
Abstract
A series of micro-hydrogel particles consisting of hyperbranched polyamidoamine (HPAMAM) without any supporting core materials was synthesized via the inverse suspension condensation polymerization of A2 and B4 monomers, N,N′-methylenebisacrylamide (MBA) and ethylenediamine (EDA). The particles were found to be highly effective when used to remove heavy metal ions, such as cadmium, copper, lead, nickel, zinc, and cobalt, from water, and they could be separated from the water by a simple filtration process. The results of this study demonstrate that crosslinked HPAMAM particles, which can be prepared by a simple and environmentally friendly process, are an attractive absorbent for water purification.
Similar content being viewed by others

Introduction
Water pollution by toxic heavy metal ions is a serious global nuisance owing to the harmful effects of heavy metals on the environment and human health1, 2. In order to address this issue, various methods for the removal of heavy metal ions in aqueous solutions, including precipitation, ion exchange, adsorption, membrane filtration, coagulation, and electrochemical treatments, have been developed3,4,5,6,7,8,9,10,11,12,13,14,15,16. Among these methods, adsorption techniques using adsorbent materials capable of capturing pollutants are easy to apply at a reasonable cost3,4,5,6,7,8,9,10,11. Specifically, chelating resins are an important category of adsorbent materials3,4,5,6,7,8,9. The majority of chelating resins consist of polystyrene beads, but a toxic chloromethylation reaction is necessary to make chelating resins from polystyrene17, 18. Accordingly, research related to the production of eco-friendly chelating resins has received much attention19, 20.
Polyamidoamine (PAMAM) dendrimers and hyperbranched polymers have attracted considerable interest in recent years. The highly branched structures together with a large number of functional groups in the main chains and end groups make these materials ideal for applications in host-guest encapsulation, nanoreactors, and delivery devices21,22,23,24,25,26,27,28,29,30. Amine and amide functional groups of PAMAM dendrimers can serve as nanoscale high-capacity containers for heavy metal ions such as Cu2+, Cd2+, and others24,25,26,27,28,29,30. However, PAMAM dendrimers require tedious multi-step synthesis to obtain a high molecular weight. Therefore, the analogous, hyperbranched polyamidoamine (HPAMAM), has been utilized in various categories in place of PAMAM dendrimers31,32,33. Drawbacks of PAMAM dendrimers and HPAMAM include their small size, the hydrodynamic diameter of the dendrimers, and the radius of gyration of the hyperbranched polymers, which is typically less than 10 nanometers21, 25, 28. Thus, additional processes such as ultrafiltration are necessary for separation from an aqueous solution24,25,26,27,28.
Generally, hyperbranched polymers can be synthesized via the one-step polymerization of an ABx (X ≥ 2) monomer34,35,36. However, given that ABx monomers includes different (A and B) functional groups with two types of reactivity, multiple synthesis routes must be carried out to form a monomer in the ABx form. To overcome this problem, a method involving the polycondensation of a mixture of multifunctional monomers with a single type of reactive group with the slow feeding of one monomer into the other has been developed36,37,38. Although synthetic methods using a mixture of multifunctional monomers opens a way of preparing hyperbranched polymers from readily available multifunctional compounds, the polymerization of multifunctional monomer mixtures such as A2 and B4 monomers (A2 + B4 polycondensation) produces crosslinked polymers, even with the slow feeding of one monomer into the other, when the conversion reaches a gelation point (the critical extent of reaction).
In this study, we used both the advantages and limitations of the A2 + B4 polycondensation method for hyperbranched polymers to make micro-sized HPAMAM hydrogel particles through the dispersal of an aqueous HPAMAM solution obtained by A2 + B4 polycondensation into an immiscible liquid and then by carrying out the polycondensation until reaching the point of critical gelation39,40,41,42,43,44,45. This simple method allowed us to make micro-sized hydrogel particles which wholly consisted of hyperbranched polyamidoamine (HPAMAM) without any additional crosslinking reagents. The crosslinked HPAMAM hydrogel particles were found to be highly effective when used to remove heavy metal ions from an aqueous solution and could easily be separated from water by a simple filtration process. We also demonstrated that the properties of HPAMAM particles, in this case the particle size, gel fraction, swelling ratio, and absorption capacity for metal ions, could be tuned by controlling the monomer feed composition, the amount of stabilizer used, and the agitation speed.
Results
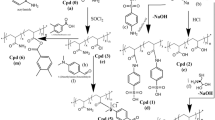
Michael addition reactions of commercially available A2 (N,N′-methylenebisacrylamide, MBA) and B4 (ethylenediamine, EDA) monomers were carried out in deionized (DI) water to investigate whether the synthesis of hyperbranched polyamidoamines is feasible, as shown in Fig. 1. Tentatively, a reaction of equal mol amounts of MBA and EDA (molar ratio of the reactive sites of MBA to EDA is 0.5) was attempted to consume all of the vinyl groups while preserving the primary amine groups as an end group. A water-insoluble polymer gel (1) was produced after 30 min of the simple mixing of the two monomers. Generally, the polymerization of a multifunctional monomer such as A2 and B4 produces crosslinked polymers, and the AB2 type of monomer is necessary to make a hyperbranched polymer. However, if one monomer (A2) is added slowly to the reaction mixture, the remaining reactive groups become only B, which prevents the formation of a gel-inducing species containing several A and B groups. Slow feeding of the A2 monomer (with a deficient concentration of vinyl groups in an aqueous solution) achieved by the relatively low solubility of MBA in water allows a preferential reaction of the vinyl group with readily available primary amines of EDA to form covalent bonds of the secondary amine. The formation of crosslinking points (i.e., tertiary amines) is suppressed in the early stage of polymerization mainly due to the low concentration of secondary amines as compared to that of the primary amines. However, the mixture of multifunctional monomers (A2 + B4) becomes a gel after a critical gelation point. The time-dependent conversion of the MBA monomer was monitored by assessing the 1H NMR spectra in the middle of the polymerization using D2O as a solvent to follow the reaction (Fig. 2).

Synthesis of crosslinked hyperbranched polyamidoamine particles (a) in an aqueous solution, and (b) via inverse suspension polymerization.

(a)1H NMR spectra of HPAMAM obtained at various polymerization times: (a) 0 min, (b) 5 min, (c) 10 min, and (d) 40 min (400 MHz, D2O), (b) polymerization time-dependent conversion of MBA monomer (inset: equation for the calculation of the conversion of MBA).
The intensity of the peak around 6 ppm (I 6.02–6.13) corresponding to the integral values of the protons (A) attached to the vinyl groups (6.02–6.13 ppm) decreased gradually compared to the intensity of the peak around 4.6 ppm (I 4.58–4.68) corresponding to the integral values of the protons (B) of methylene (-CONHCH 2 CONH-, 4.5–4.7 ppm). The conversion reached 80% after 5 min of polymerization, 88% after 10 min and >99% after 40 min. It appears that the addition reaction occurred very rapidly in the initial period of polymerization and that the retro-Michael addition was negligible.
The critical gelation points predicted by Flory’s and Carothers’ equation for equal amounts of bifunctional monomer MBA and tetrafunctional monomer EDA are 0.82 and 1.00, respectively. Therefore, the gel formed above 82% but below 100% conversion, and gel formation was observed after 10 min at RT.
Based on the NMR analysis results, the preparation of micro-hydrogel particles which consisted of hyperbranched polyamidoamines was attempted via inverse suspension polymerization. The polymerization mixture in an aqueous solution was suspended within the organic phase which contains a stabilizer but without any initiators or cross-linking agents. Further conversion of the polymerization mixture (a homogeneous precursor solution) is expected to induce a gel through crosslinking when it reaches a critical gelation point. Samples of HPAMAMs are designated as P-M x S y R z , where x, y, and z denote the molar feed ratio of MBA to EDA, the weight concentration of Span 60, and the rpm speed of agitation, respectively. To investigate the effects of experimental parameters on the morphology, absorption properties, and other characteristics, the crosslinked HPAMAM particles were prepared under various reaction conditions (Table S1, ESI).
The representative micro-hydrogel particle P-M 8/8 S 0.5 R 1k was characterized by FT-IR and thermogravimetric analysis (TGA). The FT-IR spectra showed peaks of –NH- stretching (3000~3350 cm−1), -CO- stretching (1649 cm−1), and -CONH- stretching (1537 cm−1), respectively. The C=CH 2 stretching peak (3080 cm−1) of MBA nearly disappeared (Fig. S1, ESI). The 5% weight loss temperature of P-M 8/8 S 0.5 R 1k was 240 °C (Fig. S2, ESI).
Discussion
The cross-linking density of the particles is closely related to their properties as absorbent media for water treatment applications, as this parameter affects the swelling ratio, heavy metal absorption capabilities, and degree of solvent resistance. The cross-linking density of HPAMAM particles is easily controlled by changing the initial feed ratio of the two monomers. For practical applications, the stability of particles in an aqueous solution is also important to minimize the loss of the capacity to absorb metal ions. Dried HPAMAM particles were placed in distilled water and stirred for at least two days to dissolve any unreacted monomers and oligomers, after which the HPAMAM particles were filtered and dried under a vacuum. The optimum feed ratio of the monomers was determined by measuring the gel fraction (Table S1 and Fig. S3, ESI). The soluble fraction of particles decreased to saturated values (less than 3 wt% of the particles) when the molar feed ratio of MBA to EDA approached 1.5. It seems that the cross-linking density of the HPAMAM particles obtained at a molar feed ratio of 1.5 is sufficiently stable in water to be recycled. From this study, investigations of the effect of a stabilizer and the speed of agitation were carried out with HPAMAM particles prepared with a molar feed ratio of 1.5 (MBA to EDA).
The morphology of the synthesized micro-hydrogel particles was investigated with an optical microscope and a scanning electron microscope (SEM), as shown in Fig. 3 and Figs S4–7 (ESI). Interestingly, the diameter of spherical particles did not change regardless of the feed ratio of the monomers (50~250 μm), indicating that the particle size was not affected by the cross-linking density. However, increasing the weight concentration of the stabilizer or the speed of agitation decreased the diameter, as expected.

Optical images of (a) P-M8/8S0.5R1k, (b) P-M12/8S0.5R1k, (c) P-M12/8S5.0R1k, and (d) P-M12/8S0.5R1.5k.
The swelling ratio of the particles (g/g) was obtained by measuring the weight gain of the particles after soaking them in distilled water. The Cu2+ absorption capacity (g/g) was measured by analyzing the change in the concentration of copper in the CuCl2 solution (10,000 ppm) with an inductively coupled plasma optical emission spectrometer (ICP-OES). As expected, the swelling ratio of the particles decreased with an increase in the amount of MBA because more cross-linked HPAMAM particles were formed as the stoichiometric ratio of the two functional groups (amine and vinyl groups) became two (one amine group can react with two acrylic groups; Fig. S8a, ESI). The Cu2+ absorption capacity also decreased with an increase in the amount of MBA up to the stoichiometric point, presumably due to the smaller number of free amine groups in the particles (Fig. S8b, ESI).
Interestingly, the particle sizes that varied with the amount of stabilizer and the speed of agitation did not affect the swelling ratio or the Cu2+ absorption capacity of the particles (Fig. 4 and Fig. S9, ESI), presumably because the binding of metal ions occurred not only on the surface but also in the inside of the particles. An energy dispersive X-ray spectroscopy (EDX) analysis of the cutting plane of the particles clearly showed the presence of Cu atoms inside (Fig. 5). Greater absorption of metal ions by the particles was realized through the suction of metal ions into the inside of particles fully consisting of HPAMAM.

Effect of (a) the amount of stabilizer and (b) the agitation speed on the Cu2+ absorption capacity and particle size.

EDX and XPS analyses of HPAMAM particles of P-M 12/8 S 0.5 R 1k after Cu2+ absorption: (a) collected area on the bare surface, (b) collected area on the cutting plane, (c) element analysis data, and (d) XPS spectra of the N1s region of P-M 12/8 S 0.5 R 1k before (black line) and after (red line) Cu2+ absorption.
X-ray photoelectron spectroscopy (XPS) results showed decreased binding energy of N1s upon Cu2+ absorption by the particles, as shown in Fig. 5(d). As described in the literature, chelation between the electron-rich amine and amide groups with electron-deficient metal atoms is the key principle of absorption46, 47.
The absorption capacity of other heavy metal cations, including Cd2+, Cu2+, Pb2+, Ni2+, Zn2+, and Co2+, were studied with P-M 8/8 S 0.5 R 1k . The HPAMAM particles showed excellent metal absorption capacities (g/g) of more than 0.13 with all of the heavy metals tested in this study. An especially outstanding result was noted with Cd2+ (0.27 g/g, Fig. 6). HPAMAM particles have excellent Cu2+ and Cd2+ absorption capacities compared to those of other different types of absorbents reported recently (Table 1 and Table S2, ESI). The PAMAM dendrimer has better Cu2+ absorption capacity, but these dendrimers are difficult to apply owing to the costly synthesis and the additional separation process.

(a) Absorption capacities of various heavy metal ions and (b) a recycle test of P-M 8/8 S 0.5 R 1k .
The reusability of particles for heavy metal absorption was investigated with P-M 8/8 S 0.5 R 1k (Fig. 6(b)). The desorption of Cu2+ was easily achieved through a treatment with a 0.1 N HCl solution. At a low pH, the chelation between metal ions and nitrogen atoms is weakened because competitive H+ ions occupy the amine groups, whereas the protonated amine groups are deprotonated with an increase in the pH value. During the first recycle step, the Cu2+ absorption capacity declined to around 0.02 degrees, presumably due to the undetached copper ions which arose during the desorption process. Nevertheless, recycled HPAMAM particles showed excellent Cu2+ absorption capacities which exceeded 0.15 with up to five times of recycling.
In order to investigate the possibility of mass production, a scale up test was attempted. When the experimental scale was increased to 10 L of the organic phase, spherical polymer particles 50~250 μm in size were obtained steadily with the expected yield. All of the PAMAM particles showed a consistent swelling ratio and Cu2+ an absorption capacity which exceeded 0.15 (Table S3 and Fig. S10, ESI).
Copper ion removal by the micro-hydrogel particles packed within a glass column (a P-M 12/8 S 0.5 R 0.3k −500 particle-packed column (Fig. 7)) was examined with a continuous flow of a Cu2+ solution. With the flow of the Cu2+ solution, PAMAM particles absorbed Cu2+ ions in the aqueous solution, and their color became blue starting from the lower part of the column. Even under a high flow rate (5.0 mL/min), the Cu2+ ions in the aqueous solution were completely removed up to the limit of detection.

Demonstration of a metal ion absorption test using a column packed with P-M 12/8 S 0.5 R 0.3k -500 particles (inset: in and out concentrations of Cu2+ in water with various flow rates).
In summary, micro-hydrogel particles consisting wholly of HPAMAM were readily synthesized by an eco-friendly polyaddition reaction of A2 and B4 types of monomers via inverse suspension polymerization. The particles were found to be highly efficient during the removal of heavy metal ions, in this case cadmium, copper, lead, nickel, zinc, and cobalt, from water. Cu2+ absorption capacity was found to be as high as 0.17 g/g, which is three times that of other polystyrene-based chelating polymer adsorbents. This superb absorption capacity stems from the good hydrophilic properties and high density levels of the amine and amide functional groups without any supporting materials. In addition, easy recycling through control of the pH, a successful scale-up operation, as well as the complete removal of metal ions from water through a column packed with the HPAMAM particles demonstrate that the crosslinked HPAMAM particles, obtained by an eco-friendly process, can serve as a cost-effective and efficient absorbent for water purification applications.
Methods
Polymerization of HPAMAM
EDA (0.5259 g; 8.75 mmol), MBA (1.3491 g; 8.75 mmol), and water (3.75 mL; 50 wt/v% of the monomers) were placed in a round bottomed-flask (RBF) and stirred for 6 h at 60 °C under nitrogen atmosphere. The reaction mixture was cooled to room temperature, and precipitated into acetone. The supernatant was drained out of the vial and the remaining sticky polymer was dried under vacuum for 24 h at 60 °C. Yield = 90%.
Polymerization Kinetics of HPAMAM
The polymerization kinetics of HPAMAM was studied by monitoring the polymerization with1H NMR. 1H NMR spectroscopy a were recorded on a Bruker Fourier Transform Avance 400 (400 MHz) spectrometer. A typical procedure is as follows. EDA(0.8415 g; 14.00 mmol), MBA (2.1585 g; 14.00 mmol) and deuterium oxide (10 mL; 30 wt% of the monomers) were placed in a RBF and stirred for 6 h at 60 °C1.H NMR spectra were recorded at 0, 5, 10, 20, 30, 40, 50, 60 min.
Synthesis of HPAMAM Particles by Inverse Suspension Polymerization
P-M 8/8 S 0.5 R 1k The inverse suspension polymerization reactions were carried out as follows. The suspension stabilizer Span® 60 (0.0094 g; 0.5 wt% of the monomers) was dissolved in toluene (15 mL; 4 times of water) and degassed for 30 min under vigorous agitation. EDA (0.5259 g; 8.75 mmol), MBA (1.3491 g; 8.75 mmol), and water (3.75 mL; 50 wt/v% of the monomers) were placed and stirred in a RBF until complete dissolution of the solid. The aqueous solution was introduced into the toluene solution for inverse suspension polymerization at 45 °C under nitrogen with the stirring speed of 1,000 rpm. After 6 hr of polymerization, the polymerization mixture was poured into methanol, filtered and washed several times alternatively with methanol and acetone, and then dried under vacuum for 24 hr at 60 °C.
P-M 9/8 S 0.5 R 1k to P-M 16/8 S 0.5 R 1k P-M9/8S0.5R1k – P-M16/8S0.5R1k were prepared according to the method for P-M8/8S0.5R1k except the feed composition of MBA and EDA. The molar ratios of the monomers (MBA/EDA) were 1.125, 1.25, 1.375, 1.5, 1.625, 1.75, 1.875, and 2.0, to produce P-M9/8S0.5R1k, P-M10/8S0.5R1k, P-M11/8S0.5R1k, P-M12/8S0.5R1k, P-M13/8S0.5R1k, P-M14/8S0.5R1k, P-M15/8S0.5R1k, and P-M16/8S0.5R1k respectively.
Scale Up Test
P-M 12/8 S 0.5 R 1k -30, 100, 150, 300, P-M 12/8 S 0.5 R 0.3k -500, 750, 2000, 10000 Toluene – 30–10000 mL scale were prepared according to the method for P-M8/8S0.5R1k, except the amount of reactant. The amount of each reactant was a fixed ratio. P-M12/8S0.5R1k-30, 100, 150, 300 were stirred by stirring bar at 1,000 rpm. P-M12/8S0.5R0.3k-500, 750, 2000, 10000 were stirred by mechanical stirrer at 300 rpm.
References
Denkhaus, E. & Salnikow, K. Nickel essentiality, toxicity, and carcinogenicity. Crit. Rev. Oncol. Hematol. 42, 35–56 (2002).
Patra, M., Bhowmik, N., Bandopadhyay, B. & Sharma, A. Comparison of mercury, lead and arsenic with respect to genotoxic effects on plant systems and the development of genetic tolerance. Environ. Exp. Bot. 52, 199–223 (2004).
Mohan, D. & Pittman, C. U. Activated carbons and low cost adsorbents for remediation of tri- and hexavalent chromium from water. J. Hazard. Mater. 137, 762–811 (2006).
Kurniawan, T. A., Chan, G. Y. S., Lo, W. H. & Babel, S. Physico-chemical treatment techniques for wastewater laden with heavy metals. Chem. Eng. J. 118, 83–98 (2006).
Dabrowski, A., Hubicki, Z., Podkocielny, P. & Robens, E. Selective removal of the heavy metal ions from waters and industrial wastewaters by ion-exchange method. Chemosphere 56, 91–106 (2004).
Mao, N., Yang, L., Zhao, G., Li, X. & Li, Y. Adsorption performance and mechanism of Cr(VI) using magnetic PS-EDTA resin from micro-polluted waters. Chem. Eng. J. 200–202, 480–490 (2012).
Kumar, P. A., Chakraborty, S. & Ray, M. Removal and recovery of chromium from wastewater using short chain polyaniline synthesized on jute fiber. Chem. Eng. J. 141, 130–140 (2008).
Tan, M. X., Sum, Y. N., Ying, J. Y. & Zhang, Y. G. A mesoporous poly-melamine-formaldehyde polymer as a solid sorbent for toxic metal removal. Energy Environ. Sci. 6, 3254–3259 (2013).
Çavuş, S. & Gürdaǧ, G. Noncompetitive Removal of Heavy Metal Ions from Aqueous Solutions by Poly[2-(acrylamido)−2-methyl-1-propanesulfonic acid- co -itaconic acid] Hydrogel. Ind. Eng. Chem. Res. 48, 2652–2658 (2009).
Fu, F. & Wang, Q. Removal of heavy metal ions from wastewaters: A review. J. Environ. Manage. 92, 407–418 (2011).
Babel, S. & Kurniawan, T. A. Low-cost adsorbents for heavy metals uptake from contaminated water: a review. J. Hazard. Mater. 97, 219–243 (2003).
Kozlowski, C. A. & Walkowiak, W. Removal of chromium(VI) from aqueous solutions by polymer inclusion membranes. Water Res. 36, 4870–4876 (2002).
Charerntanyarak, L. Heavy metals removal by chemical coagulation and precipitation. Water Sci. Technol. 39, 135–138 (1999).
Gode, F. & Pehlivan, E. A comparative study of two chelating ion-exchange resins for the removal of chromium(III) from aqueous solution. J. Hazard. Mater. 100, 231–243 (2003).
Camel, V. Solid phase extraction of trace element. Spectrochim. Acta Part B 58, 1177 (2003).
Chen, G. Electrochemical technologies in wastewater treatment. Sep. Purif. Technol. 38, 11–41 (2004).
Yang, L. et al. Preparation and adsorption performance of a novel bipolar PS-EDTA resin in aqueous phase. J. Hazard. Mater. 180, 98–105 (2010).
Rodrigo, R., Toro, C. A. & Cuellar, J. Preparation of chloromethylated and aminated gel-type poly(styrene-co- divinylbenzene) microparticles with controlled degree of functionalization. J. Appl. Polym. Sci. 130, 4054–4065 (2013).
Singru, R. N., Gurnule, W. B., Khati, V. A., Zade, A. B. & Dontulwar, J. R. Eco-friendly application of p-cresol – melamine – formaldehyde polymer resin as an ion-exchanger and its electrical and thermal study. Desalination 263, 200–210 (2010).
Azarudeen, R. S., Ahamed, M. A. R. & Burkanudeen, A. R. Chelating terpolymer resin: Synthesis, characterization and its ion-exchange properties. Desalination 268, 90–96 (2011).
Esfand, R. & Tomalia, D. A. Poly(amidoamine) (PAMAM) dendrimers: From biomimicry to drug delivery and biomedical applications. Drug Discov. Today 6, 427–436 (2001).
Boas, U. & Heegaard, P. M. H. Dendrimers in drug research. Chem. Soc. Rev. 33, 43–63 (2004).
Piao, Y., Wu, T. & Chen, B. One-Step Synthesis of Graphene Oxide–Polyamidoamine Dendrimer Nanocomposite Hydrogels by Self-Assembly. Ind. Eng. Chem. Res. 55, 6113–6121 (2016).
Crooks, R. M., Zhao, M., Sun, L., Chechik, V. & Yeung, L. K. Dendrimer-encapsulated metal nanoparticles: Synthesis, characterization, and applications to catalysis. Acc. Chem. Res. 34, 181–190 (2001).
Diallo, M. S., Christie, S., Swaminathan, P., Johnson, J. H. & Goddard, W. A. Dendrimer enhanced ultrafiltration. 1. Recovery of Cu(II) from aqueous solutions using PAMAM dendrimers with ethylene diamine core and terminal NH2 groups. Environ. Sci. Technol. 39, 1366–1377 (2005).
Jawor, A. & Hoek, E. M. V. Removing Cadmium Ions from Water via Nanoparticle-Enhanced Ultrafiltration. Environ. Sci. Technol. 44, 2570–2576 (2010).
Rether, A. & Schuster, M. Selective separation and recovery of heavy metal ions using water-soluble N-benzoylthiourea modified PAMAM polymers. React. Funct. Polym. 57, 13–21 (2003).
Diallo, M. S. et al. Poly (amidoamine) Dendrimers: A New Class of High Capacity Chelating Agents for Cu (II) Ions. Environ. Sci. Technol. 33, 820–4 (1999).
Niu, Y. et al. Synthesis of Silica-Gel-Supported Sulfur-Capped PAMAM Dendrimers for Efficient Hg(II) Adsorption: Experimental and DFT Study. Ind. Eng. Chem. Res. 55, 3679–3688 (2016).
Bukowska, A., Bukowski, W., Bester, K. & Flaga, S. Linkage of the PAMAM type dendrimer with the gel type resin based on glycidyl methacrylate terpolymer as a method of preparation of the polymer support for the recyclable palladium catalyst for Suzuki–Miyaura cross-coupling reactions. RSC Adv. 5, 49036–49044 (2015).
Perignon, N. et al. Hyperbranched Polymers Analogous to PAMAM Dendrimers for the Formation and Stabilization of Gold Nanoparticles Nelly Pe. Macromolecules 40, 3034–3041 (2007).
Perignon, N., Mingotaud, A.-F., Marty, J.-D., Rico-Lattes, I. & Mingotaud, C. Formation and Stabilization in Water of Metal Nanoparticles by a Hyperbranched Polymer Chemically Analogous to PAMAM Dendrimers. Chem. Mater. 16, 4856–4858 (2004).
Hobson, L. J. & Feast, W. J. Poly(amidoamine) hyperbranched systems: synthesis, structure and characterization. Polymer. 40, 1279–1297 (1999).
Jikei, M. & Kakimoto, M. Hyperbranched polymers: a promising new class of materials. Prog. Polym. Sci. 26, (2001).
Voit, B. I. & Lederer, A. Hyperbranched and Highly Branched Polymer Architectures s Synthetic Strategies and Major Characterization Aspects. Chem. Rev. 5924–5973 (2009).
Gao, C. & Yan, D. Hyperbranched polymers: from synthesis to applications. Prog. Polym. Sci. 29, 183–275 (2004).
Jikei, M. et al. Synthesis of Hyperbranched Aromatic Polyamide from Aromatic Diamines and Trimesic Acid. Macromolecules 32, 2061–2064 (1999).
Emrick, T., Chang, H. & Fre, J. M. J. An A2 + B3 Approach to Hyperbranched Aliphatic Polyethers Containing Chain End Epoxy Substituents. 6380–6382 (1999).
Dimonie, M. V. et al. Inverse suspension polymerization of acrylamide. Eur. Polym. J. 18, 639–645 (1982).
Duan, L., Chen, M., Zhou, S. & Wu, L. Synthesis and characterization of poly(N-isopropylacrylamide)/silica composite microspheres via inverse pickering suspension polymerization. Langmuir 25, 3467–3472 (2009).
Jiang, W. et al. Spherical polystyrene-supported chitosan thin film of fast kinetics and high capacity for copper removal. J. Hazard. Mater. 276, 295–301 (2014).
Luciani, C. V., Choi, K. Y. & Xiao, Z. Inverse free radical suspension polymerization as a potential means to encapsulate biologically active materials. Chem. Eng. Technol. 33, 1833–1840 (2010).
Li, G., Yang, M., Ma, P. & Zhang, G. Nanoparticles change drop breakup mechanism to obtain icecream-like microparticles in inverse suspension polymerization. RSC Adv. 6, 4622–4626 (2016).
Dowding, P., Vincent, B. & Williams, E. Preparation and Swelling Properties of Poly(NIPAM) ‘Minigel’ Particles Prepared by Inverse Suspension Polymerization. J. Colloid Interface Sci. 221, 268–272 (2000).
Wang, G., Li, M. & Chen, S. Inverse Suspension Polymerization of Sodium Acrylate. Appl. Polym. Sci. 65, 789 (1997).
Ren, Z. et al. FTIR, Raman, and XPS analysis during phosphate, nitrate and Cr(VI) removal by amine cross-linking biosorbent. J. Colloid Interface Sci. 468, 313–323 (2016).
Huang, C.-C. & He, J.-C. Electrosorptive removal of copper ions from wastewater by using ordered mesoporous carbon electrodes. Chem. Eng. J. 221, 469–475 (2013).
Igberase, E., Osifo, P. & Ofomaja, A. The adsorption of copper (II) ions by polyaniline graft chitosan beads from aqueous solution: Equilibrium, kinetic and desorption studies. J. Environ. Chem. Eng. 2, 362–369 (2014).
Bai, Y. & Bartkiewicz, B. Removal of Cadmium from Wastewater Using Ion Exchange Resin Amberjet 1200H Columns. Polish J. Environ. Stud. 18, 1191–1195 (2009).
Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (NRF-2015R1A2A1A10055222) and the Disaster and Safety Management Institute funded by Ministry of Public Safety and Security of Korean Government (MPSS-CG-2015-01).
Author information
Authors and Affiliations
Contributions
S.Y.K., S.L. and Y.E. proposed the research. S.L. and Y.E. synthesized and characterized the polymer. S.L., Y.E., J.P., J.L. prepared the manuscript. S.Y.K. supervised the whole project and revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S., Eom, Y., Park, J. et al. Micro-hydrogel Particles Consisting of Hyperbranched Polyamidoamine for the Removal of Heavy Metal Ions from Water. Sci Rep 7, 10012 (2017). https://doi.org/10.1038/s41598-017-10066-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10066-x
This article is cited by
-
Development of a nitrogen-rich hyperbranched polymer as adsorbent for enrichment and determination of auxins in plants
Analytical and Bioanalytical Chemistry (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
