
Abstract
The transcriptional repressor REST regulates many neuronal genes by binding RE1 motifs. About one third of human RE1s are recently evolved and specific to primates. As changes in the activity of a transcription factor reverberate on its downstream targets, we assessed whether REST displays fast evolutionary rates in primates. We show that REST was targeted by very strong positive selection during primate evolution. Positive selection was also evident in the human lineage, with six selected sites located in a region that surrounds a VNTR in exon 4. Analysis of expression data indicated that REST brain expression peaks during aging in humans but not in other primates. Because a REST coding variant (rs3796529) was previously associated with protection from hippocampal atrophy in elderly subjects with mild cognitive impairment (MCI), we analyzed a cohort of Alzheimer disease (AD) continuum patients. Genotyping of two coding variants (rs3796529 and rs2227902) located in the region surrounding the VNTR indicated a role for rs2227902 in modulation of hippocampal volume loss, indirectly confirming a role for REST in neuroprotection. Experimental studies will be instrumental to determine the functional effect of positively selected sites in REST and the role of REST variants in neuropreservation/neurodegeneration.
Similar content being viewed by others

Introduction
The Repressor Element 1 Silencing Transcription factor (REST, also known as neuron restrictive silencer factor, NRSF) is a transcriptional regulator that binds a specific 21 bp motif (Repressor Element 1–RE1) in the regulatory regions of target genes1, 2. To regulate expression, REST interacts with chromatin modifiers (HDAC complex) by recruiting corepressor mSin3 and CoREST3, 4.
REST acts as a negative regulator of neuronal gene expression during both embryogenesis and adult neurogenesis5, 6, and also plays a role in modulating synaptic plasticity7.
In the normal aging brain, REST is the most activated transcription factor and functions as a neuroprotective modulator8. In fact, REST levels increase with age in the prefrontal cortex (PFC) and hippocampus of healthy adults. REST expression correlates with the up-regulation of protective stress response genes, as well as with the repression of genes that promote cell death. These findings support the notion that REST is necessary during aging to maintain neuronal viability and to preserve cognitive functions8.
Moreover, REST and its target genes have been implicated in the pathogenesis of a number of different neurodegenerative diseases, including Alzheimer’s Disease (AD) clinical continuum, frontotemporal dementia, and dementia with Lewy bodies8, 9. In these pathologies, REST is depleted in the nucleus of PFC and hippocampal neurons and colocalizes in autophagosomes together with pathological misfolded proteins (e.g. Aβ, phosphorilated Tau, TDP-43, α-synuclein).
In elderly subjects with mild cognitive impairment (MCI), a disorder that has been associated with risk for dementia, a missense REST variant (rs3796529) was associated with baseline hippocampal volume and with the rate of hippocampal grey matter (GM) density loss, suggesting that the minor allele of rs3796529 confers a protective effect on hyppocampal morphology10. This finding was confirmed in a larger MCI and AD cohort; in particular the minor allele of rs3796529 was associated with larger hippocampal CA1 volumes11.
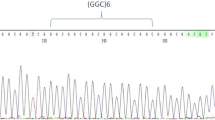
A polymorphic variable number tandem repeat (VNTR) in the coding region of REST was described and characterized as having two major alleles of 4 or 5 48-nucleotide repeats12. A haplotype containing the VNTR was associated with general cognitive skills in elderly subjects of European ancestry. In particular, individuals carrying the 4 repeat allele displayed higher cognitive abilities12.
REST is conserved in vertebrates and a genome-wide search for RE1 elements indicated that, during vertebrate evolution, novel RE1s have arisen and generated species-specific REST targets13. In particular, about one third of human RE1 elements are specific to primates. Although ancient RE1 motifs recruit REST with higher affinity, the recently evolved primate-specific RE1s have preserved the ability to recruit REST in vivo 13. In line with these data, a comparison of human and mouse embryonic stem cells identified several human-specific REST targets. These genes are enriched for memory and learning functions14.
These observations, together with the central role played by REST in neurodevelopment and neuropreservation, led to the suggestion that the expansion of REST targets contributed to the development of primate-specif traits in terms of brain function or cognition13.
Previous analyses of FOXP2, another transcription factor that is highly expressed in the brain, indicated that positive selection acted in concert on the FOXP2 gene and on its target sequences15. This is conceivable as changes in the activity or expression of a transcription factor reverberate on its downstream targets. We thus analyzed the evolutionary history of REST in primates. Results herein show that REST was targeted by very strong positive selection during primate evolution, and most positively selected sites cluster in the region surrounding the VNTR. This also holds true for sites that were specifically selected in the human lineage. We also report that a REST haplotype comprising a variant in linkage disequilibrium with the VNTR modulates right hippocampal volume in an Italian cohort of AD subjects.
Results
REST evolution in primates
To analyze the evolutionary history of REST in primates, we obtained and aligned coding sequence information for 21 primate species available in public databases (Supplementary Table S1). Because of misalignment in the region corresponding to the human polymorphic VNTR (exon 4), this 240 bp region was filtered.
We calculated the average nonsynonymous substitution/synonymous substitution rate ratio (dN/dS, also referred to as ω) using the single-likelihood ancestor counting (SLAC) method16: dN/dS for REST amounted to 0.41 (95% confidence intervals: 0.38–0.45). Although this result indicates purifying selection (i.e. dN/dS < 1) as the major driving force in shaping REST gene diversity, the dN/dS value is higher than those observed for most mammalian genes17.
We thus tested whether positive selection (i.e. dN/dS > 1) acted on a subset of REST codons by applying likelihood ratio tests (LRT) implemented in the codeml program18, 19. Specifically, we used LRTs that compare models of gene evolution allowing (NSsite models M2a and M8, positive selection models) or disallowing (NSsite models M1a and M7, null models) a class of codons to evolve with dN/dS > 1.
Both null models were rejected in favor of the positive selection models; the same result was obtained using different codon frequency models (F3X4 and F61) (Table 1). In order to identify specific sites subject to positive selection, we applied the BEB, FUBAR, and REL analyses (see Methods). To limit false positives, only sites detected using at least two methods were considered as positive selection targets. A total of 22 positively selected sites were identified, 5 of which were detected by all three methods (Fig. 1A). All selected sites were located in a relatively large region surrounding the VNTR.

REST evolutionary analysis. (A) Schematic representation of REST protein structure. Zinc-finger domains (ZnF), a variable number tandem repeat (VNTR), protein repressor domains that recruit corepressor mSin3 and CoREST, and Lysine and Proline rich domains (Lys-rich; Pro-rich) are indicated on the structure. Positively selected sites in the primate phylogeny, in human and gorilla lineages are reported in red, green, and magenta, respectively. Asterisks denote positively selected sites identified by 3 different methods. Missense variants genotyped in AD continuum subjects are shown in blue. (B) Evolutionary fingerprinting of the primate REST genes. The estimate of the distribution of synonymous (α) and nonsynonymous (β) substitution rates is plotted on a log-log scale. The ellipses reflect a Gaussian-approximated variance in each individual rate estimate, and colored pixels show the density of the posterior sample of the distribution for a given rate. The diagonal line represents the neutral expectation (dN/dS = 1), points above the line correspond to positive selection (dN/dS > 1), and points below the line to purifying selection (dN/dS < 1). (C) Violin plots of selection coefficients (median, white dot; interquartile range, black bar) for the REST gene in Homininae. Selection coefficients (γ) are classified as strongly beneficial (100, 50), moderately beneficial (10, 5), weakly beneficial (1), neutral (0), weakly deleterious (−1), moderately deleterious (−5, −10), strongly deleterious (−50, −100), and inviable (−500).
In order to evaluate the presence of episodic positive selection acting on a subset of primate branches, we applied the BUSTED test (branch-site unrestricted statistical test for episodic diversification)20. No evidence of episodic diversifying selection was detected (BUSTED p value = 0.188).
We next performed an evolutionary fingerprinting analysis21, which partitions sites into selective classes and estimates dN/dS for such classes. The best fitting model had 3 rate classes, two of them accounting for the majority of codons (Fig. 1B). Specifically, 73% of REST codons experienced negative selection (dN/dS = 0.13) and 26% were targeted by positive selection (dN/dS = 1.38). No sites were found to be evolving neutrally (Fig. 1B).
REST evolution in Homininae
In order to study the evolution of the REST gene in Homininae and to gain insight into the more recent selective events in specific lineages, we applied a population genetics-phylogenetics approach (gammaMap)22. gammaMap leverages intra-species variation and inter-specific diversity to estimate the distribution of selection coefficients (γ) along coding regions.
In humans, the polymorphic REST exon 4 VNTR contains 4 or 5 repeats12. Analysis of genomes of Homininae indicated that chimpanzees have 5 repeats, whereas the gorilla REST gene carries only four repeat copies (see Methods).
As above, the 240 bp region corresponding to the VNTR was masked before running gammaMap.
Analysis of the overall distribution of selection coefficients indicated a similar evolutionary pattern in humans, chimpanzees, and gorillas (Fig. 1C): most codons displayed γ values below −10, indicating that purifying selection drove the evolution of a major proportion of sites in REST.
We next used gammaMap to identify specific codons evolving under positive selection (defined as those having a cumulative probability > 0.80 of γ > 0) in each lineage. Six sites were found to represent positive selection targets in humans (Table 2, Fig. 1A). Only two selected sites were detected in gorillas and none in the chimpanzee lineage. Six selected sites fall in a proline-rich region, the other two sites fall in the lysine-rich region (Fig. 1A).
REST brain expression in primates
During human lifespan, REST expression increases in the nucleus of aging PFC and hippocampal neurons8. To investigate the effect of age on REST expression in other primates and compare it with humans, we retrieved data from a study that analyzed mRNA expression in two brain regions (PFC and cerebellar cortex) in humans, macaques, and chimpanzees23. In order to assess whether REST expression deviates from a null expectation of constant levels throughout lifespan, we applied a bootstrap approach to calculate confidence intervals. Results indicated that in the cerebellar cortex, REST expression has a similar pattern in the three species, with a higher expression after birth and a progressive decline with aging (Fig. 2A). Conversely, different expression dynamics were observed in the PFC, with humans showing a decline after birth followed by a plateau in childhood and a steep increase after adolescence. In chimpanzees and macaques REST expression tends to increase throughout most of the lifespan (Fig. 2B).

Expression profile of REST in the prefrontal cortex (superior frontal gyrus) and cerebellar cortex at different age points. Data are derived by Somel et al.23 and refer to humans (red), macaques (black), and chimpanzees (blue). Each point represents an individual (25 humans, 31 macaques, and 12 chimpanzees) and lines show lowess fittings47. Lighter lines represent the 2.5 and 97.5 confidence intervals calculated using 1000 bootstrap replicates. Age (x-axis) is reported in log2 scale and REST expression level in log scale.
REST polymorphisms and hippocampal volume loss
We analyzed the possible correlation between REST variants and hippocampal volume loss in a group of 99 Italian subjects with AD continuum: 60 “clinical AD” and 39 “preclinical AD” subjects according to disease stage24. All these subjects underwent MRI evaluation and bilateral hippocampal volumetries were computed from MRI T1-3D high-resolution images as well-established structural imaging markers for AD staging24.
Two REST variants were genotyped in all patients: rs3796529, located in the VNTR and previously reported as protective for hippocampal atrophy in MCI and AD10, 11, and rs2227902, in linkage disequilibrium with the exon 4 VNTR in European populations12.
To address their contribution to hippocampal atrophy, linear regression analysis was performed using age (at MRI examination), sex, APOE genotype, and volumetric scaling factor calculated by SIENAX as covariates.
We observed no association with hippocampal volume for rs3796529. Conversely, the minor T allele at rs2227902 appeared to be associated with right hippocampus volume loss (Table 3).
Haplotype analysis using the same covariates detected a haplotype significantly associated with right hippocampus volume reduction (p value = 0.008). The predisposing haplotype includes the rs2227902 T allele and the major allele (C allele) at rs3796529.
Discussion
Primate genomes have evolved to display many more REST binding sites compared to other mammals, and several recent RE1 elements regulate neuronal genes13. Herein we show that, during primate evolution, REST evolved under strong positive selection. Indeed, using the integration of multiple methods to limit false positives, we identified 22 sites that were targeted by diversifying selection. Additional selected sites in REST were specific for the human or gorilla lineages. We exclude that the relatively large number of positively selected sites is due to a relaxation of functional constraint rather than to positive selection. In fact, evolutionary fingerprinting identified two major selective classes of negatively and positively selected sites, with no indication of a class of sites close to evolutionary neutrality. Moreover, the distribution of selection coefficients in Homininae indicated that the majority of codons are evolving under purifying selection.
None of the selected sites we identified involve the zinc-finger motifs that directly interact with DNA. Conversely, several selected sites are located within the lysine-rich and proline-rich regions of the protein. Additional sites cluster in a C-terminal portion of unspecified function. An interesting possibility is that selected sites modulate REST binding to other co-factors, in turn resulting in a species-specific regulation of gene expression. Recent reports have suggested that REST should be regarded as a platform for the assembly of multiple factors, which differ in a cell type- and genomic context-dependent manner5, 25, 26. Different configurations of REST co-factors have been observed26 and the REST interactome consists of at least 200 proteins in human cell lines27. Unfortunately, the molecular details of these interactions are largely unknown, making it impossible to assess whether selection acted to modulate protein-protein interaction affinities.
In this respect, it is worth drawing a parallel with FOXP2, another transcription factor that was targeted by positive selection in the human lineage and is central for neurodevelopment. Two human-specific substitutions were identified in FOXP2 28, 29. Although these changes are not located in the DNA binding region, they do confer differential transcriptional regulation in vitro 30. Because human and chimpanzee FOXP2 proteins bind two co-factors (FOXP1 and FOXP4) with similar affinity30, it is unclear whether the two selected sites modulate transcription of downstream targets by differential binding to other co-factors or via distinct mechanisms.
In any case, by analogy with FOXP2, the positively selected sites in REST may well modulate the transcription of downstream targets, although the underlying molecular details remain to be elucidated. This might also explain the dynamic expansion that occurred at the level of RE1 elements during primate evolution, as changes in transactivation activity are expected to expose target sequences to novel evolutionary drives, as again suggested for FOXP2 and its binding sites15.
We identified six sites in REST that were targeted by positive selection specifically in the human lineage. These changes arose before the split of the Homo sapiens lineage from Neanderthals, indicating that the ensuing phenotypic changes were shared between modern and archaic humans.
Given the peculiarity of human cognition and the observed association of REST variants with cognitive abilities (at least in adults)8, it is tempting to speculate that the human-specific changes contributed to the evolution of human intelligence. However, some sobering remarks should be mentioned. First, positively selected changes in non-human primates were previously described at genes involved in developmental dyslexia29. Clearly, these changes did not provide these primates with reading abilities, indicating that inference of the phenotypic effects of selected sites is challenging. Second, although REST has been extensively studied in relation to neurogenesis and neuroprotection, this transcription factor is ubiquitously expressed and contributes to the regulation of important physiological functions, including vascular smooth muscle cell proliferation31, regulation of fetal cardiac gene expression32, maintenance of a pluripotent state of embryonic stem cells5, and oncogenesis33. Third, several works indicated that Herpes Simplex Virus 1 (HSV-1), a neurotropic human virus, has evolved the ability to exploit the CoREST/REST repressor complex to regulate its gene expression34. Because viruses related to HSV-1 infect non-human primates35, the positive selection signal we identified may result from a conflict between primate hosts and herpes simplex viruses.
Therefore, the ultimate phenotype(s) targeted by natural selection may be related to these diverse functions and not necessarily to neurodevelopment or neuroprotection. As for this latter, the effect of REST has been mainly described in elderly subjects8 and natural selection is inefficient or weak for phenotypes with post-reproductive onset.
Although the selective pressures responsible for shaping REST evolution, as well as the phenotypic consequences of such selection remain to be determined, it is clear that this transcription factor plays an important role during human aging.
REST levels increase progressively in the nuclei of hippocampal and brain cortical neurons in healthy aging humans. This leads to the upregulation of protective genes and to the downregulation of genes related to neuronal degeneration8. In fact, REST levels in PFC neurons from elderly subjects positively correlate with cognitive and memory preservation8. Interestingly, our analysis of REST expression in chimpanzees and macaques indicated that the age-dependent up-regulation of REST expression in the PFC, but not in the cerebellum, is specific for humans.
In neurodegenerative diseases, REST has often been observed within cytoplasmatic autophagosomes, and its levels show no increase in the nuclei of hippocampal neurons. As hippocampal atrophy is one of the prominent features of AD, and REST expression is reduced in hippocampal neurons of AD patients8, it was suggested that REST may be associated with the rate of change in hippocampal volume.
Recently, the major allele of the P797L variant (rs3796529) was associated with right hippocampal loss in MCI subjects10 and in AD11. This variant is located in the REST region where several selected sites also map. This association was however questioned by data from the ENIGMA consortium36.
Thus, we investigated the role of two REST variants (rs3796529 and rs2227902) as modulators of hippocampal volume loss in AD continuum. Contrary to the report from Nho and colleagues10, 11, we observed no effect of the rs3796529 variant on hippocampal volume loss/preservation; instead, the minor allele of rs2227902 was significantly associated with right hippocampal volume reduction in AD. Nonetheless, a haplotype carrying the minor allele of rs2227902 and the major allele of rs3796529 was found to predispose to hippocampal volume loss, partially in agreement with the results by Nho and coworkers10, 11.
However, we note that the size of the AD population we analyzed is small and, consequently, the results reported herein will need independent validation in additional cohorts. Also, the functional effect of the P797L change is presently unknown and no study analyzed the biochemical differences (if any) of REST molecules carrying 4 or 5 copies of the VNTR. Addressing these issues will likely be instrumental to gain insight into the role of REST variants in neuropreservation/neurodegeneration.
Overall, these findings, together with previous data, confirm a role of REST in hippocampal atrophy/preservation in neurogenerative disorders. These observation also indicate REST as a promising target for neuroprotective strategies for neurodegenerative disorders, in particular for AD.
Methods
Evolutionary analysis in Primates
Primates coding sequences were retrieved from the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov, last accessed October 31, 2015). A list of species and of GenBank accession numbers is available as Supplementary Table S1.
DNA alignments were performed using the RevTrans 2.0 utility37. Genetic variability that is generated by recombination can be mistaken as positive selection38; thus, to limit false positives, alignments were screened for the presence of recombination breakpoints using Genetic Algorithm Recombination Detection (GARD)39. GARD is a program that uses phylogenetic incongruence among segments of a sequence alignment to detect the best-fit number and location of recombination breakpoints39. No significant breakpoint (p value < 0.01) was detected.
The average non-synonymous substitution/synonymous substitution rate (dN/dS; ω) was estimated using SLAC (Single Likelihood Ancestor Counting)16, a tool from the Hyphy package based on a codon substitution matrix and ancestral state reconstruction.
We used the PAML (Phylogenetic Analysis by Maximum Likelihood) software to detected positive selection19. The codeml NSsite models that allow (M2a, M8) or disallow (M1a, M7) a class of sites to evolve with ω > 1 were fitted to the data using different codon frequencies model: the F3X4 model (codon frequencies estimated from the nucleotide frequencies in the data at each codon site) and the F61 model (frequencies of each of the 61 non-stop codons estimated from the data)18, 19. For these analyses, phylogenetic trees were reconstructed using the program phyML with a maximum-likelihood approach, a General Time Reversible (GTR) model plus gamma-distributed rates and 4 substitution rate categories40.
Positively selected sites were identified using the following methods: (1) the Bayes Empirical Bayes (BEB) analysis (with a cutoff of 0.90), which calculates the posterior probability that each codon is from the site class of positive selection (under model M8)41; (2) the Random Effects Likelihood (REL)16, which models variation in nonsynonymous and synonymous rates across sites according to a predefined distribution, with the selection pressure at an individual site inferred using an empirical Bayes approach; (3) Fast Unbiased Bayesian AppRoximation (FUBAR)42, an approximate hierarchical Bayesian method that generates an unconstrained distribution of selection parameters to estimate the posterior probability of positive diversifying selection at each site in a given alignment (with a cutoff ≥0.90).
To be conservative, we considered a site under positive selection if it was detected by at least two methods.
To investigate whether episodic positive selection acted on the primate phylogeny, we applied the branch-site unrestricted statistical test for episodic diversification (BUSTED)16, 20. BUSTED is designed to detect the action of episodic positive selection that is acting on a subset of branches in the phylogeny at a proportion of sites within the alignment.
GARD, BUSTED, REL, FUBAR and SLAC, as well as evolutionary fingerprinting21 analyses were performed either through the DataMonkey server43 (http://www.datamonkey.org) or run locally (through the HyPhy suite)44.
Population genetics-phylogenetics analysis
For the population genetics-phylogenetics analysis, genotype data from the Phase 1 of the 1000 Genomes Project were retrieved from the dedicated website (http://www.1000genomes.org/)45; in particular, SNP information were retrieved for individuals of three human populations: African (Yoruba), European, and East Asian (Chinese). Ancestral sequences were reconstructed by parsimony from the human, chimpanzee, orangutan, and macaque sequences.
For the chimpanzee and gorilla analyses, we used SNP information from 25 and 27 individuals, respectively46.
Analyses were performed with gammaMap22, that uses intra-specific variation and inter-specific diversity to estimate the distribution of population-scaled selection coefficients (γ) along coding regions. gammaMap classifies γ values into 12 categories, ranging from strongly beneficial (γ = 100) to inviable (γ = −500), with γ equal to 0 indicating neutrality. In the analysis, we assumed θ (neutral mutation rate per site), k (transitions/transversions ratio), and T (branch length) to vary among genes following log-normal distributions. For p (the probability that adjacent codons share the same population-scaled selection coefficient) we assumed a uniform distribution. We set the neutral frequencies of non-STOP codons to 1/61. For population-scaled selection coefficients we considered a uniform Dirichlet distribution with the same prior weight for each selection class. For each gene, two Markov Chain Monte Carlo runs of 100,000 iterations each were run with a thinning interval of 10 iterations. Runs were compared to assess convergence and merged to obtain posterior probabilities. To be conservative, we declared a codon to be targeted by positive selection when the cumulative posterior probability of γ ≥ 1 was >0.80.
VNTR analysis
A VNTR (Variable Number Tandem Repeat) with four or five copies has been described in the exon 4 of human REST gene12. In order to verify if the VNTR is polymorphic in chimpanzee, the genomic DNA of twelve Pan troglodytes was used as a template for REST exon 4 amplification and sequencing. We also analyzed VNTR status in one Gorilla gorilla. In particular, the DNAs, kindly provided by the Gene Bank of Primates (Primate Genetics, Germany), were amplified by PCR (primer Forward:CTGCTCAGATGGACCCTCCT; primer Reverse:GTGCCCTTTCACTCTGCATGT), and then treated with ExoSAP-IT (USB Corporation Cleveland Ohio, USA). Purified PCR products were directly sequenced on both strands with a Big Dye Terminator sequencing Kit (v3.1, Thermo Fisher Scientific), and run on an Applied Biosystems ABI 3130 XL Genetic Analyzer (Thermo Fisher Scientific).
REST expression in primates
REST mRNA expression levels in the prefrontal and cerebellar cortices were retrieved from a large-scale study that analyzed humans, macaques, and chimpanzees23.
To evaluate whether REST expression in the three primate species is different during lifespan, we applied a resampling approach. Specifically, for each species, we performed 1000 random reshuffles of expression values; this corresponds to a null hypothesis of constant REST expression during lifespan. For each reshuffle, a lowess fitting47 was calculated and confidence intervals were retrieved form the 2.5% and 97.5% percentiles of the resulting distributions.
Patients, genotyping, MRI data acquisition, and statistical analysis
A total of 99 Italian subjects with AD continuum were consecutively recruited at the Neurology Department of IRCCS Don C. Gnocchi Foundation in Milano, Italy.
The study was in accordance with the principles of the 1975 Declaration of Helsinki and was approved by the Institutional Review Boards at the Don C. Gnocchi Foundation ONLUS.
All patients gave written informed consent.
Subjects were clinically classified and distinguished according to stage24 in: 60 “clinical AD” (23 males, 37 females) and 39 “preclinical AD” (19 males, 20 females). The mean age of “clinical AD” patients was 76.41 years (5.53 standard deviation; age range 63–89 years) and of “preclinical AD” subjects was 74.10 (6.06 standard deviation; age range 62–85). All subjects underwent complete medical and neurological evaluation, laboratory analysis, MRI scan, and other investigations – when necessary (e.g. EEG, SPET scan, CSF examination, etc.) – to exclude reversible causes of dementia.
In all subjects, we analysed two variants in REST: rs3796529, previously associated with hippocampal volume loss10, 11, and rs2227902. This latter is in linkage disequilibrium with VNTR in exon 4 in the European ancestry population and has been associated with general cognitive ability12.
The two REST variants were genotyped by allelic discrimination real-time PCR, using predesigned TaqMan probe assays (ThermoFisher Scientific). Reactions were performed using TaqMan Genotyping Master Mix in an ABI 9700 analyzer (Applied Biosystems). Genotyping rate was >0.97 for both variants.
All subjects underwent also MRI examination (1.5 T Siemens AVANTO). Hippocampal volume data have been extracted for each subject from high-resolution T1 3D images (MPRAGE; TR/TE = 1900/3.37 ms, FoV = 192 mm × 256 mm, in-plane resolution 1 mm × 1 mm, slice thickness = 1 mm, number of axial slices = 176) using FSL-FIRST segmentation method48. Intracranial brain volume data were obtained using SIENAX49, part of FSL50. In particular, SIENAX was used to estimate brain tissue volume normalised for subject skull size.
Genetic association with hippocampal volumes was performed separately for right and left hippocampal volumes. In particular, we applied linear regression analysis using genotypes or haplotypes as the independent predictor variables with age (at MRI examination), sex, APOE genotype, and volumetric scaling factor calculated by SIENAX as covariates. False Discovery Rate (FDR) correction was applied to account for the two tested variants. All analyses were performed using PLINK51.
References
Chong, J. A. et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80, 949–957, doi:0092-8674(95)90298-8 (1995).
Schoenherr, C. J., Paquette, A. J. & Anderson, D. J. Identification of potential target genes for the neuron-restrictive silencer factor. Proc. Natl. Acad. Sci. USA 93, 9881–9886 (1996).
Ballas, N. et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron 31, 353–365, doi:S0896-6273(01)00371-3 (2001).
Andres, M. E. et al. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 96, 9873–9878 (1999).
Ballas, N., Grunseich, C., Lu, D. D., Speh, J. C. & Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657, doi:S0092-8674(05)00285-0 (2005).
Gao, Z. et al. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J. Neurosci. 31, 9772–9786, doi:10.1523/JNEUROSCI.1604-11.2011 (2011).
Rodenas-Ruano, A., Chavez, A. E., Cossio, M. J., Castillo, P. E. & Zukin, R. S. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci. 15, 1382–1390, doi:10.1038/nn.3214 (2012).
Lu, T. et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454, doi:10.1038/nature13163 (2014).
Baldelli, P. & Meldolesi, J. The Transcription Repressor REST in Adult Neurons: Physiology, Pathology, and Diseases(1,2,3). eNeuro 2, eCollection 2015 Jul-Aug, doi:10.1523/ENEURO.0010-15.2015 (2015).
Nho, K. et al. Protective variant for hippocampal atrophy identified by whole exome sequencing. Ann. Neurol. 77, 547–552, doi:10.1002/ana.24349 (2015).
Nho, K. & Saykin, A. J. Reply: To PMID 25559091. Ann. Neurol. 78, 499–500, doi:10.1002/ana.24417 (2015).
Miyajima, F. et al. Additive effect of BDNF and REST polymorphisms is associated with improved general cognitive ability. Genes Brain Behav. 7, 714–719, doi:10.1111/j.1601-183X.2008.00409.x (2008).
Johnson, R. et al. Evolution of the vertebrate gene regulatory network controlled by the transcriptional repressor REST. Mol. Biol. Evol. 26, 1491–1507, doi:10.1093/molbev/msp058 (2009).
Rockowitz, S. & Zheng, D. Significant expansion of the REST/NRSF cistrome in human versus mouse embryonic stem cells: potential implications for neural development. Nucleic Acids Res. 43, 5730–5743, doi:10.1093/nar/gkv514 (2015).
Ayub, Q. et al. FOXP2 targets show evidence of positive selection in European populations. Am. J. Hum. Genet. 92, 696–706, doi:10.1016/j.ajhg.2013.03.019 (2013).
Kosakovsky Pond, S. L. & Frost, S. D. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222msi105 (2005).
Sironi, M., Cagliani, R., Forni, D. & Clerici, M. Evolutionary insights into host-pathogen interactions from mammalian sequence data. Nat. Rev. Genet. 16, 224–236, doi:10.1038/nrg3905 (2015).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–159 (2007). 110.1093/molbev/msm088.
Murrell, B. et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol.msv035 (2015).
Pond, S. L., Scheffler, K., Gravenor, M. B., Poon, A. F. & Frost, S. D. Evolutionary fingerprinting of genes. Mol. Biol. Evol. 27, 520–536, doi:10.1093/molbev/msp260 (2010).
Wilson, D. J., Hernandez, R. D., Andolfatto, P. & Przeworski, M. A population genetics-phylogenetics approach to inferring natural selection in coding sequences. PLoS Genet. 7, e1002395, doi:10.1371/journal.pgen.1002395 (2011).
Somel, M. et al. MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates. PLoS Biol. 9, e1001214, doi:10.1371/journal.pbio.1001214 (2011).
Dubois, B. et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 12, 292–323, doi:10.1016/j.jalz.2016.02.002 (2016).
Rockowitz, S. et al. Comparison of REST cistromes across human cell types reveals common and context-specific functions. PLoS Comput. Biol. 10, e1003671, doi:10.1371/journal.pcbi.1003671 (2014).
Greenway, D. J., Street, M., Jeffries, A. & Buckley, N. J. RE1 Silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells 25, 354–363, doi:2006-0207 (2007).
Lee, N. et al. Interactomic analysis of REST/NRSF and implications of its functional links with the transcription suppressor TRIM28 during neuronal differentiation. Sci. Rep. 6, 39049, doi:10.1038/srep39049 (2016).
Enard, W. et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418, 869–872, doi:10.1038/nature01025 (2002).
Mozzi, A. et al. The evolutionary history of genes involved in spoken and written language: beyond FOXP2. Sci. Rep. 6, 22157, doi:10.1038/srep22157 (2016).
Konopka, G. et al. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature 462, 213–217, doi:10.1038/nature08549 (2009).
Cheong, A. et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol. Cell 20, 45–52, doi:S1097-2765(05)01594-7 (2005).
Kuwahara, K. Role of NRSF/REST in the regulation of cardiac gene expression and function. Circ. J. 77, 2682–2686, doi:DN/JST.JSTAGE/circj/CJ-13-1210 (2013).
Negrini, S., Prada, I., D’Alessandro, R. & Meldolesi, J. REST: an oncogene or a tumor suppressor? Trends Cell Biol. 23, 289–295, doi:10.1016/j.tcb.2013.01.006 (2013).
Zhou, G., Du, T. & Roizman, B. The role of the CoREST/REST repressor complex in herpes simplex virus 1 productive infection and in latency. Viruses 5, 1208–1218, doi:10.3390/v5051208 (2013).
McGeoch, D. J., Rixon, F. J. & Davison, A. J. Topics in herpesvirus genomics and evolution. Virus Res. 117, 90–104, doi:S0168-1702(06)00021-9 (2006).
Jiang, Q. & Liu, G. REST rs3796529 variant does not influence human subcortical brain structures. Ann. Neurol. 79, 334–335, doi:10.1002/ana.24590 (2016).
Wernersson, R. & Pedersen, A. G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 31, 3537–3539 (2003).
Anisimova, M., Nielsen, R. & Yang, Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164, 1229–1236 (2003).
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H. & Frost, S. D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901, doi:10.1093/molbev/msl051 (2006).
Guindon, S., Delsuc, F., Dufayard, J. F. & Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 537, 113–137 doi:10.1007/978-1-59745-251-9_6 (2009).
Anisimova, M., Bielawski, J. P. & Yang, Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 19, 950–958 (2002).
Murrell, B. et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205, doi:10.1093/molbev/mst030 (2013).
Delport, W., Poon, A. F., Frost, S. D. & Kosakovsky Pond, S. L. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457, doi:10.1093/bioinformatics/btq429 (2010).
Pond, S. L., Frost, S. D. & Muse, S. V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679, doi:bti079 (2005).
1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature 467, doi:1061-107310.1038/nature09534 (2010).
Prado-Martinez, J. et al. Great ape genetic diversity and population history. Nature 499, 471–475, doi:10.1038/nature12228 (2013).
Cleveland, W. S. LOWESS: A program for smoothing scatterplots by robust locally weighted regression. The American Statistician 35, 54 (1981).
Patenaude, B., Smith, S. M., Kennedy, D. N. & Jenkinson, M. A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56, 907–922, doi:10.1016/j.neuroimage.2011.02.046 (2011).
Smith, S. M. et al. Accurate, robust, and automated longitudinal and cross-sectional brain change analysis. Neuroimage 17, 479–489 (2002).
Smith, S. M. et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23(Suppl 1), S208–219, doi:10.1016/j.neuroimage.2004.07.051 (2004).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575, doi:10.1086/519795 (2007).
Author information
Authors and Affiliations
Contributions
M.S., R.C., and F.R.G. conceived the study; R.C., A.M., and D.F. performed the evolutionary analysis in primates and the lineage-specific selection analyses; D.F. performed analysis of REST brain expression in primates; R.N. recruited patients; F.B. and MCa performed MRI. data acquisition and analyses; A.S.C. and F.R.G. performed samples collection and APOE genetic analyses; C.P. and S.R. performed REST variant genotyping analysis, R.C., A.M., M.S. and F.R.G. analysed the data, with input from M.C., R.C., M.S., and D.F. produced the figures. R.C. and M.S. wrote the manuscript, with critical input from M.C. and from the remaining authors. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mozzi, A., Guerini, F.R., Forni, D. et al. REST, a master regulator of neurogenesis, evolved under strong positive selection in humans and in non human primates. Sci Rep 7, 9530 (2017). https://doi.org/10.1038/s41598-017-10245-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10245-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
