
Abstract
Tumor metastasis is mainly caused by somatic genomic alterations (SGAs) that perturb pathways regulating metastasis-relevant activities and thus help the primary tumor to adapt to the new microenvironment. Identifying drivers of metastasis, i.e. SGAs, sheds light on the metastasis mechanism and provides guidance for targeted therapy. In this paper, we introduce a novel method to search for SGAs driving breast cancer metastasis to the lung. First, we search for transcriptomic modules with genes that are differentially expressed in breast cell lines with strong metastatic activities to the lung and co-expressed in a large number of breast tumors. Then, for each transcriptomic module, we search for a set of SGA genes (driver modules) such that genes in each driver module carry a common signal regulating the transcriptomic module. Evaluations indicate that many genes in driver modules are indeed related to metastasis, and our methods have identified many new driver candidates. We further choose two novel metastatic driver genes, BCL2L11 and CDH9, for in vitro verification. The wound healing assay reveals that inhibiting either BCL2L11 or CDH9 will enhance the migration of cell lines, which provides evidence that these two genes are suppressors of tumor metastasis.
Similar content being viewed by others


Introduction
Breast cancer is the most common type of cancer in women, accounting for about one quarter of all cancer cases1. Metastasis takes place in the late stages of cancer development and is the major cause of mortality in patients with breast cancer and other solid tumors2, 3. As with other cancer processes, such as resisting cell death and sustaining cell proliferation, cancer metastasis is mainly caused by somatic genomic alterations (SGAs) such as somatic mutations and copy number alterations that perturb signaling pathways regulating metastasis-relevant activities, including fiber formation and focal adhesion, epithelial–mesenchymal transition, differentiation and morphogenesis, and invasion4. Somatic mutations and copy number alterations of a gene may change its 3D structure and the concentration of its gene product – protein, respectively. Hence, if the protein is a signaling protein, then the signal it carries will be disturbed. The signal perturbation causes abnormal expression of genes regulated by the signal, thus altering the behavior of cells, such as turning normal cells into cancerous ones or enabling tumor cell invasion and metastasis.
An important way to study the cancer metastasis mechanism is to find gene subsets or gene signatures that are involved in metastasis-relevant activities by contrasting the expression data of cancer cells with and without metastasis. For example, Minn et al. compared gene express profiles of MDA-MB-231 subpopulations with different degrees of lung metastatic activities to identify a set of genes that likely mediates breast cancer metastasis to the lung2. In another study, Bos et al. isolated subpopulations with strong brain metastatic activity from parental CN34 and MDA-MB-231 cell lines and then found genes that mediate the metastasis of breast cancer to the brain by comparing expression data of cell lines with different strengths of brain metastatic activity5. Harrell et al. found the gene signatures of breast metastasis to the brain, lung and liver by comparing expression data of primary tumors and their matched metastases or sets of synchronous metastases from the same patients6. However, while such studies shed light on the genes involved in the molecular mechanisms of metastasis, they do not necessarily reveal the drivers or pathways that originally activate the process of metastasis.
From the point of view of targeted therapy or precision medicine, a more important task in cancer metastasis research is to search for drivers, i.e. SGAs that initiate cancer metastatic progression, where those drivers provide ideal candidates for targeted therapy. There are abundant publications in this line of research. For example, Wagenblast et al. found that the genes SERPINE2 and SLPI are drivers of metastasis by exploring the impact of enforced expression of these two genes on metastasis7. Julien et al. investigated the role of gene PTP1B in mammary tumorigenesis by inhibiting it in vivo. They found that the rate of tumor development, number of tumors and lung metastases were significantly reduced in NDL2 transgenic mice both in PTPN1-/- mice and in PTP1B inhibitor treated mice8. Xue et al. showed that loss of the gene PAR3 promoted breast cancer metastasis in vitro and in vivo 9. Tan et al. found that the RANKL-RANK signal regulated the cancer metastasis of T cells10. All of the above examples used wet-lab techniques to search for drivers of metastasis. Currently, a large amount of gene expression, somatic mutation, copy number alteration etc. data for cell lines with metastasis2, 5, breast tumors with relapse in the brain, lung, liver etc.5, 6, 11 is becoming available. Hence, using computational methods to search for drivers of metastasis has become feasible. Computational methods can provide short lists of candidates that are very likely to be drivers of metastasis, which can then be verified by biologists, thereby greatly speeding up the research into understanding the disease mechanism of cancer metastasis.
Drivers of metastasis can be used as candidates for targeted therapy. However, not all drivers are “druggable”12, 13; furthermore, as FDA-approved drugs for targeted therapy is limited, only a small number of drivers in tumors can be directly treated by approved agents 14. One way to address this problem is to find out what drivers are on the same pathway. Then if a driver in a tumor cannot be drugged directly, we can target other genes/proteins that carry the same signal carried by the “undruggable” driver. For example, human RAS genes are a notorious oncogene family, where no effective inhibitor has reached the clinic despite more than 30 years of effort13. One current major effort for anti-RAS drug discovery is aiming to block the activities of proteins that are components of the signaling pathway downstream of RAS 13. In this study, we will develop a new computational framework that not only finds drivers of breast cancer metastasis to the lung, but also groups drivers carrying common signals together.
The de novo discovery of pathways underlying specific biological processes, e.g., metastasis, remains a challenging task. First, few studies of cancer metastasis collect omics data that reflect different aspects of the metastatic process. For example, while collecting only genomic data from metastasized tumors does reveal which genomic alterations are specifically driving metastasis, identifying differentially expressed genes (DEGs) does not reveal the driver gene or pathway that regulates the DEGs. In this study, we designed a framework that integrates the results of transcriptomic studies of metastasis with existing large-scale comprehensive omics data to enable a search for the SGAs driving expression of genes involved in metastasis. A second challenge to de novo discovery of such pathways is there are few well-established computation methods to search for SGA drivers that causally regulate the expression of a set of genes. Therefore, in this study, we developed a novel de novo computational framework to search for SGAs driving breast cancer metastasis to the lung.
In this framework, first, we found out what genes were differentially expressed in a subpopulation of breast cell lines with strong metastatic activity to the lung. Then, we used differentially expressed genes to search for gene transcriptomic modules such that the genes in each transcriptomic module are most likely to be regulated by a common signal. Next, we used the expression status of each transcriptomic module as the readout of a signal to search for genes whose SGA events have strong information with respect to the expression status of transcriptomic modules. Finally, we chose two genes from our results for in vitro verification (Please refer to the overall scheme in the METHODS section).
Results
Gene transcriptomic modules
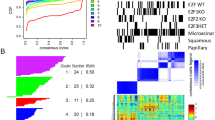
We first found the differentially expressed genes (DEGs) by comparing the MDA-MB-231 parental cell line or its subpopulations that have relatively weaker lung metastasis activities with other subpopulations that have relatively stronger lung metastasis activities. Then the number of DEGs was further refined by excluding genes that were up-regulated in cell lines and down-regulated in lung tumors (TCGA) or down-regulated in cell lines and up-regulated in lung tumors. Next, we searched for gene transcriptomic modules from DEGs such that the genes in each gene transcriptomic module were co-expressed in a large number of TCGA breast tumors and also regulated by a common transcription factor (refer to METHODS for details). Hence, it is most likely that the genes in each gene transcriptomic module are regulated by a common signal. For each gene transcriptomic module, we obtained two tumor subsets T abn and T nor , where T abn included BRCA tumors that had abnormal expressions of genes in the module and tumors in T nor had expressions of genes in the module similar to normal samples. We found 21 gene transcriptomic modules. We checked if the expression statuses of the transcriptomic modules impacted patients’ clinical outcomes. As the TCGA BRCA clinical data did not have enough death events, we used METABRIC15 data to evaluate the impact of our gene transcriptomic modules on patients’ survival. The results showed that 11 of 21 modules had significant impact (both p-value and q-value less than 0.05) on patient survival (refer to Fig. 1 and Fig. S1). Moreover, usually, if a transcriptomic module was down-regulated in high metastatic cell lines, then patients with a low expression of the transcriptomic module had a worse clinical outcome (refer to Fig. 1A). On the other hand, if a transcriptomic module was up-regulated in high metastatic cell lines, then patients with a high expression of the transcriptomic module had a worse clinical outcome (refer to Fig. 1B). Hence, our transcriptomic modules are biologically meaningful as cancer metastasis is the major cause of mortality in breast cancer patients2, 3.

The expression statuses of gene transcriptomic modules have impact on patients’ survival. H, M, L represents tumor group with high, middle, low expression of the transcriptomic module, respectively. A “*” after H/L signifies that the module is up-/down-regulated in high metastatic cell lines.
Driver modules
We assume that each gene transcriptomic module is regulated by a common signal. The abnormal expression of genes in a gene transcriptomic module in a tumor is caused by at least one SGA event that perturbs the common signal regulating the module. The major goal of the project is identifying drivers carrying common signals, where drivers provide candidates for targeted therapy; in the case where a driver g in a tumor is not “druggable”, we can instead target one of the other drivers that carry the same signal that driver g does. For this purpose, we developed a signal-based method that searched for sets of genes such that their SGA events have strong information with respect to common signals (refer to METHODS for detail).
For each gene transcriptomic module, we obtained two tumor subsets T abn and T nor , where we suppose that tumors in T abn are highly likely to have SGA events that perturb the common signal regulating the gene transcriptomic module while tumors in T nor are less likely to have SGAs that perturb this common signal. Hence, if a set of genes are drivers that carry the common signal regulating a gene transcriptomic module, then the SGA events of those genes should mostly happen among tumors of T abn and seldom occur among tumors in T nor . Corresponding to the 21 gene transcriptomic modules, we obtained 21 driver modules. Each driver module has six genes. Our results show that SGA events of genes in all driver modules are very enriched among the corresponding T abn s. The p-values calculated from a hypergeometric distribution are between 1.04E-38 and 5.47E-17, which is much better than driver modules obtained from random selection (refer to Fig. 2). These results reveal that SGA events of genes in driver modules have strong information with respect to corresponding common signals.

Enrichment p-values for random and our driver modules. SGA events of genes in our driver modules are greatly enriched among tumors in T abn s.
Evaluate driver modules via literature search
A literature search revealed that SGA events of many genes in our driver modules were related to cancer metastasis, cell invasion or migration, where many of those SGA-metastasis associations were obtained from in vitro or in vivo verifications. We kept references about cell invasion and migration as they are important processes of metastasis.
One example is driver module 1, which includes the genes LSR, PTP4A3, S100A14, SMCP, TP53, and ZWINT, where TP53 was either mutated or deleted; SMCP had both mutations and amplifications; the remaining four genes were mainly amplified (refer to Fig. 3A). García et al. found that LSR (LISCH7) mRNA in plasma was significantly associated with lymph node metastasis and with vascular invasion, and they thought that the up-regulation of the gene promotes the development of metastasis16. Bayat et al. reported that CD177(+) neutrophils migrated significantly faster through HUVECs expressing LSR in in vitro transendothelial migration experiments17. Guzinska-Ustymowicz et al. found that high expression of PTP4A3 was associated with lymph node metastasis from colorectal carcinoma18. Laurent et al. claimed that high PTP4A3 phosphatase expression correlates with metastatic risk and expressed the belief that PTP4A3 plays a causal role in the development of metastases in uveal melanoma19. Zimmerman et al. verified that a knock-out of PTP4A3 decreases migration of endothelial cells by in vitro wound healing assay20. Cho et al. demonstrated that S100A14 promotes cell migration and invasion using a wound healing assay and Martrigel invasion assay in vitro 21.

SGA pattern of driver modules. (A) SGA pattern of module 1. (B) SGA pattern of module 18.
Zhao et al. claimed that S100A14 promotes the growth and metastasis of hepatocellular carcinoma. Their claim was based on the wound healing assay and Matrigel transwell assay, which verified that S100A14 silencing suppressed cell migration and invasion while overexpression of S100A14 promoted migration and invasion of hepatocellular carcinoma cells. They also validated in vivo that inhibiting S100A14 significantly reduced lung metastasis in mouse models22. Takahashi et al. explored that SMCP might be a novel marker of CSC/CIC cells, which are highly responsible for disease recurrence after treatments and for distant metastasis23. TP53 is a well-known tumor suppressor that is associated with cell cycle arrest, apoptosis, and DNA repair etc. that also regulates cancer metastasis. Patricia et al. stated that mutant p53 proteins promote invasion, metastasis, proliferation and cell survival24. Adorno et al. validated with wound healing assays that mutant-p53 (p53 R175H) promoted the migration of H1299 cells and validated with transwell assays that mutant-p53 is required for TGFβ-driven invasion and metastasis in breast cancer MDA-MB-231 cells25. Weissmueller et al. demonstrated that mutant p53 promoted migration and metastasis in vitro and in vivo 26. Hence, five of six genes in this module have been verified to be drivers of metastasis.
Another example is driver module 18, which includes the genes BAIAP2, EEF1A2, GUK1, PPP1R3B, TEX2, and TRPS1. Except for gene PPP1R3B, which was mainly mutated or deleted, the other five genes were mainly amplified among TCGA breast tumors with abnormal expression of genes in gene transcriptomic module 18 (refer to Fig. 3B). Funato et al. stated that BAIAP2 (IRSP53) is important for the metastatic behavior of malignant tumor cells27. In28, Hoeppner et al. said that shRNA to BAIAP2 inhibited podocyte migration. Kawamura et al. verified that siRNA EEF1A2 inhibited cell migration significantly in LCSC#1 cell lines29. Xu et al. used the Matrigel transwell assay to evaluate the invasive capacities of cell lines with different expression levels of the EEF1A2 gene and found that more invasive cell lines have higher EEF1A2 expression. They also found that siRNA EEF1A2 suppressed cell invasion and migration in both a Matrigel transwell assay and wound healing assay. Hence they concluded that EEF1A2 promotes cell migration, invasion and metastasis in pancreatic cancer30. De Rocha et al. found that GUK1 was differentially expressed in metastatic pituitary carcinoma31. Hu et al. found that over-expression of TRPS1 promoted HUVEC migration significantly and knockdown of TRPS1 decreased this migration ability32. Hong et al. stated that high TRPS1 expression was significantly associated with positive lymph node metastasis33. Therefore, the SGA events of four genes in this module have been verified to be associated with cancer metastasis. We could not find references that associated genes PPP1R3B and TEX2 with metastasis, migration, or invasion directly. However, we found that Lu et al. claimed that the mutated PPP1R3B peptide represents the immunodominant epitope34 and Hayashida et al. claimed that PPP1R3 gene alterations correlated with lymph node and liver metastases35, which provides evidence of association between PPP1R3B gene and metastasis.
Further study showed that in our 21 driver modules, two modules have five genes and three modules have four genes related to metastasis, respectively. Another four modules have three genes and nine modules have two genes related to metastasis, respectively. Finally, the remaining three modules have one gene related to metastasis (refer to Table S1). Hence, our driver modules are biologically meaningful.
We also used a commercial software – “Ingenuity Pathway Analysis (IPA)” to check the functional coherence of our driver modules. Results showed that many of our driver modules were significantly (p-value were much less than 0.05) related to certain functions related to metastasis activities. For example, four genes, ATG7, ERBB2, NME1, ODF2 in driver module 2 were associated with the formation of cellular protrusions; three genes, GNA13, GRHL2, TNFRSF10B in driver module 6 were related to the morphogenesis of fibroblast; and three genes, ATAD5, CLCF1, PIK3CA in driver module 8 were associated with the proliferation of B lymphocytes.
Verify new drivers of metastasis through in vitro wound healing assay
We further chose two genes, BCL2L11 and CDH9, from our driver modules to verify if they are drivers of metastasis. To the best of our knowledge, no experiments have been performed to verify the relation of these two genes to cancer metastasis. The protein encoded by BCL2L11 belongs to the BCL-2 protein family and has been shown to interact with other members of the BCL-2 protein family and to act as an apoptotic activator. BCL2L11 is also called BIM. Much research about this gene has been focused on the association of this gene with cell death or apoptosis36,37,38. BIM can abrogate the functions of all anti-apoptotic proteins in the Bcl-2 family, Mcl-1 and A139. There exists research that mentioned the association of BIM with metastasis or migration40, 41. However, we could not find any research that used wet-lab experiments to verify the association of BCL2L11/BIM with metastasis or migration directly. CDH9 belongs to the cadherin superfamily and the proteins encoded by this gene mediate calcium-dependent cell-cell adhesion, where it is believed that the reduction in cell adhesion correlates with tumor metastasis42, 43. However, the role of CDH9 on tumor invasion and metastasis is unclear, and we could not find any reference that associated CDH9 with metastasis, migration, or invasion.
In our results, the gene BCL2L11 is in driver module 9 and is either mutated or deleted among tumors in the corresponding T abn . The gene CDH9 is in driver module 17 and mainly mutated among tumors. Hence, we suppose that both genes are metastasis suppressors. In this project, we used in vitro wound healing assays to test the association of the genes BCL2L11 and CDH9 with cell migration. We found that the siRNA of either of these two genes significantly promoted cell migration of the HCC1937, HCC1806, and MDA-MB-231 cell lines (Fig. 4 and Fig. S2). Hence, both BCL2L11 and CDH9 are highly likely to be suppressors of cancer metastasis, which agrees with our hypothesis. To the best of our knowledge, we believe that we are the first to verify that gene BCL2L11 is a suppressor of cancer metastasis. We are also the first to report and verify that CDH9 is a suppressor of cancer metastasis.

Wound healing assays of HCC1937 cell line with siBCL2L11, siCDH9, and scramble. (A) Western blot showing the impact of siRNA treatment on the expression of gene BCL2L11. (B) Western blot showing the impact of siRNA treatment on the expression of gene CDH9. (C,D) Impact of siBCL2L11 or siCDH9 on the cell migration in cell line HCC1937.
Discussion
Many methods have been developed for finding cancer drivers, with each having its advantages and limitations. The frequency-based methods, which search for genes with higher frequency of mutations than those that may be found by chance, are simple to use and easily recognize important drivers with very high SGA frequency44. People have developed various ways to improve estimation of the background mutation rate, which is key to avoid false negatives and prevent false positives45, 46. However, current frequency-based methods are not intended to identify if drivers are on the same pathway.
Mutual exclusivity is a well-known property that is currently widely used to search for drivers47,48,49,50,51,52. However, even if SGA events perturbing the same pathway have a tendency to be mutually exclusive, this does not mean that SGA events that are mutually exclusive must perturb the same pathway. Furthermore, when the tumor size is large, using only mutual exclusivity to search for drivers can easily group mutually exclusive genes that come from different pathways together. One improvement is combining gene expression information, based on the hypothesis that genes in the same pathway should be co-expressed to a certain degree, while using mutual exclusivity to search for the pathways48. Very recently, Kim et al. provided a new algorithm to estimate statistical significance of mutual exclusivity relationships52. Their new method could find some new drivers, such as TTN (muscle protein Titin), a very long gene that was highly mutated but generally assumed to be artifacts. We also developed an improved method that took into account signal information51. However, as this method still uses mutual exclusivity as the major criteria and does not allow any overlap, it misses some important drivers that might have few overlaps with other important drivers.
In this paper, our new method sets the signal information as the major basis in searching for driver modules, but at the same time, we also need the solutions to have a certain degree of mutual exclusivity. In addition to the constraint on the co-expression of genes in each transcriptomic module, we also need those genes to be regulated by a common transcription factor, which increases the likelihood that genes in each transcriptomic module are regulated by a common signal. Hence, each driver module in our results should have very strong information with respect to a common signal. We supposed that genes in a driver module carry a common signal regulating the expression of genes in its corresponding transcriptomic module, and the SGA events of genes in the driver module will perturb the common signal and cause expression changes of genes in the transcriptomic module. We believe that it is the SGA events of genes in the driver modules that cause the expression changes of genes in the transcriptomic modules and not vice versa because SGA events are more stable when compared to the gene expression levels within cells. However, using the TCGA expression and mutation data, any computational method can only identify associations between the SGA events of genes in the driver and the expression changes of genes in the transcriptomic module. Real causal relations need to be verified by experiments. We did not perform these kinds of assays in this work. However, we did verify that the siRNA of two genes in our driver modules have a direct impact on metastatic activities, which proved the feasibility of our new framework.
Methods
The expression data of cell lines MDA-MB-231 and its subpopulations were obtained from Supplementary Data S1 in Minn’s paper2. Data on somatic mutation, copy number alteration, and gene expression from BRCA tumors and normal samples (Note: no mutation and copy number alteration data for normal samples), and data on gene expression from LUSC and LUAD tumors and the corresponding normal controls were downloaded from TCGA53,54,55. The transcription factor (TF) and target genes data were obtained from Osmanbeyoglu’s paper56, which was originally obtained from MSigDB57 by removing motifs that have similar sets of targets. The overall scheme of the method is shown in Fig. 5.

The overall scheme of the framework.
Search for gene transcriptomic modules
The parental MDA-MB-231 cell line and its subpopulations were generated through a series of selections in vivo, where the parental MDA-MB-231 cell line induced LM0 subpopulations; LM0 subpopulations induced LM1 subpopulations; and LM1 subpopulations induced LM2 subpopulations2. Each new generation had more lung metastatic activity than its previous generation. We wanted to catch expression changes of genes in each generation of subpopulations by comparing the expression data of a subpopulation with its direct parental cell lines, such as 1834 cells (LM0) vs. the parental MDA-MB-231 cells or 3481 cells (LM1) vs. 1834 cells (LM0). We considered a gene to be differentially expressed if its expression changed at least 2-fold. We made a union of all up-regulated genes and did the same to the down-regulated gene. Then we removed genes that were both up- and down-regulated in different cases. We obtained a total of 1136 up-regulated and 1154 down-regulated genes. These were the initial candidates for finding gene transcriptomic modules. We set a relatively relaxed threshold in this step to avoid excluding genes that were actually significantly expressed but might be excluded due to error. We use large population tumor data to refine the members in the gene transcriptomic modules in later steps.
We believed that if a gene that is related to breast metastasis to the lung is up-regulated in highly metastatic cell lines, then the gene should not be down-regulated in lung tumors. It is similar for genes that were down-regulated in metastatic cell lines. Therefore, we further removed genes from the initial candidate list if they were up-regulated in cell lines and down-regulated in R% of LUSC or R% of LUAD tumors. We also removed genes from the list if they were down-regulated in cell lines and up-regulated in R% of LUSC or R% of LUAD tumors (we chose R to be 20 in our application, people can adjust this number according to their data and requirement). To decide if a gene was up- or down-regulated in an LUSC tumor, we first found the distribution of the gene in normal controls for LUSC tumors, i.e. the mean μ and standard deviation δ of the expression values in normal controls. Then we used the μ, δ, and a p-value of 0.05 as threshold to decide if a gene was up-regulated or down-regulated in LUSC tumors. We used the same process to identify the up-/down-regulated genes in the LUAD and BRCA tumors. After this step, we had 740 up-regulated genes and 390 down-regulated genes.
We then searched for gene transcriptomic modules where we required that genes in each module were regulated by a common transcription factor (TF) and co-expressed in a large number of breast tumors. Hence, genes in each transcriptomic module are highly likely to be regulated by a common signal. For the up-regulated gene candidates and each TF, we made a bipartite graph such that nodes on one side were up-regulated gene candidates regulated by the TF and nodes on the other side were TCGA BRCA tumors. We added an edge between a gene and a tumor if the gene was differentially expressed in the tumor. Then we searched for a dense bipartite subgraph such that it has at least n genes and at least m tumors. Furthermore, each gene in the subgraph must be connected to at least r × m tumors in the subgraph and each tumor in the subgraph must connected to at least r × n genes in the subgraph. For the dense bipartite subgraph, we obtained a gene subset, which we called the gene transcriptomic module, and a tumor subset, which we denoted as T abn . We also generated another tumor subset called T nor , such that each tumor in T nor has at most t × n genes in the gene transcriptomic module that are up-regulated. Our objectives in this step were that: 1) genes in the gene transcriptomic module are regulated by a common signal; 2) tumors in T abn have SGAs to perturb this common signal; 3) tumors in T nor do not have SGAs to perturb this common signal. In this work, we set m, n, r, and t to be 50, 8, 0.75, and 0.5 respectively, these parameters can be adjusted according to need. We used a similar process for the down-regulated gene candidates.
Select candidates for driver modules
Before selecting candidates for driver modules, we did a preprocessing of SGA data. The purpose of preprocessing was to exclude invalid SGA events, i.e. SGA events that did not affect the functions of their gene products, proteins, or the expression levels of their corresponding genes. We first excluded all silent mutations. We then removed copy number amplifications/deletions that did not have significant expression increase/decrease for their corresponding genes.
We assumed that tumors in T abn have SGAs to perturb the common signal regulating a transcriptomic module while tumors in the corresponding T nor do not. Therefore, if a gene g is on the pathway carrying the common signal, then SGA events of gene g in BRCA tumors should occur mostly in tumors in T abn and only in very few tumors that are mistakenly put into T nor because of a computational error. Enrichment analysis, then, is a reasonable tool to use to measure the information between SGAs of a gene and the common signal regulating the transcriptomic module. We used hypergeometric distribution and T nor as control background to evaluate the SGA enrichment of each gene among tumors in T abn . We excluded any gene with an SGA enrichment p-value larger than 0.01. We chose a strict p-value threshold as we wanted to increase the likelihood that the included gene candidates are on the pathway carrying the common signal. Another reason is that we only searched for driver modules with small sizes, such as 6, or 7. A small candidate pool is sufficient and can also improve our final solution.
Search for driver modules
A reasonable hypothesis is that if a set of genes carries a common signal regulating a transcriptomic module, then the SGA events of all genes in this set should also be enriched among tumors in the corresponding T abn . Furthermore, SGA events of genes on the same pathway usually show mutual exclusivity, i.e. each tumor usually has at most one SGA event to perturb a common signal47,48,49,50,51. We developed a genetic algorithm to search for driver modules such that the SGA events of genes in each driver module are highly enriched and mutually exclusive among tumors in a corresponding T abn . We first used a greedy algorithm to generate 2 S/3 solutions of size k (sets of k SGA genes) and a random process to generate another S/3 solutions of size k (the first generation of chromosomes for the genetic algorithm). The greedy algorithm repeatedly chose genes with the current best weight and without the conflict of mutual exclusivity, where any genes that had been used before were removed from re-selection of new solutions with a probability of 0.5. To create the next generation of solutions, we emulated every pair of current solutions. First we randomly chose a position to make the crossover, which obtained two new solutions. Then, we randomly replaced two genes for one new solution and one gene for the other new solution. Finally, we chose the top S solutions from the current solutions and new solutions to make the new generation solutions. For the weight function of the solution, if the size of a solution was less than k or the mutually exclusive ratio of the solution was larger than the threshold r me , we set the weight of the solution to be 1; otherwise, we set the weight to be the SGA event enrichment p-value of the solution. The mutually exclusive ratio was defined as the ratio of number of tumors in T abn that are covered by the SGA events of genes in the solution to the total SGA event count of genes in the solution among tumors in T abn . We tested several settings and presented solutions with the setting of S = 20, k = 6, r me = 0.8. We ran the algorithm for 6,000 generations.
Cell migration test for new drivers
Cell lines and cell culture
HCC1937, MDA-MB-231 and HCC1806 SPORE cell lines were from the American Type Culture Collection (Manassas, VA, USA). MDA-MB-231 was cultured in DMEM supplemented with 10% fetal bovine serum (FBS). HCC1937 and HCC1806 were maintained in RPMI 1640 supplemented with 10% FBS. The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
Antibodies
The specific antibody against BCL2L11 and the secondary antibodies of horseradish peroxidase–conjugated goat anti-mouse and anti-rabbit were from Cell Signaling (Beverly, MA). The CDH9 antibody was from Thermo Fisher Scientific. The β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO).
SiRNA transfection
Smartpool: on-targetplus BCL2L11, CDH9 and control siRNA were purchased from Dharmacon.
Human BCL2L11 siRNA - SMARTpool, L-004383-00-0005
Human CDH9 siRNA - SMARTpool, L-013169-00-0005
Non-targeting Pool, D-001810-10-05
SiRNAs were transfected into HCC1937, MDA-MB-231, and HCC1806 cells using Lipofectamine RNAiMAX Reagent (Life technologies).
Immunoblotting
The HCC1937, MDA-MB-231 and HCC1806 cells were washed with PBS and collected in a boiling sample buffer 3 days after siRNA transfection. Cellular proteins were resolved by SDS–PAGE (12% acrylamide) and transferred to PVDF membranes (Merck Millipore Ltd). After blocking with 5% non-fat milk in PBST (PBS and 0.1% Tween 20), the membranes were incubated overnight in a cold room with the primary antibodies and for 1 h with the horseradish peroxidase–conjugated secondary antibody. Bound antibodies were detected using Clarity Western ECL substrate (Bio-Rad).
Migration assays
A wound healing assay was used to analyze the cell migration of transfectant cells. 4 × 105 of cells were seeded in 35 mm dishes. Cells were transfected with siRNAs 24 h later and cultured for 2 days to a confluence of 90%. The cells were then starved with 0.1% FBS overnight and scratched with a sterile 200-μl micropipette tip to form a straight wound. The cells were washed three times with PBS and cultured in normal medium for an additional 24 h. An Olympus IX83 microscope was used to measure the wound closure. Images were recorded at the time points of 0, 6, 12 and 24 h after wounding. The distances invaded by the cells at the front of the wound were measured from the control and the experimental samples. Cell migration was assayed by calculating the migrated distance and comparing with time 0.
Statistic test
The differences between the control and treated groups was evaluated using a t-test. Statistical significance was calculated based on three experiments. A p-value of < 0.05 was considered statistically significant.
References
Stewart, B. W., Wild, C. International Agency for Research on Cancer & World Health Organization. World cancer report 2014. (International Agency for Research on Cancer, WHO Press, 2014).
Minn, A. J. et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524, doi:10.1038/nature03799 (2005).
Tavazoie, S. F. et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152, doi:10.1038/nature06487 (2008).
McCorkle, J. R. et al. The metastasis suppressor NME1 regulates expression of genes linked to metastasis and patient outcome in melanoma and breast carcinoma. Cancer genomics & proteomics 11, 175–194 (2014).
Bos, P. D. et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009, doi:10.1038/nature08021 (2009).
Harrell, J. C. et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast cancer research and treatment 132, 523–535, doi:10.1007/s10549-011-1619-7 (2012).
Wagenblast, E. et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 520, 358–362, doi:10.1038/nature14403 (2015).
Julien, S. G. et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nature genetics 39, 338–346, doi:10.1038/ng1963 (2007).
Xue, B., Krishnamurthy, K., Allred, D. C. & Muthuswamy, S. K. Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nature cell biology 15, 189–200, doi:10.1038/ncb2663 (2013).
Tan, W. et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 470, 548–553, doi:10.1038/nature09707 (2011).
Chang, H. Y. et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proceedings of the National Academy of Sciences of the United States of America 102, 3738–3743, doi:10.1073/pnas.0409462102 (2005).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: Mission possible? Nature reviews. Drug discovery 13, 828–851, doi:10.1038/nrd4389 (2014).
Baker, N. M. & Der, C. J. Cancer: Drug for an ‘undruggable’ protein. Nature 497, 577–578, doi:10.1038/nature12248 (2013).
Rubio-Perez, C. et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer cell 27, 382–396, doi:10.1016/j.ccell.2015.02.007 (2015).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352, doi:10.1038/nature10983 (2012).
Garcia, J. M. et al. Prognostic value of LISCH7 mRNA in plasma and tumor of colon cancer patients. Clinical cancer research: an official journal of the American Association for Cancer Research 13, 6351–6358, doi:10.1158/1078-0432.CCR-07-0882 (2007).
Bayat, B. et al. Neutrophil transmigration mediated by the neutrophil-specific antigen CD177 is influenced by the endothelial S536N dimorphism of platelet endothelial cell adhesion molecule-1. Journal of immunology 184, 3889–3896, doi:10.4049/jimmunol.0903136 (2010).
Guzinska-Ustymowicz, K., Pryczynicz, A. & Kemona, A. PTP4A3 expression increases strongly in lymph node metastases from colorectal carcinoma. Anticancer research 29, 3913–3916 (2009).
Laurent, C. et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer research 71, 666–674, doi:10.1158/0008-5472.CAN-10-0605 (2011).
Zimmerman, M. W. et al. Protein-tyrosine phosphatase 4A3 (PTP4A3) promotes vascular endothelial growth factor signaling and enables endothelial cell motility. The Journal of biological chemistry 289, 5904–5913, doi:10.1074/jbc.M113.480038 (2014).
Cho, H. et al. The role of S100A14 in epithelial ovarian tumors. Oncotarget 5, 3482–3496, doi:10.18632/oncotarget.1947 (2014).
Zhao, F. T., Jia, Z. S., Yang, Q., Song, L. & Jiang, X. J. S100A14 promotes the growth and metastasis of hepatocellular carcinoma. Asian Pacific journal of cancer prevention: APJCP 14, 3831–3836 (2013).
Takahashi, A. et al. Ectopically expressed variant form of sperm mitochondria-associated cysteine-rich protein augments tumorigenicity of the stem cell population of lung adenocarcinoma cells. PloS one 8, e69095, doi:10.1371/journal.pone.0069095 (2013).
Muller, P. A. & Vousden, K. H. p53 mutations in cancer. Nature cell biology 15, 2–8, doi:10.1038/ncb2641 (2013).
Adorno, M. et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137, 87–98, doi:10.1016/j.cell.2009.01.039 (2009).
Weissmueller, S. et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 157, 382–394, doi:10.1016/j.cell.2014.01.066 (2014).
Funato, Y. et al. IRSp53/Eps8 complex is important for positive regulation of Rac and cancer cell motility/invasiveness. Cancer research 64, 5237–5244, doi:10.1158/0008-5472.CAN-04-0327 (2004).
Hoeppner, M. P. et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PloS one 9, e91172, doi:10.1371/journal.pone.0091172 (2014).
Kawamura, M. et al. The prognostic significance of eukaryotic elongation factor 1 alpha-2 in non-small cell lung cancer. Anticancer research 34, 651–658 (2014).
Xu, C., Hu, D. M. & Zhu, Q. eEF1A2 promotes cell migration, invasion and metastasis in pancreatic cancer by upregulating MMP-9 expression through Akt activation. Clinical & experimental metastasis 30, 933–944, doi:10.1007/s10585-013-9593-6 (2013).
da Rocha, A. A. et al. Hepatocyte growth factor-regulated tyrosine kinase substrate (HGS) and guanylate kinase 1 (GUK1) are differentially expressed in GH-secreting adenomas. Pituitary 9, 83–92, doi:10.1007/s11102-006-9277-1 (2006).
Hu, J. et al. TRPS1 expression promotes angiogenesis and affects VEGFA expression in breast cancer. Experimental biology and medicine 239, 423–429, doi:10.1177/1535370214523904 (2014).
Hong, J., Sun, J. & Huang, T. Increased expression of TRPS1 affects tumor progression and correlates with patients’ prognosis of colon cancer. BioMed research international 2013, 454085, doi:10.1155/2013/454085 (2013).
Lu, Y. C. et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. Journal of immunology 190, 6034–6042, doi:10.4049/jimmunol.1202830 (2013).
Hayashida, Y. et al. PPP1R3 gene (protein phosphatase 1) alterations in colorectal cancer and its relationship to metastasis. Oncology reports 13, 1223–1227 (2005).
Sharma, A. et al. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell death & disease 4, e628, doi:10.1038/cddis.2013.155 (2013).
Toh, W. H., Nam, S. Y. & Sabapathy, K. An essential role for p73 in regulating mitotic cell death. Cell death and differentiation 17, 787–800, doi:10.1038/cdd.2009.181 (2010).
Yan, J. et al. Deregulated MIR335 that targets MAPK1 is implicated in poor outcome of paediatric acute lymphoblastic leukaemia. British journal of haematology 163, 93–103, doi:10.1111/bjh.12489 (2013).
Zhang, L., Ming, L. & Yu, J. BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy 10, 207–217, doi:10.1016/j.drup.2007.08.002 (2007).
Woods, N. T., Yamaguchi, H., Lee, F. Y., Bhalla, K. N. & Wang, H. G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer research 67, 10744–10752, doi:10.1158/0008-5472.CAN-07-3148 (2007).
Liu, L. et al. Macrophage migration inhibitory factor (MIF) interacts with Bim and inhibits Bim-mediated apoptosis. Molecules and cells 26, 193–199 (2008).
Cavallaro, U. & Christofori, G. Cell adhesion in tumor invasion and metastasis: loss of the glue is not enough. Biochimica et biophysica acta 1552, 39–45 (2001).
Behrens, J. The role of cell adhesion molecules in cancer invasion and metastasis. Breast cancer research and treatment 24, 175–184 (1993).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075, doi:10.1038/nature07423 (2008).
Youn, A. & Simon, R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics 27, 175–181, doi:10.1093/bioinformatics/btq630 (2011).
Dees, N. D. et al. MuSiC: identifying mutational significance in cancer genomes. Genome research 22, 1589–1598, doi:10.1101/gr.134635.111 (2012).
Miller, C. A., Settle, S. H., Sulman, E. P., Aldape, K. D. & Milosavljevic, A. Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC medical genomics 4, 34, doi:10.1186/1755-8794-4-34 (2011).
Zhao, J., Zhang, S., Wu, L. Y. & Zhang, X. S. Efficient methods for identifying mutated driver pathways in cancer. Bioinformatics 28, 2940–2947, doi:10.1093/bioinformatics/bts564 (2012).
Ciriello, G., Cerami, E., Sander, C. & Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome research 22, 398–406, doi:10.1101/gr.125567.111 (2012).
Vandin, F., Upfal, E. & Raphael, B. J. De novo discovery of mutated driver pathways in cancer. Genome research 22, 375–385, doi:10.1101/gr.120477.111 (2012).
Lu, S. et al. Identifying Driver Genomic Alterations in Cancers by Searching Minimum-Weight, Mutually Exclusive Sets. PLoS Comput Biol 11, e1004257, doi:10.1371/journal.pcbi.1004257 (2015).
Kim, Y. A., Madan, S. & Przytycka, T. M. WeSME: uncovering mutual exclusivity of cancer drivers and beyond. Bioinformatics, doi:10.1093/bioinformatics/btw242 (2016).
Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70, doi:10.1038/nature11412 (2012).
Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525, doi:10.1038/nature11404 (2012).
Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, doi:10.1038/nature13385 (2014).
Osmanbeyoglu, H. U., Pelossof, R., Bromberg, J. F. & Leslie, C. S. Linking signaling pathways to transcriptional programs in breast cancer. Genome research 24, 1869–1880, doi:10.1101/gr.173039.114 (2014).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740, doi:10.1093/bioinformatics/btr260 (2011).
Acknowledgements
The research was supported by the following NIH grants: R00LM011673, R01LM 010144, R01LM 011155, and U54HG008540. The authors would like to thank Dr. Gonghong Yan for providing help with the wound healing assays.
Author information
Authors and Affiliations
Contributions
G.Y.: Collected data; designed and performed wound healing assay. V.C.: helped with the literature search. X.L.: gave advice for the study. S.L.: Conceived the study, collected data, performed the data mining analyses, and drafted the manuscript. All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, G., Chen, V., Lu, X. et al. A signal-based method for finding driver modules of breast cancer metastasis to the lung. Sci Rep 7, 10023 (2017). https://doi.org/10.1038/s41598-017-09951-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09951-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
