
Abstract
Due to its rapid spread and association with the numerous outbreaks, the global spread of East Asian lineage of Mycobacterium tuberculosis strains presents a global concern. Although there were many attempts to describe its population structure, no consensus has been reached yet. To define unbiased classification that will facilitate future studies of this lineage, we analyzed the performance and congruence of eight different genotyping schemes based on phylogenetic analysis of 1,398 strains from 32 countries using whole-genome sequencing (WGS) data. We confirm that East Asian lineage comprises two major clades, designated proto-Beijing, which harbors unusual 43-signal spoligoprofile, and Beijing, with well-known spoligoprofile (deleted signals from 1 to 34). We show that different genotyping methods give high consistency results in description of ancient Beijing strains while the classification of modern Beijing strains is significantly divergent due to star-shaped phylogeny. Using WGS data we intersect different studies and for the first time provide balanced classification with well-defined major groups and their genetic markers. Our reconstructed phylogenetic tree can also be used for further analysis of epidemiologically important clusters and their ancestors as well as white spots of unclassified strains, which are prospective areas of research.
Similar content being viewed by others

Introduction
A nearly complete absence of horizontal gene transfer makes Mycobacterium tuberculosis (Mtb) population structure strictly clonal and hierarchical. Presently, it consists of seven phylogenetic lineages and usually smaller and more geographically delimited genetic families each of which is defined by unique event polymorphisms (SNP or deletions)1. Lineage 2 (or East Asian lineage) is arguably most widespread and the Beijing genotype family is its major component (13% of global M. tuberculosis population)2.
For the first time, the Beijing genotype was described in strains from the Beijing area in China which coined the name3. The strains were characterized by very similar IS6110-RFLP patterns and peculiar spoligotyping profile of only nine hybridization signals (35 to 43). Ten years later, a more inclusive definition of the Beijing genotype was suggested4; in particular, the spoligoprofile should have an absence of hybridization of signals from 1 to 34 and a presence of at least three of the spacers from 35 to 43 (according to the standard spoligotyping scheme). The Beijing genotype can also be identified based on phylogenetic analysis of the high-resolution 24 VNTR loci within the context of known reference strains (www.MIRU-VNTRplus.org).
The Beijing family was long believed to be a homogeneous group of strains, first of all based on the similarity of their IS6110-RFLP profiles (see representative examples in ref. 5), which in turn justified efforts to find more user-friendly and no less discriminatory molecular markers for these strains. The first evolutionarily meaningful subdivision of the Beijing genotype into large-scale phylogenetic lineages of ancient/atypical and modern/typical strains was proposed by Mokrousov in ref. 6. Further, in 2005, the same authors proposed a large-scale subdivision within Beijing genotype7 based on previously described insertion of IS6110 in the NTF region8. The simplicity of analysis made the NTF-based approach widely used to discriminate between phylogenetic lineages of the Beijing genotype, although occasionally described discrepancies have cast a shadow of doubt over this marker9.
More recently, Tsolaki et al.10 and Gagneux et al.11 proposed a subdivision of Mtb into phylogenetic lineages mainly defined by large genomic deletions (regions of difference (RD)); the lineage 2 (defined by RD105) included the Beijing genotype (defined by RD207). Later, strains with RD105 but with unusual, complete 43-signal spoligoprofile12 were published and thus presented a minor part of the lineage 2 beyond the Beijing genotype proper. It should be mentioned that RD markers10, 11 did not distinguish between ancient and modern Beijing.
The advanced whole genome sequencing (WGS) technologies facilitated and accelerated global and local studies, and contributed to both gaining insight into high-resolution population structure and building the evolutionary scale framework of the Beijing genotype, in particular, and lineage 2 on a whole. Three kinds of studies should be noted: (i) based on polymorphisms in a (relatively) limited set of genes of the 3 R (i.e. Replication, Repair, Recombination) system13, 14; (ii) based on gene polymorphisms derived from comparison of the complete genomes15; and (iii) based on WGS data analysis16,17,18. Three mutator genes of the 3 R system (mutT2, mutT4 and ogt) were used by Rad et al. to subdivide Beijing genotype into five groups and mutT2 mutation in the modern Beijing group was supposedly associated with mutator phenotype13. The 3R-based approach was pursued on the expanded set of 56 such genes by Mestre et al.14 who subdivided Beijing strains into 26 groups; this was useful to follow the evolutionary pathway of this lineage but challenging in its interpretation since many subtypes included only single isolates. In its turn, SNP-based classification of Filliol et al.15 distinguished 11 types but some variable positions are presently regarded as uncertain and unsuitable9. Furthermore, the situation is complicated due to the fact that variable positions were given according to the earlier and what is worse, non-supervised version of the genome of reference strain H37Rv. Finally, typing schemes derived from WGS data suggest existence of 516, 417, and 818 groups within lineage 2; here, a terminological discrepancy may be noted. Coll et al.16 species-wide study was based on WGS data and their subdivision was linked to the framework of the previously proposed phylogenetic lineages10, 11. On the other hand, Luo et al.17 focused on the East Asian lineage (lineage 2) and developed their classification and terminology based on independent analysis of the WGS data. A similar study was carried out by Merker et al.18 but they focused on the evolution of the Beijing genotype only and identified 3 and 5 subgroups within ancient and modern strains, respectively.
The major practical problem of such studies is that newly developed schemes partly or completely disregard previous knowledge and existing and established schemes. Consequently, a lack of any or adequate correlation between the old and the new studies makes it virtually impossible to assess the discriminatory capacity and evolutionary robustness of the particular typing methods. This latter issue is very important regarding public health. Phenotypic variation of strains from different genetic groups is a well-known and well-recognized fact and correct interpretation of typing results is a prerequisite for studies evaluating pathobiologically relevant properties such as drug resistance, virulence, transmissibility, mutator capacity etc. Additionally, there are open questions on specific polymorphisms involved in the formation of a subpopulation of the pathogen as well as validation and generalization of the existing techniques developed on the geographically delimited and partly biased datasets in new world regions.
In this study, we investigated M. tuberculosis lineage 2 classification methods and their performance and congruence using whole genome sequencing data of 1,398 strains. We correlated newly discovered and “old” molecular markers and superposed new schemes onto already long-used phylogenetic framework of the lineage 2 (~Beijing genotype). In practical terms, we aimed to facilitate communication between different research groups. We sought to clarify the evolutionary pathway of this epidemiologically and clinically significant lineage, and in particular, to highlight its known and as yet unknown epidemic clusters.
Results
Population structure and phylogenetic analysis
We performed the analysis of the evolutionary pathway of the Mtb lineage 2 on the 5,239 isolates from NCBI and ENA. Major phylogenetic lineages were determined based on SNP analysis16, 19, 20 (lin1 = 8.09%; lin2 = 29.18%; lin3 = 17.56%; lin4 = 43.42%; lin5 = 0.59%; lin6 = 0.67%; lin7 = 0.07%; unclassified = 0.49%). In total, we processed the whole-genome sequencing data for 1,398 lineage 2 strains from 32 countries and 13 independent studies and included them in our analysis (Table S1).
We identified 48,275 SNPs relative to the reference H37Rv strain. Strains of lineage 2 distinguish from others lineages by 117 specific SNPs, which is comparable to 106 and 124 SNPs from Coll et al.16 and Rose et al.20, respectively (Table S2). Overall, 1,601 SNPs were found per sample on average (range from 1165 to 1870), Overall, 1,601 SNPs were in average found per sample (range from 1165 to 1870), which corresponds to the SNP density of one SNP per 2.7 kb and consistent with previous estimations21. After excluding the repetitive, mobile elements, PE-PPE-PE_RGRS, drug-resistance associated genes and artifact SNPs linked to indels, we used remaining 39,786 SNPs to reconstruct a maximum-likelihood phylogeny of Mtb lineage 2 (Fig. 1).

Genotyping methods comparison
In the current study, we focused on eight previously published genotyping methods/schemes7, 10, 11, 13,14,15,16,17,18. Six of them were based on SNPs analysis of different genome loci, while other two methods used regions of difference and insertion of IS6110 in NTF region as genetic markers respectively (Table 1). We classified strains from our collection by every method and assigned eight unique identifiers (one for each classification scheme) to each strain (Table S1). The groups defined by particular methods and identified in the studied dataset, are shown in Table 1.
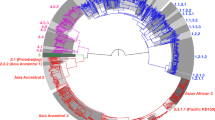
On the next step, we depicted identified groups on the phylogenetic tree and obtained highly congruent phylogenetic relationships (Fig. 1). According to analysis, lineage 2 can be divided into two major phylogenetic clades represented by 20 and 1,378 samples, respectively. First clade was called lineage 2.1 (proto-Beijing clade) by Luo et al. According to Tsolaki et al.10/Gagneux et al.11 these strains belonged to group 1 and could be characterized by the RD105 deletion. In the meantime the majority of samples harbored a larger deletion (“extended RD105”), which affected genes Rv0068-Rv0075. Only one strain, used in Luo et al.17, was characterized by the typical deletion in this region. As for Mestre et al.14 scheme, we were able to identify an ancestral group relative to Bmyc1 which we labeled as proto-Bmyc1. At the same time, we could not find Bmyc1 among identified groups (Table S3). It is interesting, that strains from proto-Beijing clade belonged to ST21 SCG3a according to Filliol et al.15 (Table S4). According to in silico spoligotyping all lineage 2.1 strains carried an uncommon for Beijing strains spoligopattern, characterized by the presence of spacers 1 to 43 (Table S1).
The second major phylogenetic clade, which included the majority of samples (N = 1,378), was labeled lineage 2.2 according to Coll et al.16 and Luo et al.17 and represented the “classic” Beijing lineage, from which strains were characterized by the RD207 deletion. Earliest branch within the clade (50 samples) had intact mutT2, mutT4 and ogt genes and was classified as a member of group 2, lineage 2.2.2, Bj-MG1 and Asia Ancestral 1 by Tsolaki et al.10/Gagneux et al.11, Coll et al.16, Luo et al.17 and Merker et al.18 studies respectively. As for Filliol et al.15, we assigned these strains to ST11. However, we were unable to distinguish ST11 from ST26, since we excluded position 909,166, located in the duplicated fragment of genome, from our analysis22. According to Mestre et al.14, strains of this branch could also be divided into Bmyc2 (45 samples) and Bmyc3 groups, with Bmyc2 as the ancestor of Bmyc3 (Table S3).
Further evolutionary step consists in acquiring the RD181 deletion and could be assigned to group 3 and lineage 2.2.1 by Tsolaki et al.10/Gagneux et al.11 and Coll et al.16, respectively. In the meantime, the transition from STK/ST3 to ST25 (by Filliol et al.15) and from Bmyc4 to Bmyc6 (by Mestre et al.14) was followed by acquiring a mutation in mutT4 (codon 48), and, subsequently, Bmyc6 gave rise to Bmyc26 and Bmyc25. Asia Ancestral 2 group, identified in Merker et al.18, and Bmyc4 almost completely matched (Table S1).
The next major monophyletic group, which included 62 samples, characterized by mutations both in mutT4 (codon 48) and ogt (codon 37), perfectly matched Bmyc25, Bj-MG2 and Asia Ancestral 3 identified by Mestre et al.14, Luo et al.17 and Merker et al.18, respectively. At the same time, Coll et al.16 and Tsolaki et al.10/Gagneux et al.11 did not discriminate this group. In addition, strains from this group, which harbored a mutation in ogt (37 codon), were assigned to ST25 from Filliol et al.15 classification. However, this group also included Beijing strains with intact ogt gene.
Finally, the largest monophyletic group (1,212 samples) in our collection, belonged to previously defined modern Beijing and harbored mutations in mutT2 (codon 58), mutT4 (codon 48) and ogt (codon 12), according to Rad et al.13 along with IS6110 insertion in the NTF region, according to Mokrousov et al.7. Surprisingly, multiple insertion sites for IS6110 were found in the NTF region even in some ancient Beijing strains hence this region may be defined as an IS6110 hot spot (Text S1). In summary, modern Beijing included 8 separate groups - from 215 to 518 depending on the study and some of these groups were described in at least two studies (Fig. 1).
Unified classification
To address the question of the goodness of each classification, we calculated HGDI, as a proxy of allelic diversity23, 24 (Table S5). Merker’s and Mestre’s classifications showed the highest, in relative terms, HGDI (0.77 and 0.56, respectively) and therefore we combined them to provide an unbiased unified classification of lineage 2, which we suggest to use for future studies (Fig. 1). The major drawback of these classifications was their inability to distinguish proto-Beijing strains, which severely restricted their applicability for lineage 2 classification. As we have mentioned already, we were not able to identify Bmyc1 (Mestre et al.14 scheme), although we found an ancestral group, which we labeled proto-Bmyc1. Equally important, Merker et al.18 included only Beijing strains carrying classical Beijing spoligotype in their study, which, by definition, excluded proto-Beijing strains.
Additional analysis of the two selected classification revealed their consistency between themselves and with other classifications. We found “well-defined” groups identified both in Merker et al.18 and Mestre et al.14 schemes, as well in other classifications (e.g. Asia Ancestral 3, which was also identified as Bj-MG2 or Bmyc25), and included such groups in our classification. Next, Asian African 1, Central Asia, Pacific RD150, Bmyc13 and Bmyc18 are also a part of our classification, since they represent clear-cut clusters with group-specific SNPs, which can be used for their identification. For the Bmyc13 group, we have studied countries of patients’ origin and found that this cluster represents Asia and Africa as well; thus we labeled it Asian African 3, following Merker’s naming. As for Bmyc18, even though this group comprises three samples and clustered within Asian African 2, we included it in our classification and labeled Asian African 2/RD142, since it harbors an RD142 deletion and has been described in four independent studies. However, we excluded Bmyc26 and Bmyc6 groups, since these groups represent an intermediate phase between ancient and modern Beijing, and we did not find any cluster-specific SNPs, which can be used for their detection. Thus we present a method for lineage 2 classification, which includes both proto-Beijing and Beijing clades, and has a higher discriminatory power (HGDI 0.79), comparing to other genotyping methods. Within Beijing clade we classify ten groups, where three of them belong to ancient Beijing group (Asia Ancestral 1, Asia Ancestral 2, Asia Ancestral 3) and seven belong to modern Beijing (Asian African 1, Asian African 2, Asian African 2/RD142, Asian African 3, Pacific RD150, Europe/Russia B0/W148 outbreak and Central Asia) (Fig. 1).
SNPs intersection
During the analysis of the genotyping schemes and the phylogenetic tree we found that authors of the different genotyping systems tend to use the same SNPs for groups discrimination, but at the same time one group can be detected using different SNPs. For this reason, we summarized the specific SNPs from each of the studies, found intersections between them and suggest a set of markers to be used with our classification (Fig. 2, Figure S1, Table S6).

Mtb lineage 2 dendrogram representing the major phylogenetic groups and informative genetic markers.
A mutation at pos. 497491(G > A) was used both in Coll et al.16 and Luo et al.17 studies for lineage 2 determination in general and is advisable to use for the first check of sample group. On a next step, two mutations can be used for detection proto-Beijing strains. Position 3309880 (G > A) (Luo et al.17) is preferable to pos. 1881090 (C > T) (Coll et al.16) when detecting proto-Beijing strains, since the latter allows to discriminate strains with the extended-RD105 deletion only.
Two mutations common for all Beijing strains were found and reported: pos. 2505085 (G > A) (Coll et al.16) and pos. 4280708 (G > A) (Filliol et al.15), and therefore can be used for Beijing strains detection. Asia Ancestral 1 group (Bj-MG 1, ST11) can be recognized using different mutations: pos. 1047873 (C > T), reported by Mestre et al.14, and pos. 346693 (G > T), reported by Luo et al.17, Coll et al.16 and Merker et al.18.
The following groups are characterized by the RD181 deletion and can be detected by numerous polymorphisms from different studies, although all of them are inherited by the derived/downstream sublineages. However, next group, Asia Ancestral 2, harbors a unique mutation at pos. 886115 (T > C), which can be used for detection. As mentioned above, Asia Ancestral 3 represents a well-defined group, which was identified by four different studies. However, all of them used the same mutation in ogt gene (codon 37, pos. 1477522 C > A).
As for the following groups, all of them belong to modern Beijing clade. This group carries numerous specific SNPs and can be recognized with mutations in mutT2 (codon 58, pos. 1286766 G > C) and ogt genes (codon 12, 1477596 C > T). The remaining groups, defined within modern Beijing clade, harbor unique mutations and can be easily identified. Full list of markers, used in different studies and phylogenetic tree with major clades are shown in Fig. 2, Figure S1 and Table S6.
Application to epidemiological studies
The increased availability of whole-genome sequencing resulted in its large-scale use for Mtb genotyping and partial replacement of traditional genotyping methods. These technologies allow studying the origin and the evolution of the pathogen, the accumulation of antibiotic resistance and disease outbreaks. For estimation of abundance of our collection and its applicability for further studies we analyzed seven tuberculosis outbreaks. They represented strains circulating in the former Soviet Union (FSU) countries (Clade B, Clade A and CAO clusters18, 25), Gran Canaria (strain GC123726), South Korea (strain K27), and USA (strain 210 and HN87828, 29). In our study, we could detect and classify all the above mentioned clusters and outbreaks strains (Figure S2).
According to our analysis, Strain K belonging to Bmyc3 and all 5 samples from our collection, that were assigned to Bmyc3, were isolated from patients from South Korea (Table S1). GC1237 strain, included in Mestre et al.14, belongs to Bmyc25 (Bj-MG2, Asia Ancestral 3). However, from the phylogenetic analysis we identified several subgroups within Bmyc 25, and GC1237 was a member of only one of that clades. We assigned Strain 210 and HN878 strain to Bmyc18, Asian Africa 2, and ST8 (Mestre et al.14, Merker et al.18, and Filliol et al.15 schemes, respectively). As we have mentioned above, ST8 represents a subgroup within ST22, harboring the RD142 deletion. In total, we identified 105 samples from 12 countries from this group in our collection (Figure S2, Table S1).
Next, we analyzed strains from Clade B, Clade A and CAO. Collectively, these clusters were recently defined as East European sublineage of the Beijing family that is prevalent in Russia and other FSU countries25. Based on our analysis, we detected 15 sublineage-specific SNPs which differ these strains from other modern Beijing. Subsequently, East European sublineage is subdivided into 2 groups, one giving rise to Clade B (289 strains) and other giving rise to Clade A (177 strains) and CAO (79 strains) (Figure S2). These clusters show a restricted genetic diversity (Clade B mean pairwise distance of 28 SNPs, ±SD 10 SNPs, CAO mean pairwise distance of 22 SNPs, ±SD 6 SNPs, Clade A mean pairwise distance of 16 SNPs, ±SD 9 SNPs) and on average harbor 50–60 unique cluster-specific SNPs (Table S6), which contributes to the hypothesis of their recent formation. To check this hypothesis, and estimate the time of origin of the East European sublineage, we used GTR substitution model and a strict molecular clock prior of 1 × 10−7 substitutions per nucleotide per year and estimated that the most recent common ancestor of Clade B, Clade A and CAO arose around 180 years ago (95% highest posterior density (HPD) 140–210), which is also corroborated with recent estimation by Eldholm et al.30.
Discussion
Due to the high prevalence, successfulness and the ease of detection, there have been many attempts to classify strains belonging to lineage 2 (East-Asian) and to clarify their evolutionary pathway. Studies of epidemic strains, aimed at examining transmission chains and identifying the factors contributing to their dissemination, are no less interesting. However, the major difficulties that a nonspecialist faces when goes through the tuberculosis literature is the fluctuating and evolving nomenclature concerning this lineage. In our study, we combined all of this data using our collection of 1,398 samples and presented the unified classification and evolutionary pathway of lineage 2 of Mtb.
At least 8 different genotyping schemes were proposed by leading scientific groups, based on different genomic markers, such as, IS6110-RFLP, RDs, SNPs datasets, VNTR and WGS studies7, 10, 11, 13,14,15,16,17,18 (Table 1). In our study, we confirmed that lineage 2 comprises 2 major clades, designated proto-Beijing, which harbors unusual spoligoprofile, and Beijing, with classic, well-known spoligoprofile. There is no consensus on dating the origin of this lineage and the opinions and hypotheses are contrasting. One such hypothesis is based on the assumption that the age of the human Mtb sensu stricto is 70,000 years1 and the age of lineage 2 is 30,000 years17, other estimates time to most recent common ancestor (TMRCA) of Beijing lineage as 6,000 years18. While citing these papers, many authors illustrate differences in TMRCA estimations. However, after detailed analysis, it is clear, that Merker et al.18 did not consider proto-Beijing strains in their study, so these two different estimations cannot be compared in a straightforward way. In its turn, Beijing clade is divided into ancient and modern groups, in which smaller groups can be clearly identified (Fig. 1).
Analysis of ancient Beijing strains population structure using different classifications revealed high consistency of different genotyping methods. We were able to identify clear-cut clusters, which represent groups identified by two or more independent studies (e.g. Asia Ancestral 1, Asia Ancestral 2, Asia Ancestral 3 according to our classification). Such groups harbor many unique SNPs and can be easily identified using any of them. On the other hand, we also found groups representing intermediate phases, for which we did not find cluster-specific SNPs. One of such groups is Bmyc26, which illustrates transition from the ancient to the modern Beijing. This transition is usually determined by mutations in mutT2 (codon 58) and ogt (codon 12) genes. However, we also identified strains with mutation in mutT2 and intact ogt gene and labeled these strains Bmyc26/10 as an intermediary group between Bmyc26 and Bmyc10 (Table S3). These strains were isolated from patients from China and were firstly described in Liu et al.31.
In its turn, the boundary between the ancient and the modern Beijing is less distinctly traced in the study of Coll et al.16 and Tsolaki et al.10/Gagneux et al.11 thus this makes their genotyping schemes less suitable for the differentiation of Beijing strains (Fig. 1). The key point is that both of these classifications are fully matched each other and their major drawback is that both ancient and modern strains of Beijing subtype can be labeled as Group 3/lineage 2.2.1. On the other hand, in silico genotyping based on IS6110 in the NTF locus showed controversial results: multiple insertion sites of this element among ancient Beijing strains demonstrate this region to be IS6110 insertion hotspot (Text S1). At the same time, we did not find any strains from ancient Beijing group that contained the IS6110 insertion in a typical spot for modern Beijing, as it was described by Nakanishi et al.9. However, that study included more than 1000 samples belonging to ancient Beijing group in contrast to only 191 such strains in the present study. As for modern Beijing group, all samples belonging to it harbored IS6110 in the left part of the NTF region and in general had less additional insertion elements in non-typical sites. These results suggested that particular IS6110 insertion in the NTF region occurred independently in modern strains.
Phylogenetic relationships of modern Beijing strains are less clear compared to the ancient Beijing family. We noted a large number of contradictions and inconsistencies between different authors. Further analysis revealed that this situation was caused by star-shape phylogeny of the modern Beijing when new groups evolve independently. Consequently, during their attempts to clarify the evolutionary pathway, authors were only able to suggest different genotyping schemes, and variety of groups they received depended on the collection diversity. Therefore, in order to obtain a balanced classification, it is necessary to consider all classifications and carefully study identified groups, their unique SNPs and other genetic markers.
As in ancient Beijing group, individual groups were independently identified in several different studies (e.g. Pacific RD150 and Asian African 2). Pacific RD150 group is a part of the CC5 complex and is phylogeographically specific for the Pacific region18. Additionally we note that strains with the RD150 deletion are a clear-cut, distinct group. In this sense, our study differs from Faksri et al.32, who showed deletions of this region in other modern Beijing groups.
One more group is strains with the RD142 deletion. We identified only 3 such strains among our collection, but they clustered together with epidemic HN878 and strain 210 also harboring this deletion (Figure S2). In general, strains from this group clustered together as a part of a larger group, identified both by Filliol et al.15 and Merker et al.18 (ST22 and Asian African 2 respectively), but despite this, we included the Bmyc18 group (which we called Asian African 2/RD142) in our classification because of its importance in terms of epidemiology and a unique RD142 deletion.
Another epidemically important population is the East European sublineage of Beijing family25, widespread in the FSU countries and characterized by a high level of drug resistance. In this study, the origin of this sublineage was estimated to be 1847 CE (95% highest posterior density (HPD), 1809–1882 CE) and their closest relatives were within the phylogenetically basally located subgroup of modern Beijing strains. Subsequently, the sublineage is divided into two subpopulations. One of them gave rise to Clade B, which was initially designated B033 and W1485 but also named СС218, East European 217 and ECDC000234. Our estimation of the TMRCA of this lineage is 1960 CE (HPD 1944–1973 CE), which is similar to the previous estimation30. Meanwhile, due to the size of our collection, we were able not only to identify the above-mentioned clusters, but also to find and describe their closest ancestors, which is essential for understanding the reasons behind their epidemiological success. It is interesting to note that the vast majority of Clade B samples were from Russia and Belarus, with the closest related strains being isolated primarily from patients from China (Figure S2). This allows us to assume that the ancestral forms of Clade B were from China, which corroborates with the recent study of Yin et al.35.
The second branch of the East European sublineage, named the Central Asia group18 and East European 117, was a more heterogeneous population than the first branch (mean pairwise distance of 80 SNPs, ± SD 36 SNPs), which is in consistence with previous studies17, 18. However, here we should also emphasize a number of features. This group is relatively older, compared to Clade B (1877 CE, HPD 1843–1901 CE) and, according to the Merker et al. study18, Central Asia group contained 31% of MDR strains, which also tended to cluster. According to our study, more than half of the Central Asia group strains were MDR (310 out of 506), with most of them also belonged to clustering samples of epidemic populations. For example, Clade A and CAO clusters contained 196 and 50 MDR strains from 224 and 80 strains of clusters, respectively. In its turn, the exclusion of samples belonging to these clusters from the Central Asia group results in decreasing the percentage of MDR strains to 31.7% (64 of 202), which corresponds to the average for lineage 2 excluding the East European sublineage (30% MDR samples, 149 of 505). The latter suggests that using any classification within a strictly delineated group, one can find more successful strains characterized by their geographical distribution (Clade A is one of the most common clusters in Russia, CAO cluster strains are most common in Central Asia), virulence and drug resistance.
Conclusion
Our SNP-based phylogenetic analysis of a global collection of Mtb lineage 2 isolates suggests that the evolutionary pathway and branch development description, proposed by different groups, are consistent with regard to the ancient strains. At the same time, phylogeny analysis of the modern strains revealed great discrepancies and contradictions, which we examined in this study. Our results provide additional insights into phylogeny of Mtb lineage 2, since the whole-genome SNP tree revealed much more phylogenetic detail, such as strict relationship between groups, positioning of epidemiologically important groups and “blank spots” of noticeable clusters.
Our proposed classification allows identifying both proto-Beijing and Beijing strains. Beijing group is divided into two, ancient Beijing clade and modern Beijing clade, which consist of three (Asia Ancestral 1, Asia Ancestral 2, Asia Ancestral 3) and seven (Asian African 1, Asian African 2, Asian African 2/RD142, Asian African 3, Pacific RD150, Europe/Russia B0/W148 outbreak and Central Asia) (Figs 1 and 2) groups respectively.
We suggest that the classification proposed herein, as well as analysis of existing genotyping schemes evolutionary pathways should facilitate future studies of lineage 2 and will help to classify Mtb strain correctly. A more detailed study of the SNP tree together with phenotypic information might result in more accurate and robust clade assignment and may lead to better understanding of the molecular determinants and the selection forces that have contributed to the global success of the East Asian lineage.
Materials and Methods
Genome sequencing data
Whole-genome sequencing data of 5,715 Mtb isolates was obtained from National Center for Biotechnology Information (NCBI) and European Nucleotide Archive (ENA). The dataset consisted of thirteen independent WGS studies, available under accessions ERP00011136, ERP000124, ERP00019225, ERP00027637, ERP00043638, 39, ERP0017311, ERP00261740, ERP004677, ERP00698918, ERP01305441, SRA06509542, SRP05109317, and TB-ARC - Belarus.
In addition, a set of 5 samples, W-148 (CP012090.1), Strain K (CP007803.1), Strain 210 (ADAB00000000.1), GS1237 (ERR071082), and HN878 (NZ_CM001043), associated with tuberculosis outbreaks in different countries, was downloaded from NCBI. Genome H37Rv (NC_000962.3) was used as reference.
SNPs calling
We aligned reads from whole-genome sequencing to the H37Rv (NC_000962.3) genome using Bowtie 243. After, we sorted, indexed the aligned reads and converted them into a mpileup file using SAMtools44 and discarded samples with median read depth less than 30. We used VarScan45 to determine the variants in the remaining samples (n = 5,239) and MUMmer 3.20 with its nucmer and show-snps functions for the alignment of complete Mtb genomes to H37Rv46.
Bioinformatics analysis
For further analysis, we made a custom R script. We assigned the Mtb samples to the main phylogenetic lineages on the basis of lineage-specific SNPs16, 19, 20. SNPs with a variant allele frequency of less than 90% or with coverage of less than 5 reads were discarded, as they are likely to originate from mapping errors. We annotated the remaining SNPs using the H37Rv annotation and classify them into synonymous and nonsynonymous. In addition, we filtered out SNPs in repetitive, mobile elements, PE-PPE-PE_RGRS genes and drug-resistance associated genes due to complexity of such regions47. From a group of samples which differ by less than 10 SNPs, we left only the one with higher mapping rate. For further validations we used Tablet48.
To create a maximum-likelihood phylogenetic tree with R phangorn package49 we concatenated remaining SNPs (n = 39,786) for each lineage 2 strain (n = 1,398) into an artificial sequence and used iTOL50 for visualization and annotation.
BEAST evolutionary analysis
We selected 896 samples, representing Central Asia outbreak, CladeA, and CladeB, for computing dated phylogeny and divergence time using BEAST (v1.8.4)51. We used GTR model with gamma site heterogeneity model with 4 parameters and we defined lognormal prior distribution for the substitution rate (1 × 10−7, ranging from 9.3 × 10−6 to 1.7 × 10−7) thus allowing for ~0.5 SNP/genome/year51. We run chains of 107 generations, sampled every 1000 was run and assessed convergence using Tracer, ensuring all relevant parameters reached an effective population size of >100.
Spoligotyping, RD deletions, and IS6110 analysis
Isolates assigned to lineage 2 (n = 1,398) were in silico spoligotyped from raw sequence files (fastq format) using SpoTyping software52. For the analysis of presence or absence of six LSP loci (RD105, extended-RD105, RD207, RD181, RD150, and RD142) we compared the mapping depth of the targeted region with the coverage of the corresponding flanking regions. If the average coverage of the targeted region was at least two fold lower than that of both flanking regions, a deletion was called, as described before17. We used ISMapper53 for the analysis of the IS6110 insertion in NTF region and Integrative Genomics Viewer for visual control of the insertion position54.
References
Comas, I. et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 45, 1176–1182. doi:10.1038/ng.2744 Epub 2013 Sep 1171 (2013).
Parwati, I., van Crevel, R. & van Soolingen, D. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect Dis. 10, 103–111, doi:10.1016/S1473-3099(1009)70330-70335 (2010).
van Soolingen, D. et al. Predominance of a single genotype of Mycobacterium tuberculosis in countries of east Asia. J Clin Microbiol. 33, 3234–3238 (1995).
Kremer, K. et al. Definition of the Beijing/W lineage of Mycobacterium tuberculosis on the basis of genetic markers. J Clin Microbiol. 42, 4040–4049 (2004).
Bifani, P. J., Mathema, B., Kurepina, N. E. & Kreiswirth, B. N. Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains. Trends Microbiol. 10, 45–52 (2002).
Mokrousov, I. et al. Phylogenetic reconstruction within Mycobacterium tuberculosis Beijing genotype in northwestern Russia. Res Microbiol. 153, 629–637 (2002).
Mokrousov, I. et al. Origin and primary dispersal of the Mycobacterium tuberculosis Beijing genotype: clues from human phylogeography. Genome Res. 15, 1357–1364 Epub 2005 Sep 1316 (2005).
Plikaytis, B. B. et al. Multiplex PCR assay specific for the multidrug-resistant strain W of Mycobacterium tuberculosis. J Clin Microbiol. 32, 1542–1546 (1994).
Nakanishi, N. et al. Evolutionary robust SNPs reveal the misclassification of Mycobacterium tuberculosis Beijing family strains into sublineages. Infect Genet Evol. 16, 174–7, doi:10.1016/j.meegid.2013.1002.1007 Epub 2013 Feb 1022 (2013).
Tsolaki, A. G. et al. Genomic deletions classify the Beijing/W strains as a distinct genetic lineage of Mycobacterium tuberculosis. J Clin Microbiol. 43, 3185–3191 (2005).
Gagneux, S. et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 103, 2869–2873 Epub 2006 Feb 2813 (2006).
Flores, L. et al. Large sequence polymorphisms classify Mycobacterium tuberculosis strains with ancestral spoligotyping patterns. J Clin Microbiol. 45, 3393–339 Epub 2007 Aug 3315 (2007).
Ebrahimi-Rad, M. et al. Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing family. Emerg Infect Dis. 9, 838–845 (2003).
Mestre, O. et al. Phylogeny of Mycobacterium tuberculosis Beijing strains constructed from polymorphisms in genes involved in DNA replication, recombination and repair. PLoS One. 6, e16020, doi:10.11371/journal.pone.0016020 (2011).
Filliol, I. et al. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol. 188, 759–772 (2006).
Coll, F. et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 5, 4812, doi:10.1038/ncomms5812 (2014).
Luo, T. et al. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. Proc Natl Acad Sci USA 112, 8136–8141 doi:10.1073/pnas.1424063112 Epub 1424062015 Jun 1424063115 (2015).
Merker, M. et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 47, 242–249, doi:10.1038/ng.3195 Epub 2015 Jan 1019 (2015).
Homolka, S. et al. High resolution discrimination of clinical Mycobacterium tuberculosis complex strains based on single nucleotide polymorphisms. PLoS One. 7, e39855, doi:10.31371/journal.pone.0039855 Epub 0032012 Jul 0039852 (2012).
Rose, G. et al. Mapping of genotype-phenotype diversity among clinical isolates of Mycobacterium tuberculosis by sequence-based transcriptional profiling. Genome Biol Evol. 5, 1849–1862, doi:10.1093/gbe/evt1138 (2013).
Coll, F. et al. PolyTB: a genomic variation map for Mycobacterium tuberculosis. Tuberculosis (Edinb). 94, 346–354. doi:10.1016/j.tube.2014.1002.1005 Epub 2014 Feb 1015 (2014).
Wada, T., Iwamoto, T., Hase, A. & Maeda, S. Scanning of genetic diversity of evolutionarily sequential Mycobacterium tuberculosis Beijing family strains based on genome wide analysis. Infect Genet Evol. 12, 1392–1396, doi:10.1016/j.meegid.2012.1304.1029 Epub 2012 May 1399 (2012).
Hunter, P. R. & Gaston, M. A. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol. 26, 2465–2466 (1988).
Mokrousov, I. Revisiting the Hunter Gaston discriminatory index: Note of caution and courses of change. Tuberculosis (Edinb). 104, 20–23, doi:10.1016/j.tube.2017.1002.1002 Epub 2017 Feb 1016 (2017).
Casali, N. et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 46, 279–286, doi:210.1038/ng.2878 Epub 2014 Jan 1026 (2014).
Alonso, H. et al. Deciphering the role of IS6110 in a highly transmissible Mycobacterium tuberculosis Beijing strain, GC1237. Tuberculosis (Edinb). 91, 117–126, doi:10.1016/j.tube.2010.1012.1007 Epub 2011 Jan 1020 (2011).
Han, S. J. et al. Complete genome sequence of Mycobacterium tuberculosis K from a Korean high school outbreak, belonging to the Beijing family. Stand Genomic Sci. 10, 78, 10.1186/s40793-40015-40071-40794, eCollection 42015 (2015).
Sreevatsan, S. et al. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci USA 94, 9869–9874 (1997).
Yang, Z. et al. Diversity of DNA fingerprints of Mycobacterium tuberculosis isolates in the United States. J Clin Microbiol. 36, 1003–1007 (1998).
Eldholm, V. et al. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. Proc Natl Acad Sci USA 113, 13881–13886 Epub 12016 Nov 13821 (2016).
Liu, Q. et al. Genetic features of Mycobacterium tuberculosis modern Beijing sublineage. Emerg Microbes Infect. 5, e14, doi:10.1038/emi.2016.1014 (2016).
Faksri, K. et al. Genetic diversity of the Mycobacterium tuberculosis Beijing family based on IS6110, SNP, LSP and VNTR profiles from Thailand. Infect Genet Evol. 11, 1142-1149 doi:10.1016/j.meegid.2011.1104.1007. Epub 2011 Apr 1114 (2011).
Narvskaya, O. Genome polymorphism of Mycobacterium tuberculosis and its role in epidemic process. D.Sc. dissertation. Institute of Experimental Medicine, St. Petersburg, Russia. (In Russian) (2003).
De Beer, J. L., Kodmon, C., van der Werf, M. J., van Ingen, J. & van Soolingen, D. Molecular surveillance of multi- and extensively drug-resistant tuberculosis transmission in the European Union from 2003 to 2011. Euro Surveill. 19(11), 20742 (2014).
Yin, Q. Q. et al. Evolutionary History and Ongoing Transmission of Phylogenetic Sublineages of Mycobacterium tuberculosis Beijing Genotype in China. Sci Rep. 6, 34353, doi:10.1038/srep34353. (2016).
Bryant, J. M. et al. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect Dis. 13, 110, doi:10.1186/1471-2334-1113-1110 (2013).
Walker, T. M. et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 13, 137–146, doi:10.1016/S1473-3099(1012)70277-70273 Epub 72012 Nov 70215 (2013).
Glynn, J. R. et al. Whole Genome Sequencing Shows a Low Proportion of Tuberculosis Disease Is Attributable to Known Close Contacts in Rural Malawi. PLoS One. 10, e0132840, doi:10.0131371/journal.pone.0132840, eCollection 0132015 (2015).
Guerra-Assuncao, J. A. et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife. 4, doi:10.7554/eLife.05166 (2015).
Perdigao, J. et al. Unraveling Mycobacterium tuberculosis genomic diversity and evolution in Lisbon, Portugal, a highly drug resistant setting. BMC Genomics. 15(991), doi:10.1186/1471-2164-1115-1991 (2014).
Phelan, J. et al. Mycobacterium tuberculosis whole genome sequencing and protein structure modelling provides insights into anti-tuberculosis drug resistance. BMC Med. 14–31, doi:10.1186/s12916-12016-10575-12919 (2016).
Zhang, H. et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 45, 1255–1260 doi:10.1038/ng.2735 Epub 2013 Sep 1251 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9, 357–359, doi:10.1038/nmeth.1923 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079, doi:10.1093/bioinformatics/btp2352 Epub 2009 Jun 2078 (2009).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576, doi:10.1101/gr.129684.129111 Epub 122012 Feb 129682 (2012).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 Epub 2004 Jan 2030 (2004).
Phelan, J. E. et al. Recombination in pe/ppe genes contributes to genetic variation in Mycobacterium tuberculosis lineages. BMC Genomics. 17–151, doi:10.1186/s12864-12016-12467-y (2016).
Milne, I., Bayer, M., Stephen, G., Cardle, L. & Marshall, D. Tablet: Visualizing Next-Generation Sequence Assemblies and Mappings. Methods Mol Biol. 1374, 253–68, doi:10.1007/1978-1001-4939-3167-1005_1014 (2016).
Schliep, K. P. phangorn: phylogenetic analysis in R. Bioinformatics. 27, 592–593, doi:10.1093/bioinformatics/btq1706 Epub 2010 Dec 1017 (2011).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–245, doi:10.1093/nar/gkw1290. Epub 2016 Apr 1019 (2016).
Drummond, A. J., Ho, S. Y., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 Epub 2006 Mar 2014 (2006).
Xia, E., Teo, Y. Y. & Ong, R. T. SpoTyping: fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 8, 19, doi:10.1186/s13073-13016-10270-13077 (2016).
Hawkey, J. et al. ISMapper: identifying transposase insertion sites in bacterial genomes from short read sequence data. BMC Genomics. 16(667), doi:10.1186/s12864-12015-11860-12862. (2015).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192,doi:10.1093/bib/bbs1017 Epub 2012 Apr 1019 (2013).
Acknowledgements
This work was supported by 17-15-01412 grant of the Russian Science Foundation of the Russian Federation.
Author information
Authors and Affiliations
Contributions
E.S., S.K., I.M., J.B., E.I. wrote the main manuscript text. E.S. and S.K. prepared Figures and Tables. S.K. and D.I. conducted genome analysis. E.S., S.K., E.I. and V.G. designed the experiment. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shitikov, E., Kolchenko, S., Mokrousov, I. et al. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis . Sci Rep 7, 9227 (2017). https://doi.org/10.1038/s41598-017-10018-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10018-5
This article is cited by
-
The relative transmission fitness of multidrug-resistant Mycobacterium tuberculosis in a drug resistance hotspot
Nature Communications (2023)
-
Insight into pathogenomics and phylogeography of hypervirulent and highly-lethal Mycobacterium tuberculosis strain cluster
BMC Infectious Diseases (2023)
-
Molecular characteristics of Mycobacterium tuberculosis drug-resistant isolates from HIV- and HIV+ tuberculosis patients in Russia
BMC Microbiology (2022)
-
Impact of pathobiological diversity of Mycobacterium tuberculosis on clinical features and lethal outcome of tuberculosis
BMC Microbiology (2022)
-
Distribution and identification of Mycobacterium tuberculosis lineage in Kashgar prefecture
BMC Infectious Diseases (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
