the eleven available transcriptome studies were excluded because they lacked a no lens control comparison10, 12, used custom microarray chips with limited genome coverage9, 18, or profiled miRNA rather than mRNA19. Five of the remaining six studies profiled gene expression in chick retinal, RPE, and/or choroidal tissue13,14,15,16,17. The final study analyzed scleral transcriptome responses in the form deprivation mouse model11. This latter study was excluded because it differed from the others both in the tissue profiled (sclera rather than retina/RPE/choroid) and model species (all other included studies were conducted in chick, the most widely used animal model for refractive error research).
The remaining five chick studies profiled a range of posterior ocular tissues (comprised of varying combinations of retina, RPE, and/or choroid), induction time-points (ranging from 6 hours to 3 days), and optical manipulations (myopia induction via occlusion or negative lenses, or hyperopia induction via positive lenses). Despite this, published comparisons (e.g. see supplementary materials Stone et al.15 and Riddell et al.13) have identified a number of common findings across the studies, including similarities between studies profiling very different tissue compositions such as retina/RPE and amacrine cells. This suggests that, although the expression data must to some extent reflect the specific cell types profiled, at least a subset of responses during refractive error induction are sufficiently generalized or sufficiently strong to be measurable regardless of the exact tissue composition. This pattern of results is in accordance with most theories of environmentally-driven ocular growth which postulate a signal or cascade of signals that propagate across the posterior eye (presumably inducing related expression responses from multiple cell types)4, 22,23,24. Likewise, there is evidence for extensive similarities in the molecular response to FDM and LIM21.
Based on these past observations, datasets profiling different tissue compositions (retina, RPE, and choroid) and myopia-inducing manipulations (LIM and FDM) were not separated in the meta-analysis. We did, however, separate datasets profiling early (≤1 day) and later (>1 day) induction time-points (as described previously21). This decision was based on past research suggesting that early and late expression responses are dissimilar in chick14, 15, in line with the theoretical expectation that the biological processes involved in the onset and progression of refractive change are likely to be active at early time-points, while secondary responses to prolonged lens-wear may predominate at later time-points15, 20.
The separation of datasets into early and late categories resulted in four meta-analysis conditions: early myopia (4 datasets), late myopia (4 datasets), early hyperopia (4 datasets), and late hyperopia (3 datasets). The datasets included in each of these conditions were heterogeneous, encompassing a range of circadian phases, developmental ages, tissue types, control conditions (fellow eye or separate no lens), and exact optical manipulations (occlusion or lenses of various powers). We considered this heterogeneity to be a strength in the analysis design, as genes showing differential expression across these diverse datasets are presumably less likely to reflect these confounding influences. Given the diversity of the datasets, we chose to use the non-parametric rank product meta-analysis method that transforms expression values into ranks to identify consistently highly ranked genes across replicate experiments (i.e., genes that are consistently up- or down-regulated)25. The Rank Product approach has higher sensitivity and selectivity than t-based meta-analysis methods, particularly when sample sizes are small and data is heterogeneous26.
Results
Initial evaluation of the meta-analysis results
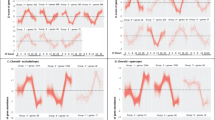
Datasets profiling gene expression in the chick retina, RPE, and/or choroid during myopia (8 datasets) and hyperopia (7 datasets) induction were split into early and late time-points, and differentially-expressed genes in each condition were then identified using rank product meta-analysis. Fifty-nine and 189 genes were differentially-expressed in early and late myopia induction conditions, respectively (Supplementary Table S1). Twenty-seven and 136 genes were differentially-expressed in early and late hyperopia induction conditions (Supplementary Table S2). Correspondence at the top (CAT) plots were used to assess the contribution of each dataset to the meta-analysis findings. These plots show the proportion of top ranked genes from each individual dataset present in the meta-analysis results as a function of list size. As shown in Fig. 1, no individual dataset disproportionately accounted for the meta-analysis findings.

Correspondence at the top (CAT) plots showing the contribution of each dataset to the meta-analysis findings. These plots evaluate the consensus between top ranking genes from each individual dataset and top ranking genes from each meta-analysis condition (early and late myopia and hyperopia). The x-axis is top ranking genes based on the rank product analyses and the y-axis is the proportion of genes in common with the meta-analysis results.
Most of the meta-analysis findings were unique (i.e., the genes differentially-expressed in the meta-analysis were not differentially-expressed in any of the individual transcriptome studies that were meta-analyzed; see Fig. 2a). To test the utility of these new results, we compared both the original transcriptome findings and the meta-analysis findings with the genes and proteins implicated in human Genome-Wide Association Studies (GWAS) and animal proteomics studies of refractive error. As reported previously21, the individual studies included in the meta-analysis implicated a number of genes that were also differentially-expressed in animal proteomics studies of refractive error and/or located within 500 kilobases (Kb) of human GWAS ocular axial length or refractive error loci (Fig. 2b). The meta-analysis replicated some of these results, and identified 18 new commonalities with GWAS and proteomics studies (Fig. 2b,c). Because the list of genes implicated in the meta-analysis was much smaller than the list of genes implicated in the individual transcriptome studies, the meta-analysis results showed more statistically significant concordance with these broader omics data types.

The meta-analysis results show improved concordance with previous animal proteomic and human GWAS findings. (a) Venn diagrams comparing differentially-expressed genes from each meta-analysis (pfp < 0.05) with differentially-expressed genes from the individual studies that were meta-analyzed (as per the original authors’ statistical criteria). Note that most of the genes identified in the rank product meta-analyses were unique (i.e., not classified as differentially-expressed in the original transcriptome studies). (b) Heat map comparing the overlap of original transcriptome study results and rank product meta-analysis results with the genes implicated in previous GWAS and proteomics studies of refractive error. The shared category includes genes implicated in both the rank product meta-analyses and the individual studies included in the meta-analyses. Meta-analysis and original study categories include the remaining genes that were uniquely implicated in either the rank product meta-analysis or the original studies. The number of genes implicated in each list is shown in parenthesis after the list name. List intersections (i.e., the number of overlapping genes), Benjamini–Hochberg adjusted p-values, and odds ratios are superimposed on the heat map grid. Statistically significant overlaps are highlighted in red. Note that, although there were generally more commonalities in total between the original transcriptome study findings and the proteomics/GWAS results, the list of genes implicated in the original studies was very large resulting in low statistical significance and odds ratios. Supplementary Table S3 provides further details on all of the overlapping genes. (c) Venn diagram showing the rank product meta-analysis genes that were also implicated in previous GWAS or proteomics studies (i.e., all overlapping genes from the right two columns of Fig. 2b). Shared findings are shown in bold italic font.
Pathway over-representation
After initial evaluations, we tested the differentially-expressed genes in each meta-analysis condition for over-representation of Reactome pathways. Several signaling pathways (particularly those related to G-protein coupled signaling) were over-represented in the genes differentially-expressed in the early myopia and hyperopia datasets (Fig. 3). In addition, genes from the NCAM signaling for neurite outgrowth pathway were over-represented in the early myopia data only. A wider range of processes were implicated in the late myopia and hyperopia, with genes from immune/inflammation (complement-coagulation cascade and class A scavenging receptors), tissue structure (extracellular matrix and muscle contraction), and arachidonic acid metabolism over-represented in both datasets (Fig. 3). Several signal transduction pathways were also implicated in the late myopia dataset, along with genes involved in phototransduction, retinoid metabolism, and mitochondrial translation. Detailed over-representation results are provided in Supplementary Table S4.

Bubble plots showing over-represented Reactome pathways in each meta-analysis condition. The size of each bubble represents the number of genes contributing to pathway over-representation (ranging from 3–37), and the z-score indicates whether genes from the pathway were primarily up-regulated (positive z-score) or down-regulated (negative z-score). The FDR q-value cut-off of 0.05 is indicated by the ‘threshold’ line. Detailed over-representation results are provided in Supplementary Table S4.
Common gene and pathway findings across conditions
We next assessed the degree of overlap between early and late myopia and hyperopia conditions at both the gene and pathway level. A large number of genes were differentially-expressed in multiple meta-analyses, with all possible conditions except early and late hyperopia showing more overlap then expected (p < 0.001) given the number of genes profiled (Fig. 4a). Most notably, the genes implicated in myopia and hyperopia conditions at each time-point were more similar than the genes implicated within each growth condition over time (see Jaccard Similarity indices in Fig. 4a). This pattern was also evident at the pathway level, where Jaccard similarity indices were 0.5 and 0.47 for early and late time-points, respectively (Fig. 4b). Full details of the overlap in gene and pathway findings are provided in Supplementary Table S5.

Common gene and pathway findings across meta-analysis conditions. Heat maps show overlap in (a) the genes differentially-expressed in each meta-analysis condition and (b) the pathways over-represented in each meta-analysis condition. The number of genes/pathways implicated in each meta-analysis condition is shown in parenthesis after the condition name. List intersections (i.e., the number of overlapping genes/pathways), Benjamini–Hochberg adjusted p-values, and Jaccard similarity indices are superimposed on the heat map grid. Note that the genes and pathways implicated in myopia and hyperopia conditions at each time-point are more similar than the genes implicated within each growth condition over time. Supplementary Table S5 provides a detailed list of the overlapping gene and pathway findings.
The genes underlying these overlapping results were primarily differentially-expressed in a growth non-specific manner, with just 15 genes displaying growth-specific profiles (i.e., concurrent up-regulation during myopia induction and down-regulation during hyperopia induction, or visa versa; Fig. 5). Induced network module analysis was used to identify relationships between these 15 growth-specific genes, and to predict additional ‘intermediate’ genes or proteins that may be involved in growth-specific processes. This analysis revealed that 5 of the 15 growth-specific genes encoded proteins that form a highly connected network involved in immune/inflammation-mediated extracellular matrix (ECM) remodeling. An additional 3 growth-specific genes encoded proteins involved a network related to cytoskeletal processes (Fig. 5). Given their interconnectivity with growth-specific genes, the intermediate genes from these two networks are good candidates for involvement in growth-specific processes (or refractive error phenotypes more broadly). Indeed, 2 of the 16 intermediate nodes (C1QA and MMP2) were also differentially-expressed in the meta-analysis (though not in a growth-specific manner). Moreover, approximately 50% of the intermediate genes have been implicated in previous targeted studies of myopia development (see Supplementary Table S6).

Network analysis demonstrates interactions between genes showing growth-specific expression patterns. The ConsensusPathDB Induced Network Module Analysis aims to connect a list of seed genes (in this case, the 15 genes showing growth-specific expression patterns) via different types of interactions (protein interactions, biochemical interactions, or gene regulatory interactions). Connections are made directly, or via an intermediate node (shown in grey). Eight of the 15 growth-specific genes were assigned to two highly connected protein networks involved in immune/inflammation-mediated extracellular matrix remodeling and cytoskeletal processes. The remaining 7 growth-specific genes are shown at the bottom of the image. Note that intermediate nodes MMP2 and C1QA were also differentially expressed in the meta-analysis, but not in a growth-specific manner. Information for each interaction in the networks is provided in Supplementary Table S7.
Comparison with the genes implicated in common ocular pathologies
We next compared the differentially expressed genes from each meta-analysis condition with the genes previously linked to disorders for which myopia or hyperopia are risk factors. Primary open angle glaucoma (POAG), choroidal neovascularization (CNV) and cataract were chosen for comparison because of their link with myopia27, 28. Sub-clinical age-related macular degeneration (AMD) and AMD itself were chosen because of their link with hyperopia29, 30.
As shown in Fig. 6a, the genes differentially expressed in early and late myopia overlapped significantly with the genes associated with sub-clinical AMD, AMD, and CNV. In addition, the genes differentially-expressed in late myopia showed significant similarities with those previously linked to cataract development (Fig. 6a). Although there were fewer similarities evident in the hyperopia conditions, the genes differentially expressed in late hyperopia did overlap significantly with both AMD and CNV associated genes. These gene commonalities as a whole were enriched for pathways related to extracellular structure (ECM organization, collagen formation, glycosaminoglycan metabolism) and the complement-coagulation system (complement cascade, response to elevated platelet cytosolic Ca2+; Fig. 6b).

Many differentially-expressed genes from the meta-analysis have previously been associated with ocular pathologies. (a) Heat map showing overlap between the genes differentially-expressed in each meta-analysis condition and the genes associated with POAG, cataract, sub-clinical AMD, AMD, and CNV. The number of genes associated with each meta-analysis condition and ocular pathology is shown in parenthesis after the list name. List intersections (i.e., the number of overlapping genes) and Benjamini–Hochberg adjusted p-values are superimposed on the grid. Statistically significant overlaps are highlighted in red. (b) Chord diagram showing genes that were both differentially-expressed in the meta-analysis and associated with an ocular pathology (i.e., all intersecting genes from Fig. 5a). Rectangles on the left indicate whether a gene was differentially expressed in early or late myopia or hyperopia conditions. Left-right connections indicate gene associations with ocular pathologies. Over-represented Reactome pathways for the list of meta-analysis/pathology gene commonalities are shown on the bottom right. Symbols preceding gene names in the chord diagram indicate gene contributions to these pathway over-representations.
Discussion
We conducted a rank product meta-analysis of differential gene expression in transcriptome datasets profiling chick retina, RPE, and/or choroid during refractive error induction. The analysis identified a refined list of differentially-expressed genes that shows better concordance relative to past transcriptome studies with both the proteins implicated in animal proteomic studies of refractive error and the candidate genes near human GWAS refractive error loci. We found that gene expression responses during myopia and hyperopia induction were highly similar at both early (≤1 day) and late (>1 day) time-points, and identified just 15 genes showing growth-specific expression profiles.
The early time-point in both growth conditions was characterized by differential-expression of genes involved in a range of signal transduction processes (particularly G-protein coupled receptor signaling), including many genes implicated in previous research (e.g. VIP31, 32, GCG33, 34, EGR15, 35, and BMP236). At the later time-point, both growth conditions were primarily characterized by differential expression of genes involved in remodeling of cellular and extracellular structure, immune and inflammatory processes, mitochondrial translation, and oxidative defense. Although differential-expression of structural genes is expected given the well-documented morphological features of refractive errors, the involvement of immune, mitochondrial, and oxidative defense genes in both growth conditions at late time-points is notable.
Most of the implicated immune genes were from the complement-coagulation pathway. This pathway has long been known for its role in inducing immune and inflammatory responses, however, recent research has also implicated complement in the promotion of cellular homeostasis (e.g. via metabolic reprogramming)37. A diverse range of complement-related genes were differentially-expressed, with components and regulators of both the classical and alternate pathways up-regulated in late myopia (CS1, PROS1, C1QB and CFD) and late hyperopia (SERPING1, C1QA, CRP, C7 and CFD). In addition, MASP1 (from the lectin pathway) was differentially-expressed in a growth-specific manner (down-regulated in late myopia and up-regulated in late hyperopia). These findings are in line with those of previous targeted studies that identified up-regulation of serum complement markers in patients with pathological myopia38 and increased C1q and C3 protein expression in the sclera of myopic guinea pigs39. Exploratory transcriptome studies have also implicated C713, CFI15, and C310 in animal myopia models. There is comparatively little evidence implicating the complement system in hyperopia, with only one transcriptome study identifying up-regulation of C7 in the chick LIH model13. Thus, to our knowledge, the present meta-analysis findings provide the first evidence for the involvement of multiple complement pathway genes in hyperopia.
It has previously been suggested that the complement system may promote ECM remodeling in the sclera during myopia induction39. The present findings are consistent with this notion, though the up-regulation of complement genes in both growth conditions at the later time-point suggests that any complement-mediated ECM remodeling is unlikely to play a causal role in directional ocular growth (at least in retina/RPE/choroid). On the other hand, our analysis did link complement with a small network of genes that were differentially-expressed in a growth-specific manner. This network was composed primarily of genes involved in immune-mediated ECM remodeling, and included connections with many intermediate genes that have been strongly linked to myopia in previous targeted studies (e.g. MMP2, TGFB1, TGFB2, FGF2, INS, and IGF2; see Supplementary Table S6). These previous findings lend support to the relevance of the immune-ECM network as a whole, suggesting the need for further research into the role of the lesser explored members (i.e., PTX3, MASP1, NOV, MBL2, C1QA, AP-1, FCN1, and FCN2) in directional ocular growth and refractive change.
In addition to complement pathway and structural genes, five genes encoding mitochondrial ribosomal proteins and one gene encoding a subunit of mitochondrial complex I (NDUFS5) were up-regulated in late myopia. These findings are in accordance with previous omics studies showing enrichment of genes and proteins from the oxidative phosphorylation pathway in chick and mouse models of myopia13, 40. Together with this past research, our findings are suggestive of an increase in mitochondrial oxidative phosphorylation in late myopia similar to that seen in some cancers41,42,43. Such an up-regulation of oxidative phosphorylation in myopic eyes is theoretically plausible given the compromised choroidal vasculature44, and the increased need for energy to fuel cell growth9, 45,46,47 and maintain ionic gradients48,49,50. It must be noted, however, that two of the mitochondrial ribosomal genes (MRPS21 and MRPL2) were also up-regulated in late hyperopia. This finding is contrary to our previous reports of bidirectional expression of metabolic pathways in early myopia and hyperopia13, presumably reflecting the restoration of normal growth rates as refractive compensation to positive lenses is achieved at later time-points.
If oxidative phosphorylation was up-regulated in late myopia and hyperopia, concurrent differential expression of antioxidant defense genes would be expected to compensate for associated changes to the production of reactive oxygen species through electron leak51, 52. Such expression changes were evident, particularly in the late myopia dataset where a wide range of genes linked with oxidative stress were either up-regulated (GPX1, GSTA3, MT-4, APOA1, OTUB, PTGDS, HSPB1, HSPB2) or down-regulated (XHD, SLC40A1, TF, SOD3, HPGDS, BCMO1). In addition to ‘classic’ antioxidant enzymes (SOD3, GPX1, GSTA3)53, 54, these genes included those involved in regulation of the p53 stress pathway (OTUB)55, lipid peroxidation (APOA1)56, 57, labile iron levels (XHD, SLC40A1, TF)58,59,60, intracellular zinc homeostasis (MT4)61, and arachidonic acid metabolism (PTGDS, HPGDS)62.
The factor/s driving these highly similar expression profiles across myopic and hyperopic groups require further investigation. The strong dissociation of responses at early and late time-points certainly suggests that most of the non-specific expression changes (i.e., those in structural, complement-coagulation, metabolic, and oxidative stress pathways) occurred secondary to other processes. It is also important to highlight that the representation of growth non-specific responses may have been increased by the meta-analysis design, which combined studies using relatively heterogeneous methodologies (e.g. different tissue compositions and exact optical manipulations). This choice was intended to identify genes showing differential expression across a broad range of myopia-inducing or hyperopia-inducing conditions, thus generating a better understanding of the generalized transcriptome responses underlying each type of refractive error. The improved concordance of our meta-analysis results with findings from human GWAS and animal proteomics studies of refractive error suggests that this approach was, at least in part, successful. However, this design presumably also biased the results towards identification of genes that respond to varied stimuli (e.g. stress-related genes) and genes that respond to the non-specific effects of lens-wear (e.g. blur or heat under the goggle)63.
As mentioned above, human orthologs of sixteen of the genes differentially-expressed in our meta-analysis fall in close proximity to GWAS refractive error peaks in humans. To date, debate surrounding the relevance of animal models to human myopia has primarily focused on similarities and differences in visual experience, refractive development and anatomy (for review see refs 4, 64,65,66,67,68). Our findings contribute to an additional small but growing body of evidence for molecular similarities between humans and animal models21, 65, 69,70,71,72,73, made possible by the recent availability of large-scale refractive error GWAS74. The two induction time-points in our meta-analysis were roughly proportionally represented in the overlap with human GWAS candidate genes. This is notable because, although some of the overlapping data originated from case-control GWAS of pathological myopia, many of the commonalities were with the CREAM75 and 23&Me76 data which identified loci associated with spherical equivalent and myopia age of onset, respectively, in large cohorts. Thus, although the chick studies included in our meta-analysis involved the rapid induction of a moderate (early time-point) to high (late time-point) degree of refractive error, it seems that findings in this model may be useful for understanding the broad spectrum of refractive errors present in human populations.
Many of the genes and pathways implicated in our meta-analysis have also been previously associated with AMD, CNV, and/or cataract in humans. Extracellular matrix remodeling77,78,79, activation of immune/inflammatory processes (particularly within the complement system)80,81,82,83,84,85,86, and oxidative stress87,88,89,90,91 have all been implicated in AMD and CNV pathogenesis. Oxidative damage is also thought to be a major factor in the pathogenesis of age-related cataract92, 93. More commonalities with human pathology genes were evident at the late (relative to the early) induction time-point. This is consistent with the greater risk of pathological complications in humans with high degrees of refractive error27,28,29,30, as well as ultrastructural studies suggesting that extreme occlusion-induced myopia can lead to pathological complications in chick49, 94,95,96. In this context, it is plausible that the observed commonalities could provide some insight into the molecular mechanisms underlying the progression from refractive errors to secondary pathologies. For example, up-regulation of oxidative defense genes in the present study is consistent with the theory that myopia may precipitate cataract development by increasing the generation of toxic lipid peroxidation by-products in the retina27, 97. However, it must be noted that the overlap of myopia and hyperopia genes with secondary pathologies was not exclusive to disorders for which they are a risk factor. For example, there is no reported association between myopia and AMD27, yet both early and late myopia conditions overlapped significantly with the genes associated with sub-clinical pre-AMD and AMD itself. Similarly, to our knowledge, hyperopia has not been linked with CNV in human populations despite the two common gene associations demonstrated here.
This lack of specificity could reflect several factors. Due to current data availability, the disease-associated gene lists could not be sourced from studies specifically investigating the pathological complications of refractive errors. The CNV genes, for example, were associated with ‘wet-AMD’ type CNV rather than myopic CNV. Similarly, the use of data from human genetic linkage and association studies made it difficult to determine whether the fold changes seen in myopic and hyperopic chicks are concordant or discordant with those seen in pathology (the latter possibly conferring a protective effect). Finally, many of the commonalities converged on pathways that displayed growth non-specific expression patterns in the meta-analysis, raising the possibility that they reflect responses to side-effects of lensing in the chick model (e.g. increased heat) rather than responses specifically related to changes in ocular growth and refractive state. Irrespective of the many questions raised, such extensive overlap between pathology-associated genes and the genes differentially-expressed in an animal model of relatively mild environmental perturbation is remarkable, and highlights optical-induced refractive error in chick as a potentially useful model for studying ‘pathology-type’ molecular responses of relevance to a range of human ocular disorders.
In summary, our rank product meta-analysis implicated highly similar genes in chick myopia and hyperopia models. The results provide novel evidence for transcriptional activation of the complement system during both myopia and hyperopia induction, and confirm existing literature implicating cell signaling, mitochondrial metabolism, and structural processes in refractive error. Further comparisons to previous research demonstrate improved concordance with human GWAS and animal proteomics studies of refractive error, and extensive similarities with the genes implicated in sight-threatening ocular pathologies.
Methods
Transcriptome study inclusion criteria
Datasets were included in the meta-analysis if they used a commercially available microarray platform or RNA-sequencing to compare gene expression in chick retina, RPE and/or choroid during optically-induced myopia or hyperopia induction, and normal development. Five studies meeting these inclusion criteria on 02/01/2017 (Table 1) were downloaded from Gene Expression Omnibus (GEO)98.
Data preprocessing
Microarray CEL files were RMA background corrected and quantile normalized using the affy package (v1.52.0)99. Probe annotations were updated (Affymetrix release 36, 04/13/16) and, because down-stream processes required a single measure for each gene, probe identifiers were then collapsed to the gene level by retaining only the probe with the highest average expression. This method was chosen as it provides good between-study consistency100. RNA-seq counts per million (CMP) data from Riddell et al.13 were imported into R and log2 transformed with an offset of 1 (i.e., log2(CMP +1)). The pre-processed microarray and RNA-seq datasets were merged into a single matrix containing only the 10,062 genes measured in all experiments, and then further divided into four conditions representing early (≤1 day) and late (>1 day) myopia and hyperopia induction (as shown in Table 2).
Rank product meta-analysis
Differentially-expressed genes (DEG) were identified using the Bioconductor RankProd package (v.3.0.0)25, 26, 101. Genes with a pfp (percentage of false positive predictions) of <0.05 were considered differentially-expressed. This pfp cut-off is theoretically equivalent to a false discovery rate of 5%101. When a single study contained multiple experimental conditions (i.e., multiple time-points within early or late categories; see Table 2), the expression values from each condition were considered as a separate dataset for the purpose of the meta-analysis.
Evaluation of the meta-analysis results
Correspondence at the top plots
Correspondence at the top (CAT) plots102 were used to evaluate the contribution of each dataset to the meta-analysis results. A rank product analysis of differential gene expression in each individual dataset was conducted as described above. The results of these individual study analyses and the meta-analysis were ordered by the rank product statistic (RP/Rsum), and the proportion of individual study results present in the meta-analysis results was plotted as a function of list size.
Comparison with previously published omics data
The meta-analysis results were compared with previously published omics data in two stages. Firstly, they were compared with the results of the individual studies that were meta-analyzed to identify the number of replicated and unique gene findings. Then, both the individual study results and the meta-analysis results were compared to the results of human GWAS and animal proteomics studies to assess whether the meta-analysis improved concordance with these broader omics data types. To facilitate these latter cross-species/cross-platform comparisons, DEG from the meta-analysis and from Riddell et al.13 were converted to human ortholog Ensembl Gene IDs as described previously21. All other data for the comparisons were derived from our previous publication21 (i.e., human orthologs of differentially-expressed proteins from proteomics studies, DEG and human orthologs of DEG from the original transcriptome studies, and genes within 500 Kilobases (Kb) and 250Kb of GWAS refractive error and axial length loci). The statistical significance of common findings across each list of genes was then assessed using the R software package GeneOverlap (version 1.10.0) as described previously21.
Pathway over-representation analysis
To facilitate interpretation, DEG from each condition were tested for over-representation of Reactome pathways using ConsensusPathDB (v.31)103. The manually-curated Reactome database was chosen for use because it is currently one of the most complete open source pathway repositories104. The 10,062 genes analyzed were used for the background list, and gene sets with an FDR q-value of <0.05 with ≥3 members were considered over-represented. Over-represented pathways with similar biological functions and identical gene contributions (i.e., redundant results) were collapsed into a single annotation, and the non-redundant pathway findings were then visualized as bubble plots using the R GOplot package105.
Testing overlap between meta-analysis conditions
Gene and pathway findings across the four conditions (early and late myopia and hyperopia induction) were compared using the R software package GeneOverlap. The statistical significance of overlapping findings was calculated relative to the 10,062 genes included in the meta-analysis (for DEG) or the 1786 Reactome pathways included in the over-representation analysis (for pathways). Pairwise overlaps between conditions were visualized as heat maps showing the intersection, Benjamini-Hochberg adjusted p-value, and Jaccard Index. The overlap analysis identified 15 genes that were differentially-expressed in a growth-specific manner (i.e., concurrently up-regulated in myopia and down-regulated in hyperopia, or visa versa). These genes were further investigated using ConsensusPathDB’s Induced Network Module Analysis that connects seed genes through different types of interactions106. The analysis was conducted using high and medium confidence filters, genetic, biochemical & gene regulatory interactions, and an intermediate node Z-score threshold of 15.
Testing overlap with secondary ocular pathologies
Author curated lists of genes associated with AMD, CNV, POAG, and cataract in humans (either via genetic linkage/association or RNA/protein expression changes) were sourced from recent publications as follows. Genes associated with AMD and/or drusen composition were sourced from Table S2 of Newman et al.79 Genes linked to CNV were sourced from Supplementary Material I of Zhang et al.86 Genes associated with POAG and its endophenotypes were sourced from Supplementary Table S1 of Iglesias et al.107 Genes associated with cataract were sourced from Cat-Map108 on 2017/02/26. In addition, a large list of genes potentially associated with sub-clinical pre-AMD (i.e., AREDS level 2) were sourced from Table S3 (disease module MD2) of Newman et al.79 Disease-associated gene lists were converted to Ensembl Gene IDs using BioMart (Ensembl release 86; Homo sapiens GRCh38.p7), and compared to human orthologs of the genes differentially-expressed in the early and late myopia and hyperopia meta-analyses. These comparisons were made using the GeneOverlap package as described previously21. The results were visualized as chord diagrams using the R GOplot package105, and overlapping genes were tested for over-representation of Reactome pathways as described above.
Data availability
The datasets analysed during the current study are available in the GEO repository, www.ncbi.nlm.nih.gov/geo/. GEO accession numbers are provided in Table 1.
References
Pan, C. W., Ramamurthy, D. & Saw, S. M. Worldwide prevalence and risk factors for myopia. Ophthal Physl Opt. 32, 3–16 (2012).
Grosvenor, T. Refractive component changes in adult‐onset myopia: evidence from five studies. Clinical and Experimental Optometry 77, 196–205 (1994).
Strang, N. C., Schmid, K. L. & Carney, L. G. Hyperopia is predominantly axial in nature. Current Eye Research 17, 380–383 (1998).
Norton, T. T. Animal models of myopia: learning how vision controls the size of the eye. Ilar Journal 40, 59–77 (1999).
Fischer, A. J., McGuire, J. J., Schaeffel, F. & Stell, W. K. Light-and focus-dependent expression of the transcription factor ZENK in the chick retina. Nature Neuroscience 2, 706–712 (1999).
Rohrer, B. & Stell, W. K. Basic fibroblast growth factor (bFGF) and transforming growth factor beta (TGF-β) act as stop and go signals to modulate postnatal ocular growth in the chick. Experimental Eye Research 58, 553–562 (1994).
Seko, Y., Shimizu, M. & Tokoro, T. Retinoic acid increases in the retina of the chick with form deprivation myopia. Ophthalmic Research 30, 361–367 (1998).
Stone, R. A., Lin, T., Laties, A. M. & Iuvone, P. M. Retinal dopamine and form-deprivation myopia. PNAS 86, 704–706 (1989).
Tkatchenko, A. V., Walsh, P. A., Tkatchenko, T. V., Gustincich, S. & Raviola, E. Form deprivation modulates retinal neurogenesis in primate experimental myopia. PNAS 103, 4681–4686 (2006).
Shelton, L. et al. Microarray analysis of choroid/RPE gene expression in marmoset eyes undergoing changes in ocular growth and refraction. Mol Vis 14, 1465–1479 (2008).
Metlapally, R. et al. Genome-Wide Scleral Micro-and Messenger-RNA Regulation During Myopia Development in the Mouse. IOVS 57, 6089–6097 (2016).
Brand, C., Schaeffel, F. & Feldkaemper, M. P. A microarray analysis of retinal transcripts that are controlled by image contrast in mice. Mol Vis 13, 920–932 (2007).
Riddell, N., Giummarra, L., Hall, N. & Crewther, S. Bidirectional expression of metabolic, structural, and immune pathways in early myopia and hyperopia. Frontiers in Neuroscience 10, 1–16, doi:10.3389/fnins.2016.00390 (2016).
McGlinn, A. M. et al. Form-deprivation myopia in chick induces limited changes in retinal gene expression. IOVS 48, 3430–3436 (2007).
Stone, R. A. et al. Image defocus and altered retinal gene expression in chick: clues to the pathogenesis of ametropia. IOVS 52, 5765–5777 (2011).
Ashby, R. S. & Feldkaemper, M. P. Gene expression within the amacrine cell layer of chicks after myopic and hyperopic defocus. IOVS 51, 3726–3735 (2010).
Schippert, R., Schaeffel, F. & Feldkaemper, M. P. Microarray analysis of retinal gene expression in chicks during imposed myopic defocus. Mol Vis 14, 1589–1599 (2008).
Rada, J. A. & Wiechmann, A. F. Ocular expression of avian thymic hormone: changes during the recovery from induced myopia. Mol Vis 15, 778–792 (2009).
Tkatchenko, A. V. et al. Large-Scale microRNA Expression Profiling Identifies Putative Retinal miRNA-mRNA Signaling Pathways Underlying Form-Deprivation Myopia in Mice. PloS one 11, e0162541, doi:10.1371/journal.pone.0162541 (2016).
Stone, R. A. & Khurana, T. S. Gene profiling in experimental models of eye growth: clues to myopia pathogenesis. Vision Res 50, 2322–2333 (2010).
Riddell, N. & Crewther, S. G. Integrated comparison of GWAS, transcriptome, and proteomics studies highlights similarities in the biological basis of animal and human myopia. IOVS 58, 660–669 (2017).
Crewther, D. P. The role of photoreceptors in the control of refractive state. Progress in Retinal and Eye Research 19, 421–457 (2000).
Rymer, J. & Wildsoet, C. F. The role of the retinal pigment epithelium in eye growth regulation and myopia: a review. Visual Neuroscience 22, 251 (2005).
Wallman, J. & Winawer, J. Homeostasis of eye growth and the question of myopia. Neuron 43, 447–468 (2004).
Hong, F. et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22, 2825–2827 (2006).
Hong, F. & Breitling, R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics 24, 374–382 (2008).
Saw, S. M., Gazzard, G., Shih-Yen, E. C. & Chua, W. H. Myopia and associated pathological complications. Ophthalmic & Physiological Optics 25, 381–391 (2005).
Wong, T. Y., Ferreira, A., Hughes, R., Carter, G. & Mitchell, P. Epidemiology and disease burden of pathologic myopia and myopic choroidal neovascularization: an evidence-based systematic review. American Journal of Ophthalmology 157, 9–25 e12, doi:10.1016/j.ajo.2013.08.010 (2014).
Lavanya, R. et al. Hyperopic refractive error and shorter axial length are associated with age-related macular degeneration: the Singapore Malay Eye Study. IOVS 51, 6247–6252 (2010).
Flitcroft, D. I. The complex interactions of retinal, optical and environmental factors in myopia aetiology. Progress in Retinal and Eye Research 31, 622–660 (2012).
Stone, R. A., Laties, A. M., Raviola, E. & Wiesel, T. N. Increase in retinal vasoactive intestinal polypeptide after eyelid fusion in primates. PNAS 85, 257–260 (1988).
Seltner, R. L. P. & Stell, W. K. The effect of vasoactive intestinal peptide on development of form deprivation myopia in the chick: A pharmacological and immunocytochemical study. Vision Res 35, 1265–1270 (1995).
Feldkaemper, M. P. & Schaeffel, F. Evidence for a potential role of glucagon during eye growth regulation in chicks. Visual Neuroscience 19, 755–766 (2002).
Zhu, X. & Wallman, J. Opposite effects of glucagon and insulin on compensation for spectacle lenses in chicks. IOVS 50, 24–36 (2009).
Simon, P., Feldkaemper, M., Bitzer, M., Ohngemach, S. & Schaeffel, F. Early transcriptional changes of retinal and choroidal TGFbeta-2, RALDH-2, and ZENK following imposed positive and negative defocus in chickens. Mol Vis 10, 588–597 (2004).
Zhang, Y., Liu, Y. & Wildsoet, C. F. Bidirectional, optical sign-dependent regulation of BMP2 gene expression in chick retinal pigment epithelium. IOVS 53, 6072–6080 (2012).
Elvington, M., Liszewski, M. K. & Atkinson, J. P. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev 274, 9–15 (2016).
Long, Q., Ye, J., Li, Y., Wang, S. & Jiang, Y. C-reactive protein and complement components in patients with pathological myopia. Optometry and Vision Science 90, 501–506 (2013).
Gao, T.-T., Long, Q. & Yang, X. Complement factors C1q, C3 and C5b-9 in the posterior sclera of guinea pigs with negative lens-defocused myopia. International Journal of Ophthalmology 8, 675 (2015).
Barathi, V. A. et al. Involvement of GABA transporters in atropine-treated myopic retina as revealed by iTRAQ quantitative proteomics. Journal of Proteome Research 13, 4647–4658 (2014).
Sotgia, F. et al. Mitochondria “fuel” breast cancer metabolism: Fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle 11, 4390–4401 (2012).
Lyng, H. et al. Gene expressions and copy numbers associated with metastatic phenotypes of uterine cervical cancer. BMC Genomics 7, 268, doi:10.1186/1471-2164-7-268 (2006).
Chen, Y., Cairns, R., Papandreou, I., Koong, A. & Denko, N. C. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PloS One 4, e7033, doi:10.1371/journal.pone.0007033 (2009).
Shih, Y.-F. et al. Reduction in choroidal blood flow occurs in chicks wearing goggles that induce eye growth toward myopia. Current Eye Research 12, 219–227 (1993).
Teakle, E. M., Wildsoet, C. F. & Vaney, D. I. The spatial organization of tyrosine hydroxylase-immunoreactive amacrine cells in the chicken retina and the consequences of myopia. Vision Res 33, 2383–2396 (1993).
Troilo, D., Xiong, M., Crowley, J. C. & Finlay, B. L. Factors controlling the dendritic arborization of retinal ganglion cells. Visual Neuroscience 13, 721–733 (1996).
Beresford, J. A., Crewther, S. G. & Crewther, D. P. Anatomical correlates of experimentally induced myopia. Australian and New Zealand Journal of Ophthalmology 26, S84–S87 (1998).
Seko, Y., Shimokawa, H., Pang, J. & Tokoro, T. Disturbance of electrolyte balance in vitreous of chicks with form-deprivation myopia. Japanese Journal of Ophthalmology 44, 15–19 (2000).
Liang, H., Crewther, S. G., Crewther, D. P. & Junghans, B. M. Structural and elemental evidence for edema in the retina, retinal pigment epithelium, and choroid during recovery from experimentally induced myopia. IOVS 45, 2463–2474 (2004).
Crewther, S. G., Liang, H., Junghans, B. M. & Crewther, D. P. Ionic control of ocular growth and refractive change. PNAS 103, 15663–15668 (2006).
Turrens, J. F. Mitochondrial formation of reactive oxygen species. The Journal of physiology 552, 335–344 (2003).
Cadenas, E. & Davies, K. J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology & Medicine 29, 222–230 (2000).
Scandalios, J. G. Oxidative stress: Molecular perception and transduction of signals triggering antioxidant gene defenses. Braz J Med Biol Res 38, 995–1014 (2005).
Jowsey, I. R., Smith, S. A. & Hayes, J. D. Expression of the murine glutathione S-transferase α3 (GSTA3) subunit is markedly induced during adipocyte differentiation: activation of the GSTA3 gene promoter by the pro-adipogenic eicosanoid 15-deoxy-Δ 12, 14-prostaglandin J 2. Biochemical and Biophysical Research Communications 312, 1226–1235 (2003).
Sun, X. X. & Dai, M. S. Deubiquitinating enzyme regulation of the p53 pathway: A lesson from Otub1. World J Biol Chem 5, 75–84 (2014).
Hayek, T., Oiknine, J., Dankner, G., Brook, J. G. & Aviram, M. HDL apolipoprotein A-I attenuates oxidative modification of low density lipoprotein: Studies in transgenic mice. Eur J Clin Chem Clin Biochem 33, 721–725 (1995).
Niki, E., Yoshida, Y., Saito, Y. & Noguchi, N. Lipid peroxidation: mechanisms, inhibition, and biological effects. Biochemical and Biophysical Research Communications 338, 668–676 (2005).
Graf, E., Mahoney, J. R., Bryant, R. G. & Eaton, J. W. Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J Biol Chem 259, 3620–3624 (1984).
Kruszewski, M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res 531, 81–92 (2003).
Chung, H. Y. et al. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age (Omaha) 20, 127–140 (1997).
Ruttkay-Nedecky, B. et al. The role of metallothionein in oxidative stress. International Journal of Molecular Sciences 14, 6044–6066 (2013).
Balboa, M. A. & Balsinde, J. Oxidative stress and arachidonic acid mobilization. Biochimica et Biophysica Acta 1761, 385–391 (2006).
Hodos, W., Revzin, A. & Kuenzel, W. Thermal gradients in the chick eye: a contributing factor in experimental myopia. IOVS 28, 1859–1866 (1987).
Edwards, M. H. Animal models of myopia: a review. Acta Ophthalmol Scand 74, 213–219 (1996).
Morgan I. G., Rose, K. A., & Ashby, R. S. Animal models of experimental myopia: limitations and synergies with studies on human myopia in Pathologic Myopia (eds. Spaide, R. F., Ohno-Matsui, K., Yannuzzi, L. A.) 39–58 (Springer New York, 2014).
Mutti, D. O., Zadnik, K. & Adams, A. J. Myopia. The nature versus nurture debate goes on. IOVS 37, 952–957 (1996).
Schaeffel, F. & Feldkaemper, M. Animal models in myopia research. Clin Exp Optom 98, 507–517 (2015).
Zadnik, K. & Mutti, D. O. How applicable are animal myopia models to human juvenile onset myopia? Vision Res 35, 1283–1288 (1995).
Cheng, C. et al. Nine loci for ocular axial length identified through genome-wide association studies, including shared loci with refractive error. Am J Hum Genet 93, 264–277 (2013).
Hawthorne, F. A. & Young, T. L. Genetic contributions to myopic refractive error: insights from human studies and supporting evidence from animal models. Exp Eye Res 114, 141–149 (2013).
Miyake, M. et al. Identification of myopia-associated WNT7B polymorphisms provides insights into the mechanism underlying the development of myopia. Nat Commun 6, 6689 (2015).
Stambolian, D. Genetic susceptibility and mechanisms for refractive error. Clin Genet 84, 102–108 (2013).
Tkatchenko, A. V. et al. APLP2 regulates refractive error and myopia development in mice and humans. PLoS Genet 11, e1005432, doi:10.1371/journal.pgen.1005432 (2015).
Williams, K. M. & Hammond, C. J. GWAS in myopia: insights into disease and implications for the clinic. Expert Review of Ophthalmology 11, 101–110 (2016).
Verhoeven, V. J. et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat Genet 45, 314–318 (2013).
Kiefer, A. K. et al. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet 9, e1003299 (2013).
Nita, M., Strzalka-Mrozik, B., Grzybowski, A., Mazurek, U. & Romaniuk, W. Age-related macular degeneration and changes in the extracellular matrix. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research 20, 1003–1016 (2014).
Cai, J. et al. Roles of NFkappaB-miR-29s-MMP-2 circuitry in experimental choroidal neovascularization. Journal of Neuroinflammation 11, 88, doi:10.1186/1742-2094-11-88 (2014).
Newman, A. M. et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med 4, 16, doi:10.1186/gm315 (2012).
Weber, B. H. et al. The role of the complement system in age-related macular degeneration. Dtsch Arztebl Int 111, 133–138 (2014).
McHarg, S., Clark, S. J., Day, A. J. & Bishop, P. N. Age-related macular degeneration and the role of the complement system. Molecular Immunology 67, 43–50 (2015).
Tan, P. L., Bowes Rickman, C. & Katsanis, N. AMD and the alternative complement pathway: genetics and functional implications. Hum Genomics 10, 23, doi:10.1186/s40246-016-0079-x (2016).
Nozaki, M. et al. Drusen complement components C3a and C5a promote choroidal neovascularization. PNAS 103, 2328–2333 (2006).
Rohrer, B., Coughlin, B., Bandyopadhyay, M. & Holers, V. M. Systemic human CR2-targeted complement alternative pathway inhibitor ameliorates mouse laser-induced choroidal neovascularization. Journal of Ocular Pharmacology And Therapeutics 28, 402–409 (2012).
Yamamoto, Y. et al. Associations of inflammatory cytokines with choroidal neovascularization in highly myopic eyes. Retina 35, 344–350 (2015).
Zhang, J. et al. Mining for genes related to choroidal neovascularization based on the shortest path algorithm and protein interaction information. Biochimica et Biophysica Acta 1860, 2740–2749 (2016).
Chowers, I. et al. The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. IOVS 47, 2135–2140 (2006).
Hahn, P., Milam, A. H. & Dunaief, J. L. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol 121, 1099–1105 (2003).
Imamura, Y. et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. PNAS 103, 11282–11287 (2006).
Dong, A. et al. Oxidative stress promotes ocular neovascularization. Journal of Cellular Physiology 219, 544–552 (2009).
Blasiak, J., Petrovski, G., Vereb, Z., Facsko, A. & Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. BioMed Research International 2014, 768026, doi:10.1155/2014/768026 (2014).
Giblin, F. J. et al. Nuclear light scattering, disulfide formation and membrane damage in lenses of older guinea pigs treated with hyperbaric oxygen. Experimental Eye Research 60, 219–235 (1995).
Wolf, N. et al. Age-related cataract progression in five mouse models for anti-oxidant protection or hormonal influence. Experimental Eye Research 81, 276–285 (2005).
Hirata, A. & Negi, A. Lacquer crack lesions in experimental chick myopia. Graefe’s Archive for Clinical and Experimental Ophthalmology 236, 138–145 (1998).
Hayes, B. P., Fitzke, F. W., Hodos, W. & Holden, A. L. A morphological analysis of experimental myopia in young chickens. IOVS 27, 981–991 (1986).
Liang, H., Crewther, D. P., Crewther, S. G. & Barila, A. M. A role for photoreceptor outer segments in the induction of deprivation myopia. Vision Res 35, 1217–1225 (1995).
Zigler, J. S., Bodaness, R. S., Gery, I. & Kinoshita, J. H. Effects of lipid peroxidation products on the rat lens in organ culture: A possible mechanism of cataract initiation in retinal degenerative disease. Archives of Biochemistry and Biophysics 225, 149–156 (1983).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research 30, 207–210 (2002).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Miller, J. A. et al. Strategies for aggregating gene expression data: The collapseRows R function. BMC Bioinformatics 12, 322, doi:10.1186/1471-2105-12-322 (2011).
Carratore, F. D. RankProd: Rank Product method for identifying differentially expressed genes with application in meta-analysis. R package version 3.0.0 (Bioconductor, 2016).
Irizarry, R. A. et al. Multiple-laboratory comparison of microarray platforms. Nat Methods 2, 345–350 (2005).
Kamburov, A., Wierling, C., Lehrach, H. & Herwig, R. ConsensusPathDB-A database for integrating human functional interaction networks. Nucleic Acids Research 37, D623–628, doi:10.1093/nar/gkn698 (2009).
Bauer‐Mehren, A., Furlong, L. I. & Sanz, F. Pathway databases and tools for their exploitation: benefits, current limitations and challenges. Molecular Systems Biology 5, 290 (2009).
Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015).
Herwig, R., Hardt, C., Lienhard, M. & Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 11, 1889–1907 (2016).
Iglesias, A. I. et al. Genes, pathways, and animal models in primary open-angle glaucoma. Eye 29, 1285–1298 (2015).
Shiels, A., Bennett, T. M. & Hejtmancik, J. F. Cat-Map: putting cataract on the map. Mol Vis 16, 2007–2015 (2010).
Acknowledgements
Supported by Australia Research Council Grant DP110103784 to DPC and SGC, and by Australian Government Research Training Program Scholarship to N.R.
Author information
Authors and Affiliations
Contributions
N.R. and S.G. conceived and designed the study. N.R. analyzed the data, wrote the manuscript, and prepared the figures in consultation with S.G.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riddell, N., Crewther, S.G. Novel evidence for complement system activation in chick myopia and hyperopia models: a meta-analysis of transcriptome datasets. Sci Rep 7, 9719 (2017). https://doi.org/10.1038/s41598-017-10277-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10277-2
This article is cited by
-
Genome-wide analysis of retinal transcriptome reveals common genetic network underlying perception of contrast and optical defocus detection
BMC Medical Genomics (2021)
-
RNA-seq and GSEA identifies suppression of ligand-gated chloride efflux channels as the major gene pathway contributing to form deprivation myopia
Scientific Reports (2021)
-
The plasminogen protein is associated with high myopia as revealed by the iTRAQ-based proteomic analysis of the aqueous humor
Scientific Reports (2021)
-
Analysis of genetic networks regulating refractive eye development in collaborative cross progenitor strain mice reveals new genes and pathways underlying human myopia
BMC Medical Genomics (2019)
-
Epigenetically dysregulated genes and pathways implicated in the pathogenesis of non-syndromic high myopia
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
