
Abstract
Signal transduction in sensory neurons of the mammalian vomeronasal organ (VNO) involves the opening of the canonical transient receptor potential channel Trpc2, a Ca2+-permeable cation channel that is activated by diacylglycerol and inhibited by Ca2+-calmodulin. There has been a long-standing debate about the extent to which the second messenger inositol 1,4,5-trisphosphate (InsP3) and type 3 InsP3 receptor (InsP3R3) are involved in the opening of Trpc2 channels and in sensory activation of the VNO. To address this question, we investigated VNO function of mice carrying a knockout mutation in the Itpr3 locus causing a loss of InsP3R3. We established a new method to monitor Ca2+ in the endoplasmic reticulum of vomeronasal sensory neurons (VSNs) by employing the GFP-aequorin protein sensor erGAP2. We also performed simultaneous InsP3 photorelease and Ca2+ monitoring experiments, and analysed Ca2+ dynamics, sensory currents, and action potential or field potential responses in InsP3R3-deficient VSNs. Disruption of Itpr3 abolished or minimized the Ca2+ transients evoked by photoactivated InsP3, but there was virtually no effect on sensory activation of VSNs. Therefore, InsP3R3 is dispensable for primary chemoelectrical transduction in mouse VNO. We conclude that InsP3R3 is not required for gating of Trpc2 in VSNs.
Similar content being viewed by others

Introduction
The mammalian olfactory system has evolved two major signaling systems for chemoelectrical transduction: one that depends on cyclic nucleotide-gated (CNG) channel activation and involving cAMP or cGMP signaling, respectively, and another that depends on transient receptor potential (TRP) channel activation, mainly involving the Trpc2 cation channel1, 2. Trpc2 is a central transduction element in sensory neurons of the mouse vomeronasal organ (VNO)3,4,5 which play important roles in the detection of socially-relevant molecular cues such as pheromones and kairomones6,7,8. The recent finding that Trpc2 is also expressed in sensory neurons of the main olfactory epithelium (MOE)9, 10 where it is required in type B cells for the detection of low environmental oxygen11 has sparked renewed interest in its function. Despite two decades of research, the second messenger signaling mechanisms underlying activation of Trpc2 and its corresponding sensory responses are still debated and there is presently no single agreed-upon mechanism for its activation7, 12,13,14,15. Moreover, the behavioral phenotypes of Trpc2 mutant mice are more complex than previously thought16. A complete understanding of Trpc2 signaling mechanisms will be required to fully appreciate the role of Trpc2 in chemical communication and social behaviors.
Here we assess the impact of the second messenger inositol 1,4,5-trisphosphate (InsP3) and the type 3 InsP3 receptor (InsP3R3, encoded by the gene Itpr3) on chemodetection and transduction of mouse vomeronasal sensory neurons (VSNs), and thus on the activation of Trpc2 channels. VSN chemodetection depends on several large G-protein-coupled receptor families (GPCRs), predominantly the vomeronasal type 1 and type 2 receptors, whose activation stimulates phospholipase C (PLC) leading to the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) into InsP3 and diacylglycerol (DAG)2, 6, 17, 18. Trpc2 was initially proposed to function as a Ca2+ store-activated, capacitative Ca2+ entry (CRAC) channel19,20,21 and several studies suggested a critical role for InsP3 in VSN chemotransduction22,23,24,25,26,27. More recently, a transduction model has been presented in which InsP3R3 is physically linked to Trpc2 and thereby would directly contribute to its activation28, 29. However, because Trpc2 channels are highly localized in VSN microvilli, at a considerable distance from Ca2+ stores, the validity of such a model has been questioned12, 30, 31. Our own studies concluded that Trpc2 is a DAG-activated, Ca2+-permeable cation channel that can be inhibited by Ca2+-calmodulin (Ca2+-CaM) and does neither require InsP3 nor Ca2+ stores for its activation in VSNs5, 7, 18, 32. Since then, there has been considerable evidence for the existence of additional, sequential or parallel VSN signaling mechanisms some of which require elevated intracellular Ca2+ levels and could thus depend on InsP3R3 activation and store-dependent Ca2+ mobilization16. These mechanisms include the activity of Ca2+-activated chloride channels33,34,35,36,37, several types of Ca2+-activated potassium channels38, as well as Ca2+-activated cation channels32, 39.
To resolve these problems and to further define the nature of the signal transduction mechanism in VSNs, we investigated VNO function in a gene-targeted mouse strain that carries a knockout mutation in the Itpr3 locus and thus lacks InsP3R340, 41. We used a recently developed method to monitor Ca2+ in the endoplasmic reticulum (ER) by employing a novel, genetically-encoded Ca2+ sensor protein specifically targeted to the ER (erGAP2)42, 43, and we performed simultaneous InsP3 photorelease and Ca2+ monitoring experiments. By applying a combination of state-of-the-art Ca2+ imaging and electrophysiological methods using wild-type and InsP3R3-deficient VSNs, we asked the following: (1) Are intracellular Ca2+ stores significantly mobilized during VSN sensory activation? (2) Does InsP3 induce Ca2+ signals in VSNs? (3) Is InsP3R3 necessary for stimulus-evoked Ca2+ signaling, the generation of sensory currents, and action potential or field potential responses in VSNs? With this approach, we provide compelling evidence that sensory activation of the mouse VNO is largely independent of InsP3R3, ruling out a crucial role for InsP3 signaling in the primary chemotransduction process of mouse VSNs.
Results
Monitoring [Ca2+]ER after VSN stimulation
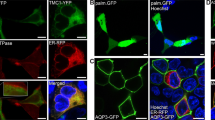
To investigate the role of InsP3 receptors (InsP3Rs) and intracellular Ca2+ stores in stimulus-evoked responses of mouse VSNs, we first monitored Ca2+ signals in the ER lumen of isolated VSNs. The ER is the main Ca2+-storage organelle of the cell, as the Ca2+ concentration in ER lumen ([Ca2+]ER) is commonly 104-fold higher than cytosolic Ca2+ ([Ca2+]C). Conventional Ca2+ indicators pose serious limitations for monitoring [Ca2+]ER in terms of Ca2+ affinity, organelle selectivity and/or dynamic range. To overcome these difficulties, we used a novel low-Ca2+ affinity sensor, erGAP2, that can be targeted specifically to the ER lumen42. erGAP2 is a fluorescent, genetically-encoded Ca2+ indicator based on the fusion of two jellyfish proteins, GFP and aequorin. erGAP2 has been optimized for measurements in high Ca2+ concentration environments (Kd for Ca2+ = 407 µM) due to two substitutions on the EF hands of the Ca2+-binding domain (Fig. 1a). Targeting to the ER is achieved through a combination of the calreticulin signal peptide and the ER retention sequence KDEL flanking the sensor (Fig. 1a). This reporter enables ratiometric imaging using two excitation peaks at 405 and 470 nm and single emission at 510 nm (Fig. 1b)43.

erGAP2 Ca2+ reporter enables imaging of [Ca2+]ER in VSNs. (a) Domain structure of the erGAP2 construct. Kz, Kozac sequence; CR, calreticulin signal sequence; GFP, green fluorescent protein; AEQ, aequorin; KDEL, ER retention signal. (b) erGAP2 is inserted into a p-HSV-1 amplicon expression vector, and HSV-1 viruses are prepared and used to infect freshly dissociated VSNs for 24 h. Ratiometric imaging is performed by double excitation at 405/470 nm and single emission at 510 nm. (c) Fluorescent image (470 nm excitation) showing expression of erGAP2 in VSNs. Freshly dissociated cells were infected with HSV-erGAP2 for 24 h. Scale bar, 50 μm. (d) erGAP2 ratiometric imaging in single VSNs. Stimulation with caffeine (Caf, 50 mM) but not with HMW or E mix caused a reduction in the F470/405 ratio. Scale bar, 10 μm. (e) Analysis of F470/405 ratio over time from the cell shown in d (arrow). (f) Simultaneous ER and cytosolic [Ca2+] recordings on VSNs infected with HSV-erGAP2 and loaded with fura-2. Imaging is performed by triple excitation with 340/380/470 nm light and single emission at 510 nm. [Ca2+]C is estimated by 340/380 ratio and [Ca2+]ER by 470 nm single excitation. Example of a cell showing cytosolic Ca2+ transients to the sulfated steroid E1050 (100 μM) and caffeine (50 mM), and a simultaneous decrease in [Ca2+]ER only in response to caffeine.
We expressed erGAP2 in freshly dissociated VSNs from C57BL/6 (wild-type, WT) mice using a herpes simplex virus type 1 (HSV-1) amplicon vector44 (Fig. 1b). A 24-h incubation period was sufficient to observe robust erGAP2 expression in infected VSNs with intact cell viability (Fig. 1c). We performed ratiometric [Ca2+]ER imaging on infected VSNs and analysed stimulus-induced fluorescence signals (Fig. 1d). We compared response patterns to several previously established VSN chemostimuli: a mix of sulfated steroids (E1100, E0893, E0588, and E1050, each at 100 µM), and the high molecular weight fraction of mouse urine (HMW; 1:300) which contains major urinary proteins (MUPs). These stimuli have been reported to activate VSNs of both the apical (sulfated steroids) and basal (HMW) layers of the VNO neuroepithelium45,46,47,48,49 and their responses are known to depend on Trpc245, 46. We also applied caffeine (Caf, 50 mM), a potent activator of the ubiquitous ryanodine receptors50, to induce Ca2+ release from the ER. We measured F470/F405 ratios in 368 cells that expressed detectable erGAP2 fluorescence. All cells showed a decrease of the F470/F405 ratio in response to a 60-s caffeine stimulus, indicative of a sharp decrease of [Ca2+]ER. In contrast, stimulation with the mix of sulfated steroids (E mix) or HMW did not induce major changes in F470/F405 ratio. Imaging of a representative cell and its F470/F405 ratio trace as a function of time is displayed in Fig. 1d and e, respectively. On average, both E mix and HMW induced no or only very minor reductions in F470/F405 ratio (−0.0117 ± 0.0033 and −0.0155 ± 0.0035, respectively) whereas caffeine produced a robust, 15-fold larger decrease in F470/F405 ratio (−0.175 ± 0.0071; Mann-Whitney test, n = 368, P = 1.6 × 10−85 and 5.6 × 10−83).
Simultaneous [Ca2+]ER and [Ca2+]C imaging in VSNs
To determine whether VSNs activated by HMW or steroid ligands show Ca2+ release from the ER, we performed simultaneous measurements of [Ca2+]ER and [Ca2+]C using combined imaging of erGAP2 and the cytosolic Ca2+ dye fura-2. For this approach, we imaged erGAP2-expressing cells loaded with fura-2 and performed triple illumination with 340 nm, 380 nm and 470 nm excitation light and single channel emission at 510 nm (Fig. 1f). Changes in [Ca2+]C were estimated by calculating the 340/380 nm ratio (R340/380), and [Ca2+]ER was estimated by using single F470 dynamics because of the partial F405 overlap with fura-2. We imaged a total of 599 erGAP2-expressing cells and applied 60-s stimuli of either caffeine, HMW, or the sulfated steroid estradiol-3,17-disulfate (E1050; 100 µM). We observed robust [Ca2+]C increases, measured as a rise in R340/380, in 15 cells responding to HMW and in 8 cells responding to E1050. These cells also showed [Ca2+]C increases in response to caffeine that were accompanied by simultaneous reductions in F470, indicative of massive Ca2+ release from the ER (Fig. 1f).
By contrast, changes in F470 in response to E1050 and HMW were nearly absent or not existent in these cells (Fig. 1f and below), indicating that [Ca2+]ER remains largely unaffected during activation with E1050 and HMW. On average, caffeine induced a 10-fold larger reduction of F470 (−3.6 ± 0.4848 a.u.; n = 23) than HMW (−0.3 ± 0.1393 a.u.; Mann-Whitney test, P = 7.1 × 10−7; n = 15) and E1050 (Mann-Whitney test, P = 7.7 × 10−7; n = 8). E1050 even induced a small F470 increase of 0.07 ± 0.3305 a.u., not significantly different from HMW (Mann-Whitney test, P = 0.38). Importantly, average caffeine-induced [Ca2+]C peaks were similar to those induced by HMW (0.4 ± 0.0982 vs. 0.56 ± 0.1288, respectively; Mann-Whitney test, P = 0.14) or even smaller than E1050 peaks (0.75 ± 01734; Mann-Whitney test, P = 0.04). Therefore, the main Ca2+ source contributing to HMW- and E1050-induced elevations of [Ca2+]C does not originate from ER stores.
Removal of extracellular Ca2+ abolishes ligand-induced VSN responses
To assess whether extracellular Ca2+ is necessary to generate elevations of [Ca2+]C following stimulation with HMW and E1050, we performed fura-2 imaging in freshly dissociated, non-infected VSNs45, 47, 51. We first established that multiple, 60-s stimulations with HMW or E1050 that were separated by 4-min interstimulus intervals are sufficient to produce robust and repeatable increases in [Ca2+]C (Fig. 2a and b). Next, we used a protocol in which a Ca2+-free extracellular medium containing 0.5 mM EGTA was applied during the second stimulation with either HMW or E1050. Ca2+ responses were completely absent during the second stimulus application in Ca2+-free medium for all cells that responded to any of the two stimuli during the first application (HMW, n = 3; E1050, n = 8; Fig. 2a and b). When extracellular Ca2+ was re-introduced, [Ca2+]C increases recovered during a third application (Fig. 2a and b). To verify that Ca2+ stores were not depleted under these conditions, we applied caffeine in Ca2+-free medium and observed robust Ca2+ transients (Fig. 2e). These responses were very similar to caffeine responses obtained in Ca2+-containing medium after a 4-min recovery period (Fig. 2e and f; Wilcoxon signed-rank test, P = 0.166) indicating that internal Ca2+ stores were not depleted. Therefore, extracellular Ca2+ and Ca2+ entry is required to generate cytosolic Ca2+ transients in response to activation with HMW and E1050, consistent with previous results47, 52.

Fura-2 Ca2+ imaging in freshly dissociated VSNs. Examples of cells imaged with fura-2 (R340/380 ratio images) and corresponding time courses showing cytosolic Ca2+ transients evoked by urine HMW fraction (1:300 dilution) (a) and E1050 (b). Cytosolic Ca2+ transients to both types of stimuli are abolished in the absence of extracellular Ca2+ (EGTA 0.5 mM), an effect that is reversible. (c) Depletion of [Ca2+]ER by a 20-min pre-incubation (only last 5 min shown) with the SERCA blocker CPA (30 µM) has no effect on E1050-induced cytosolic Ca2+ transients. (d) CPA treatment abolishes caffeine-evoked cytosolic Ca2+ signals. Recording example and group data showing that responses to 50 mM caffeine (Caf) are nearly abolished after a 20-min CPA (30 µM) incubation (only last 5 min shown), with a recovery after a 5-min washout (n = 14; Wilcoxon signed-rank test: P < 0.001). (e) Recording example and (f) group data of caffeine stimulation during and after incubation with or without 0.5 mM EGTA, showing that [Ca2+]ER is not depleted (n = 39; Wilcoxon signed-rank test, P = 0.166). (g) Recording example showing a CPA-mediated intracellular Ca2+ rise at the beginning of a 20-min incubation period (experimental design as in (d); n = 14). Scale bars, 10 µm. Median values and interquartile ranges are shown in box plots.
Depletion of intracellular Ca2+ stores does not affect ligand-induced Ca2+ responses
To further explore the impact of intracellular Ca2+ stores on stimulus-evoked VSN responses, we applied cyclopiazonic acid (CPA), a potent inhibitor of sarcoplasmic-endoplasmic reticulum Ca2+-ATPases (SERCAs), in order to deplete intracellular Ca2+ stores. VSNs were incubated with 30 µM CPA for 20 min in an extracellular medium containing Ca2+. We then performed fura-2 imaging during stimulation with HMW or E1050 in the presence of CPA. We observed robust increases of [Ca2+]C in 4/140 cells stimulated with HMW and in 7/250 cells stimulated with E1050 (Fig. 2c). Unlike other SERCA inhibitors such as thapsigargin, the effect of CPA can be reversed after washout. We used 60-s caffeine pulses (50 mM) to determine that a 5-min washing period after CPA incubation was sufficient to restore [Ca2+]ER and enable robust Ca2+ release (Fig. 2d). Caffeine-induced Ca2+ responses increased drastically after CPA washout (Wilcoxon signed-rank test, P < 0.001; Fig. 2d). We compared the cell-to-cell peak amplitudes of HMW and E1050 during CPA treatment (0.2788 ± 0.0579, HMW; 0.5153 ± 0.0745, E1050) vs. washout (0.3018 ± 0.0766, HMW; 0.4058 ± 0.0611, E1050). Although E1050 responses tended to be slightly larger in the presence of CPA, we found no significant differences between the two conditions (Wilcoxon signed-rank test, P = 0.58, HMW; P = 0.07, E1050). To determine the effect of CPA application on intracellular Ca2+, we recorded the first 5 min after CPA incubation and observed a slow and transient Ca2+ increase (n = 14; Fig. 2g), consistent with sustained, store-dependent Ca2+ release.
Together, these results indicate that depletion of intracellular Ca2+ stores does not prevent the generation of cytosolic Ca2+ transients following activation with HMW and E1050. Thus, intracellular Ca2+ stores are not critical for VSN cytosolic Ca2+ transients to these sensory stimuli and extracellular Ca2+ must be the main source of cytosolic Ca2+ for these Ca2+ responses.
InsP3R3 is expressed in mouse VSNs
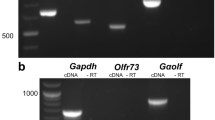
InsP3R3 immunoreactivity has been reported in rat VSNs28 and transcripts of all three InsP3R isoforms were identified in whole mouse VNO tissue53. To obtain independent evidence for the presence and function of InsP3Rs in mouse VSNs, we investigated the expression of InsP3R3 in VNO cryosections of adult WT mice using specific anti-InsP3R3 antibodies. We observed InsP3R3 immunoreactivity in the entire vomeronasal neuroepithelium, especially in VSN somata, dendrites and possibly in dendritic endings, whereas immunoreactivity was weaker in supporting cell somata and non-sensory parts of the VNO (Fig. 3a). Importantly, this labeling was absent in InsP3R3−/− VNO sections, confirming antibody specificity (Fig. 3a). Next, we performed RT-PCR on cDNA libraries prepared from single-cell RNA. We dissociated VNOs from OMP-GFP mice54 to obtain single, isolated and fluorescent VSNs that were individually collected using a microcapillary pipette. In OMP-GFP mice, GFP serves as a marker for mature (olfactory marker protein-expressing) VSNs. We picked 10 GFP+ cells and 2 GFP– cells. We prepared RNA from each cell, generated single-cell cDNA libraries10, 44, and assessed for gene expression by PCR with gene-specific primers. We amplified Omp PCR products in 8/10 GFP+ cells (Fig. 3b), indicative of a 80% success rate for this method. We further screened for Itpr3 gene expression in these 8 GFP+ cells and obtained Itpr3 PCR products in 4 cell samples (Fig. 3c), demonstrating that InsP3R3 is indeed expressed in at least a fraction of VSNs. Full-length gels are presented in Supplementary Fig. S1. We found no detectable expression of Itpr1 (InsP3R1) or Itpr2 (InsP3R2) genes in all samples, except for one of the GFP− control cells that was positive for Itpr2 (not shown). For comparison, we also sampled 18 cells from InsP3R3−/− VNO (which are GFP−), 8 of which were positive for the Omp PCR (Fig. 3b). We did not amplify Itpr3 PCR products from any of these cells (Fig. 3c). We found Itpr2 expression in 5 of the cells that were negative for Omp (not shown). These results indicate that InsP3R3 is the predominant isoform expressed in VSNs, whereas InsP3R1 and InsP3R2 are not co-expressed in these cells. InsP3R2 is likely expressed by supporting and other non-sensory cell types. There was no evidence for upregulation of Itpr1 or Itpr2 in InsP3R3−/− VSNs.

InsP3R3 is expressed in VSNs and is required for Ca2+ responses to photoreleased InsP3. (a) Immunolabeling of VNO sections with anti-InsP3R3 antibody (green) and the nuclear dye Hoechst 33342 (blue) in VNOs from WT and InsP3R3−/− mice. Scale bars, 100 µm. (b) Single-cell RT-PCR for olfactory marker protein (Omp) and (c) Itpr3 genes in single VSNs from OMP-GFP or InsP3R3−/− mice. Bands positive for Omp were amplified in 8/10 GFP + cells (OMP-GFP) and in 8/18 non-labeled cells (InsP3R3−/−). From these Omp + cells, Itpr3 RT-PCR product was amplified in 4 cells but in none of the InsP3R3−/− cells. Full-length gels are presented in Supplementary Fig. S1. (d) Photolysis of caged InsP3 in isolated VSNs. Fluo-4 confocal images of VSNs loaded with caged InsP3. Both InsP3R3+/− (top, arrowhead) and InsP3R3−/− (bottom) cells showed responses to HMW. Outlined regions of cells activated by HMW (dotted areas) were stimulated with UV light causing a Ca2+ increase in the InsP3R3+/− but not in the InsP3R3−/− cell. Scale bar, 10 µm. (e) Examples of Ca2+ responses in InsP3R3+/− (left) and InsP3R3−/− (right) cells stimulated with HMW fraction (1:100), E1050 (100 µM) and UV light. (f) Photoactivation efficiency of InsP3R3+/− cells loaded with caged InsP3 was between 30 and 40%. (g) Comparison of Ca2+ peak amplitudes (ΔF/F0) to photoreleased InsP3 in InsP3R3+/− (grey box, open dots) and InsP3R3−/− cells (black box, black dots). All cells responded at least once to a chemostimulus (HMW or E1050). Mann-Whitney test, ***P < 0.001. (h) Percentage of InsP3R3+/− and InsP3R3−/− cells responding to HMW or E1050. HMW: 9.28% (48/517 cells, InsP3R3+/−) and 13.79% (40/290 cells, InsP3R3−/−). E1050: 10.24% (38/371 cells, InsP3R3+/−) and 12.36% (32/259 cells, InsP3R3−/−). (i) Ca2+ peak amplitudes (ΔF/F0) of cells that were stimulated successively with a given chemostimulus (HMW or E1050), a UV stimulus, and a second chemostimulus. Note that UV-evoked Ca2+ responses were absent in InsP3R3−/− cells. Kruskal-Wallis test, P < 0.0001; Mann-Whitney test, **P < 0.01; ***P < 0.001. Cell numbers indicated above each bar.
Photolysis of caged InsP3 shows that InsP3R3 is required for Ca2+ release
To determine whether InsP3 is capable to evoke Ca2+ elevations in VSNs and, if so, whether this signal would require InsP3R3, we performed flash photolysis experiments with caged InsP3 in InsP3R3+/− and InsP3R3−/− VSNs (Fig. 3d–h). Freshly dissociated VSNs were co-loaded with a photoactivatable and membrane-permeant caged InsP3 propionyloxymethyl ester55 as well as the Ca2+ indicator fluo-4 (see Methods for details). We used confocal laser-scanning microscopy to monitor changes in cytosolic Ca2+ and to locally deliver ultraviolet light (UV, 355 nm) stimulation which, in turn, caused photoliberation of caged InsP3 and thereby released the active InsP3 molecule. Cells exhibiting Ca2+ transients in response to either E1050 or HMW were identified as functional VSNs and the regions circumscribed by each cell were outlined and stimulated with UV light (Fig. 3d). A typical UV stimulation for a single cell with a diameter of 10 µm consisted of 10 individual scans with a pixel dwell time of 1.54 µs, resulting in a total UV exposure time of 1.76 ms. With these conditions, InsP3R3+/− VSNs showed striking Ca2+ transients in response to UV stimulation (Fig. 3d and e) demonstrating that InsP3 is indeed capable to induce increases of cytoplasmic Ca2+ in these cells. Consistent with previous studies using InsP3 photorelease in other cell types56, uncaging efficiency in InsP3R3+/− VSNs was between 30% and 40%: 14/38 (37%) of E1050-activated cells and 15/48 (31%) of HMW-activated cells showed Ca2+ transients after UV exposure (Fig. 3f and g). By contrast, InsP3R3−/− VSNs, which showed normal response rates for HMW and E1050 (Fig. 3h), were largely insensitive to InsP3 photorelease (Fig. 3d,e,g and i), indicating that InsP3R3 is required for InsP3-induced Ca2+ release in these VSNs. For example, from a total of 32 cells responding to E1050 and 43 additional cells responding to HMW, we observed only a single cell in each group that could be activated by UV light (Fig. 3f and g). Furthermore, when we analyzed only those cells (n = 16 InsP3R3+/− VSNs and n = 13 InsP3R3−/− VSNs, respectively) that were stimulated successively with a chemostimulus (HMW or E1050), UV light, and a second chemostimulus (Fig. 3i), UV-evoked responses were completely absent in InsP3R3−/− versus InsP3R3+/− VSNs (Mann-Whitney, P < 0.001). Together, these results show that photolysis of InsP3 can induce transient Ca2+ elevations in VSNs heterozygous for the InsP3R3 mutation but that these responses are absent or strongly reduced in InsP3R3-deficient VSNs. Therefore, InsP3R3 is the predominant isoform mediating InsP3-evoked Ca2+ release in these sensory neurons.
Stimulus-induced Ca2+ signaling in InsP3R3-deficient VSNs
We performed a systematic analysis of stimulus-evoked Ca2+ responses in InsP3R3−/− VSNs. We first monitored Ca2+ signals in the ER lumen of InsP3R3−/− VSNs using ratiometric erGAP2 imaging. We analysed [Ca2+]ER in response to caffeine, HMW or E mix in 67 dissociated VSNs expressing erGAP2 via HSV-1 infection. Caffeine induced strong reductions of F470/F405 ratio, consistent with a decrease of [Ca2+]ER (Fig. 4a). Average values of these responses (−0.193) were not significantly different from WT VSNs (Mann-Whitney test, P = 0.103; Fig. 4b). Thus, ER Ca2+ loading and caffeine-induced Ca2+ release seem to be intact in these InsP3R3−/− cells. E mix and HMW induced modest changes in F470/F405 ratio (−0.0126 and −0.015 a.u., respectively), not significantly different from WT VSNs (Kruskal-Wallis test, P = 0.48; Fig. 4b).

Ligand-evoked Ca2+ signals in InsP3R3−/− VSNs. (a) F470/405 ratio of an erGAP2-expressing InsP3R3−/− VSN stimulated with HMW (1:300), E mix, caffeine (50 mM) and high K+ (100 mM). (b) [Ca2+]ER peak amplitudes in WT and InsP3R3−/− VSNs with erGAP2 imaging (n = 368 and 67 cells, respectively. n.s., not significant; Mann-Whitney test, P = 0.103, caffeine; Kruskal-Wallis test, P = 0.48; HMW and E mix). (c) Simultaneous erGAP2 and fura-2 Ca2+ imaging of an InsP3R3−/− VSN stimulated with HMW (1:300), E1050 (100 µM), caffeine, and high K+. (d) Percentage of activated cells responding to either HMW or E1050 (fura-2 imaging) in WT (n = 599) or InsP3R3−/− (n = 171) VSNs. 15 and 6 cells responded to E1050 and 8 and 7 cells responded to HMW for WT (n = 8) and InsP3R3−/− VSNs (n = 6), respectively. n.s. not significant, Mann-Whitney test, P = 0.27 (HMW) and 0.74 (E1050). (e) [Ca2+]ER peak amplitudes of the cells indicated in (d). n.s., not significant, Mann-Whitney test, P = 0.91 (HMW) and 0.12 (E1050). (f) [Ca2+]ER changes measured with erGAP2 at 470 nm on the same cells. n.s., not significant, Mann-Whitney test, P = 0.83 (caffeine), P = 0.3 (HMW), p = 0.99 (E1050). (g) Fura-2-imaged InsP3R3−/− VSN activated by HMW. Response to HMW is abolished after removal of extracellular Ca2+. (h) Analysis of [Ca2+]C peak amplitudes in WT and InsP3R3−/− VSNs to HMW and E1050. n.s., not significant, Mann-Whitney test, P = 0.47, HMW (n = 3 and 8); P = 0.76, E1050 (n = 8 and 3); HMW vs. EGTA-HMW: *P = 8.3 × 10−4 (InsP3R3−/−). (i) E1050-evoked [Ca2+]C responses of an InsP3R3−/− VSN in presence or absence of CPA. (j) [Ca2+]C peak amplitudes of WT and InsP3R3−/− VSNs elicited by HMW (n = 4 and 3) and E1050 (n = 7 and 5). n.s., not significant, Wilcoxon signed-rank test, HMW vs. CPA-HMW: P = 0.58 (WT) and P = 0.42 (InsP3R3−/−); E1050 vs. CPA-E1050: P = 0.08 (WT) and P = 0.78 (InsP3R3−/−).
Simultaneous [Ca2+]ER and [Ca2+]C measurements using erGAP2 and fura-2 imaging in InsP3R3−/− cells (Fig. 4c) produced comparable results: we imaged 171 erGAP2-expressing cells loaded with fura-2 and observed responses in 6 cells to HMW and in 7 cells to E1050 (5 separate experiments). The cell activation rate per experiment for both stimuli ranged between 4.6–6.5%, but was not significantly different from WT controls (2–3.2%; Mann-Whitney test, P = 0.27–0.74; Fig. 4d). The average amplitude of [Ca2+]C increases in response to HMW and E1050 (0.35–0.41 ratio values) tended to be slightly lower in InsP3R3−/− vs. WT cells (0.56–0.75), but this difference was not statistically significant (Mann-Whitney test, P = 0.91–0.12; Fig. 4e). InsP3R3−/− cells showed [Ca2+]C increases in response to caffeine similar to those observed in WT cells (0.24–0.4; Mann-Whitney test, P = 0.83), indicating that increases in [Ca2+]C are not different in InsP3R3−/− cells (Fig. 4e). These cells showed large [Ca2+]ER (F470) reductions for caffeine (−3.76 a.u.), but not for E1050 (+0.09 a.u.) and HMW (−0.33 a.u.) stimuli (Fig. 4f). In all cases, average changes in F470 did not differ significantly in InsP3R3−/− vs. WT cells (Mann-Whitney test, P = 0.3–0.99; Fig. 4f) indicating that genetic ablation of InsP3R3 has little or no effect on [Ca2+]ER following stimulation with HMW and E1050. Removal of extracellular Ca2+ also abolished HMW and E1050-induced [Ca2+]C increases (Fig. 4g) in a similar way for both InsP3R3−/− and WT cells (Fig. 4h). Similarly, ER Ca2+ depletion with CPA had no effect on [Ca2+]C amplitudes induced by HMW and E1050 (Wilcoxon signed-rank test, HMW: P = 0.42, n = 3; E1050: P = 0.79, n = 5) in InsP3R3−/− and WT cells (Fig. 4i and j). Although not significant, [Ca2+]C amplitudes tended to be somewhat higher in InsP3R3−/− VSNs. Interestingly, this tendency is reversed in erGAP2-infected cells after 24 h (Fig. 4e). The reasons for this are not known but argue further against a major contribution of InsP3R3-mediated Ca2+ release to the cytosolic signal under these conditions. We also note that the number of responding cells cannot be compared directly in freshly dissociated versus erGAP2-infected cells. Together, these results indicate that InsP3R3 and Ca2+ release from the intracellular stores are not critically required for ligand-evoked cytosolic Ca2+ transients and sensory activation of mouse VSNs.
InsP3R3 is not required for activation of VSN sensory currents
To strengthen our results obtained with Ca2+ imaging, we carried out whole-cell patch-clamp recordings in voltage-clamped VSNs5, 57. These experiments used acute VNO tissue slices52, 58, 59 in which the cellular VSN architecture is preserved, enabling recordings from optically identified VSNs located in apical or basal layers of the sensory epithelium. VSN sensory currents depend, in full or in part, on the activation of Trpc2 cation channels3,4,5. Trpc2 can form a protein-protein interaction complex with InsP3R3 and it has been proposed that this interaction contributes to the electrical response of VSNs to chemostimulation28, 29. If so, activation of VSN sensory currents should be severely disrupted in InsP3R3−/− mice. To assess this, we recorded sensory currents in InsP3R3+/− vs. InsP3R3−/− VSNs (Fig. 5). Cells were initially stimulated with a mixture of diluted urine from male and female mice (DU, 1:100), a rich source of natural pheromones. This stimulus induced small but reliable inward currents in 19/51 (37%) of InsP3R3+/− VSNs and in 20/48 (42%) of InsP3R3−/− VSNs, consistent with previous reports of WT VSNs33, 35, 38. The properties of urine-evoked sensory currents were analysed in more detail using 5-s urine applications, a holding potential of −70 mV, and a KCl-based intracellular solution (Fig. 5a–d). Under these conditions, we found no significant difference in peak amplitude, time-to-peak, or decay time constants between the two genotypes (Fig. 5a–d and Table 1).

Urine-evoked VSN sensory currents do not require InsP3R3. (a) Diluted urine (DU, 1:100) elicited sensory currents in both InsP3R3+/− (left) and InsP3R3−/− (right) VSNs. Holding potential, −70 mV. (b) Analysis of peak amplitudes of InsP3R3+/− (n = 13) vs. InsP3R3−/− (n = 19) VSNs. (c) Analysis of time-to-peak values of InsP3R3+/− (n = 13) vs. InsP3R3−/− (n = 18) VSNs. (d) Comparison of decay time constants (τ) of DU-evoked currents (n = 11 and 17, respectively). (e) DU-evoked currents in InsP3R3+/− vs. InsP3R3−/− VSNs recorded at a holding potential of −100 mV (K+ reversal potential). (f) Analysis of peak amplitudes at −100 mV (n = 23 and 25, respectively). (g) Time-to-peak values for all DU responses at −100 mV (n = 23 and 24, respectively). (h) Decay time constants (τ) of DU-evoked currents at −100 mV. (i) A 5-min preincubation with thapsigargin (TG, 1–5 µM) has no effect on the amplitude of DU-evoked currents (holding potential, −100 mV) in InsP3R3+/− (n = 9) or InsP3R3−/− (n = 10) VSNs. Median values and interquartile ranges are shown in box plots.
Ca2+-activated K+ channels can contribute to urine-evoked currents in VSNs38. To determine whether large K+ currents would mask the contribution of InsP3R3 at resting membrane potential, we recorded urine-evoked currents at a holding potential of −100 mV, close to the K+ reversal potential (Fig. 5e–h). This resulted in considerably larger peak amplitudes of urine-evoked currents, but again there was no significant difference between the two genotypes in peak amplitude, time-to-peak, or decay time constant (Fig. 5e–h and Table 1). Furthermore, urine-evoked sensory currents (holding potential, −100 mV) were fully preserved when the tissue was pretreated for 5 min with the SERCA inhibitor thapsigargin (1–5 µM) to deplete intracellular Ca2+ stores irreversibly, and there was no significant difference between InsP3R3+/− vs. InsP3R3−/− VSNs in their peak amplitudes (Fig. 5i and Table 1). These results indicate that Ca2+ release from intracellular stores is not essential for urine-evoked VSN sensory currents.
We also analysed sensory currents evoked by the sulfated steroid E1050 (Fig. 6 and Table 1). This stimulus routinely evoked large inward currents with peak amplitudes of several hundred picoamperes, even at a concentration of only 10 nM (Fig. 6a and b). However, we found no significant difference in peak amplitude, time-to-peak and decay time constant between the two genotypes (Fig. 6b–d and Table 1). Furthermore, current-voltage curves obtained at the peak of E1050-evoked responses (CsCl-based intracellular solution) where very similar between the two genotypes and showed a reversal potential close to 0 mV (InsP3R3+/−: Vrev = 4.3 ± 2 mV; InsP3R3−/−: 1.4 ± 1.2 mV, n = 3 each) (Fig. 6e). Therefore, InsP3R3 is not essential for the activation of VSN sensory currents.

No requirement of InsP3R3 for β-estradiol 3,17-disulfate (E1050)-evoked VSN sensory currents. (a) E1050 (10 nM) induced large inward currents in both InsP3R3+/− and InsP3R3−/− VSNs. Holding potential, −70 mV. (b) Analysis of E1050-evoked peak amplitudes of InsP3R3+/− (n = 9) and InsP3R3−/− (n = 6) VSNs. (c) Time-to-peak values of E1050-evoked current of InsP3R3+/− (n = 9) and InsP3R3−/− (n = 5) VSNs. (d) Decay time constants (τ, single exponential fits) of E1050-evoked currents responses of InsP3R3+/− (n = 9) and InsP3R3−/− (n = 5) VSNs. (e) Examples of current-voltage (I-V) curves, measured with voltage ramps (duration: 60 ms, slope: −3.3 mV/ms) elicited at the peak of E1050-evoked currents in VSNs of the two different genotypes. Control I-V curves obtained without E1050 stimulation are shown for comparison. Median values and interquartile ranges are shown in box plots.
Normal action potential responses in InsP3R3−/− VSNs
Our results of Figs 5 and 6 were further supported by extracellular loose-patch recordings from VSNs in tissue slices measuring stimulus-evoked action potential sequences (Fig. 7a–f). These represent VSN output signals that are ultimately transmitted to the olfactory forebrain. Using 1-s pulses of diluted urine as stimulus, action potential responses could be readily evoked and repeated multiple times in both InsP3R3+/− and InsP3R3−/− VSNs (Fig. 7a and b). Post-stimulus time histograms (PSTHs) obtained from group data of such recordings revealed no significant difference between the two genotypes (P = 0.07–0.97) (Fig. 7c and d). The same basic result was also observed for action potential sequences in response to E1050 (10 nM, P = 0.06–1) (Fig. 7e and f).

No requirement of InsP3R3 for VSN action potential responses or Ca2+-CaM-dependent adaptation. (a,b) Repeated DU pulses (1 s duration, 20 s interval) evoke action potential responses in InsP3R3+/− and InsP3R3−/− VSNs. (c,d) Group data showing PSTH analyses of DU-evoked action potential responses from InsP3R3+/− (n = 16) and InsP3R3−/− (n = 18) VSNs. (e,f) Group data showing PSTH analyses of action potential responses to E1050 (10 nM) from InsP3R3+/− (n = 5) and InsP3R3−/− (n = 7) VSNs. (g) Examples of VNO field potential responses to 6-s pulses of isobutylamine (0.1 µM) or SYFPEITHI (1 nM) in InsP3R3+/− and InsP3R3−/− mice. (h–j) Analyses of field potential peak responses (unpaired t-test: isobutylamine: t(12) = 0.76, P = 0.46; SYFPEITHI: t(5) = 0.76, P = 0.48) (h), ratio between plateau and peak (unpaired t-test: isobutylamine: t(12) = 2.07, P = 0.06; SYFPEITHI: t(5) = 0.09, P = 0.93) (i), and time constant of adaptation onset (single exponential fit, unpaired t-test: isobutylamine: t(8) = 1.08, P = 0.31; SYFPEITHI: t(5) = 0.14, P = 0.89) (j) for the two ligands and the two genotypes as indicated. Results are based on 4–8 independent recordings from 3–5 mice for each ligand and genotype.
InsP3R3 is not required for Ca2+-calmodulin-dependent VNO adaptation
The N-terminus of Trpc2 binds CaM in a Ca2+-dependent manner60. We showed previously that Ca2+-CaM feedback mediates sensory adaptation and inhibits DAG-activated Trpc2 currents in VSNs32. For other TRPC channels such as Trpc3, an activation model has been proposed in which an InsP3R binds to a site that partially overlaps with the Ca2+-CaM site, thereby displacing CaM and thus causing channel activation61. We tested whether activation and Ca2+-CaM-dependent VSN adaptation is altered in the absence of InsP3R3 by performing extracellular field potential recordings from the surface of the sensory epithelium using an intact VNO wholemount preparation32, 52, 58, 62, 63 (Fig. 7g–j). We recorded isobutylamine-evoked potentials (0.1 µM), which depend on type 1 vomeronasal receptors62, and potentials to SYFPEITHI (1 nM), a major histocompatibility complex (MHC) binding peptide58. SYFPEITHI-evoked responses require type 2 vomeronasal receptors and the G protein Gαo47, 59 but persist in Trpc2-deficient mice64. We found that both ligands evoked robust, phasic-tonic field potentials in InsP3R3+/− and InsP3R3−/− VNO that underwent time-dependent desensitization with prolonged stimulation (Fig. 7g). We analysed field potential peak amplitudes, the ratio between plateau and peak as a measure of the extent of adaptation, and the adaptation onset time constant32 (Fig. 7h–j). Despite a slight trend for lower plateau-peak ratios with isobutylamine, there was no significant difference between the two genotypes for both stimuli in any of these parameters (unpaired t-test: P = 0.09–0.93). These results indicate that InsP3R3 is neither required for the activation of ligand-evoked field potentials in the VNO nor for their Ca2+-CaM-dependent desensitization.
Discussion
The role of the second messenger InsP3 in vertebrate olfaction has been discussed controversially for almost 30 years, but no gene deletion studies have been performed to address this critical problem in any of the olfactory subsystems for any vertebrate species. To overcome this limitation, we undertook a series of investigations employing InsP3R3-deficient mice. We focused on sensory neurons of the VNO because these have been shown previously to express InsP3R3 but not InsP3R1 or InsP3R228. In agreement with these results, our knockout-controlled immunolabeling and single-cell RT-PCR experiments confirmed the presence of InsP3R3 RNA and protein in mouse VSNs but found no evidence for other InsP3Rs in these cells. We used a confocal, laser-scanning-controlled approach to photorelease InsP3 and simultaneously monitor cytosolic Ca2+ in isolated VSNs. These experiments demonstrated for the first time that InsP3 evokes transient, intracellular Ca2+ rises in VSNs and that InsP3R3 is functionally required for this effect, providing a robust foundation for investigating the role of InsP3R3 in sensory activation of the mouse VNO. We applied a wide range of vomeronasal chemostimuli and used multiple recording techniques and VNO preparations to address a potential role of InsP3R3. Our approach included Ca2+ imaging as well as voltage-clamp, loose-patch, and field-potential recordings in isolated VSNs, acute VNO tissue slices, and a VNO wholemount preparation. All of these experiments led to the same basic result, namely that InsP3R3 is dispensable for sensory activation of the VNO and, therefore, does not contribute crucially to the primary, chemoelectrical transduction process of VSNs. We also demonstrated that InsP3R3 is not required for Ca2+-CaM-dependent VSN adaptation. As a whole, these experiments call for a revision of current schemes suggesting that InsP3R3 has a critical function in VSN sensory activation. We note that our experiments do not rule out a potential role for InsP3 signaling or InsP3R3 function in secondary or modulatory signaling events of mammalian VSNs, nor do they rule out distinct VSN signaling mechanisms in lower vertebrates65. We also cannot exclude that primary signaling of noncanonical VSNs, such as those expressing formyl peptide receptor66 or odorant receptor genes67, would require InsP3R3. Future experiments will be needed to address these questions.
Our results have important implications for the gating mechanism of Trpc2 cation channels and, hence, for the TRP channel field in general. Most importantly, our results rule out any gating model in which a physical link of InsP3R3 with Trpc2, either directly or indirectly, mediates the opening of the Trpc2 channel in VSNs. Consequently, these results also rule out any model in which the displacement of inhibitory Ca2+-CaM by InsP3R3 causes Trpc2 activation. The present results are fully consistent with and strengthen further our previous findings that Trpc2 is a DAG-activated, Ca2+-permeable cation channel that can be inhibited by Ca2+-CaM and does neither require InsP3 nor Ca2+ release from intracellular stores for its activation in VSNs5, 7, 18, 32. These experiments leave the activation of Trpc2 by DAG, possibly in conjunction with PIP2, as the most plausible VSN primary transduction mechanism. It remains to be seen whether this model applies also to other Trpc2-expressing cells outside the VNO where cells likely exhibit different cellular architecture or express other Trpc2 splice variants and interacting proteins. It has been suggested that modes of Trpc2 activation are cell-specific and require different interactions and activators in different cell types14. Our results should also be of general interest to the activation models of other DAG-sensitive TRP channels such as TRPC3, TRPC6, and TRPC768, 69.
There is considerable evidence that Ca2+-activated chloride channels in VSNs, for which TMEM16A/anoctamin1 is an essential component37, serve to amplify the sensory response and cause further VSN depolarization because of elevated intracellular Cl− concentrations33, 35, 70, 71. In dorsal root ganglia, activation of anoctamin1 by localized Ca2+ signals requires coupling with the type 1 InsP3 receptor72. Similarly, it has been proposed that activation of VSN chloride channels is triggered by Ca2+ release from intracellular stores35. Our electrophysiological recordings in InsP3R3-deficient VSNs found no evidence for a significant reduction of the size of sensory responses. This argues strongly that chloride channel activation in VSN sensory responses is not mediated by InsP3R3-dependent Ca2+ release.
Several interesting results emerge from our study with respect to the role of sulfated steroids as VSN ligands. Initially, these molecules were tested at relatively high concentrations, at 100 or 200 µM, using extracellular spike recordings46 or Ca2+ imaging48, but the primary sensory currents evoked by these ligands have not been analyzed previously. Using patch-clamp recordings under voltage-clamp, we found that VSNs in VNO slices generate surprisingly large currents at only 10 nM of E1050 (Fig. 6) and also evoke action potentials at this concentration (Fig. 7). These low thresholds are supported by a previous study identifying V1rj receptors that selectively respond to sulfated steroids (including E1050) at 10 nM73. Our response rates for E1050 are also consistent with a report that found approximately 0.2–5.5% of the VSNs to be responsive to individual sulfated steroid components including E105074. We cannot yet determine whether the differences in amplitude or response kinetics reflect specific differences in the underlying mechanisms of the sensory currents but, importantly, there was no obvious effect of the InsP3R3 deletion on responses to E1050, neither at low nor at high stimulus conditions.
Our experiments demonstrate for the first time the suitability of a novel, genetically-encoded Ca2+ sensor protein, erGAP2, for monitoring Ca2+ dynamics in the ER of mammalian VSNs. erGAP2 is a ratiometric low-affinity Ca2+ sensor of the GFP-aequorin protein family that has been optimized for measurements in high Ca2+ concentration environments and that can be targeted to intracellular organelles42, 43. These experiments, together with pharmacological manipulations of store-dependent Ca2+ mobilization, provided clear evidence that Ca2+ release from the ER does not play a critical role in the primary transduction mechanism of mouse VSNs, results that are fully consistent with previous conclusions5.
In summary, the present study provides important new insights into the primary mechanisms underlying chemoelectrical signal transduction of the mammalian VNO and are necessary for advancing our understanding of Trpc2 activation. Our data show conclusively that InsP3R3 is not required for these functions. These results advance substantially our understanding of the molecular mechanisms mediating sensory activation of the mammalian VNO.
Materials and Methods
Mice
All animal protocols complied with the ethical guidelines for the care and use of laboratory animals issued by the German Government and were approved by the Animal Welfare Committee of Saarland University School of Medicine. All methods were carried out in accordance with the relevant guidelines and regulations. Mice were housed in ventilated cages under a 12:12 hour light/dark cycle with food and water available ad libitum. Generation of mice that carry a knockout mutation in the Itpr3 locus and thus lack InsP3R3 (InsP3R3−/−) has been described40, 41. This strain was intercrossed with C57BL/6 mice for at least twelve times before use41. C57BL/6 mice (denoted as wild-type, WT) or InsP3R3+/− littermates were used as reference mice. Some experiments were performed on OMP-GFP mice (heterozygous for the mutation) in which all cells expressing olfactory marker protein (OMP) are genetically labeled and show robust GFP fluorescence54. All experiments were performed on adult, 6–18 weeks old mice (both sexes).
Live-cell Ca2+ imaging
Ca2+ imaging of freshly dissociated VSNs was performed as described45, 47, 51. VNO epithelium was detached from the cartilage and minced in PBS at 4 °C. The tissue was incubated (20 min at 37 °C) in PBS supplemented with papain (0.22 U/ml) and DNase I (10 U/ml; Fermentas), gently extruded in DMEM (Invitrogen) supplemented with 10% FBS, and centrifuged at 100 × g (5 min). Dissociated cells were plated on coverslips previously coated with concanavalin-A type V (0.5 mg/ml, overnight at 4 °C; Sigma). Cells were used immediately for fura-2 imaging after loading them with fura-2/AM (5 µM; Invitrogen) for 60 min, or they were infected with HSV-1 encoding erGAP2 virus and incubated at 37 °C in FBS-supplemented DMEM medium for 24 h before imaging44. Coverslips containing VSNs were placed in a laminar-flow chamber (Warner Instruments) and constantly perfused at 22 °C with extracellular solution Hank’s balanced salt solution (HBSS, Invitrogen) supplemented with 10 mM Hepes (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid). Cells were alternately epi-illuminated at 405 and 470 nm for erGAP2 imaging, or at 340 and 380 nm for fura-2 imaging, and light emitted above 510 nm was recorded using a C10600-10B Hamamatsu camera installed on an Olympus IX71 microscope. For simultaneous recordings of [Ca2+]ER and [Ca2+]C, erGAP2-expressing cells were incubated for 60 min with fura-2/AM and sequentially excited at 340, 380 and 470 nm. We recorded emitted light at wavelengths >510 nm. The ratio F340/380 was used as an index of [Ca2+]C and F470 as an index of [Ca2+]ER. Images were acquired at 0.25 Hz and analysed using ImageJ (NIH), including background subtraction, region of interest (ROI) detection and signal analyses. ROIs were selected manually and always included the whole cell body. Peak signals were calculated from the temporal profiles of image ratio/fluorescent values. Results are based on recordings from 3–5 mice for each condition and genotype.
Chemostimulation
Chemostimuli for Ca2+ imaging were prepared fresh daily and diluted in extracellular solution giving the following final concentrations: HMW fraction, 1:300 dilution; sulfated estrogen mix (E mix): E1050 (1,3,5(10)-estratrien-3, 17β-diol disulphate), E1100 (1,3,5(10)-estratrien-3, 17β-diol 3-sulphate), E0893 (1,3,5(10)-estratrien-3, 17α-diol 3-sulphate) and E0588 (17β-dihydroequilin D 3-sodium sulphate), each at 100 µM (Steraloid); E1050, 100 µM; caffeine, 50 mM; KCl, 100 mM. Sulfated estrogens were initially prepared in dimethyl sulfoxide (DMSO) and further diluted in extracellular solution. To obtain HMW fraction, 0.5 ml of fresh urine was collected from adult C57BL/6 males (8–12 weeks old, sexually naïve)47 and size-fractionated by centrifugation (14,000 × g for 30 min) using Microcon 10-kDa molecular mass cutoff ultrafiltration columns (Millipore). The centrifugation retentate was washed with 0.5 ml of PBS three times and resuspended in 0.5 ml of PBS. Ca2+-free solution was prepared by adding 0.5 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid) to the extracellular solution. In some experiments, cells were incubated for 20 min at RT in extracellular solution containing 30 µM cyclopiazonic acid (CPA) to deplete intracellular Ca2+ stores before application of HMW or E1050.
Photorelease of InsP3
VNO cells were dissociated and plated on coverslips as described above and loaded with 3 μM caged InsP3/PM [D-2,3-O-isopropylidene-6-O-(2-nitro-4,5-dimethoxy)benzyl-myo-inositol 1,4,5-trisphosphate-hexakis (propionoxymethyl)ester; Enzo Life Sciences, Switzerland] mixed with the same volume of Pluronic F127 in DMSO (10%) in Hepes-HBSS buffer. Cells were loaded with caged InsP3/PM for 30 min at room temperature in the dark followed by an additional 30 min incubation of 2.5 μM fluo-4/AM (Invitrogen) and InsP3/PM. Stock solutions were made in DMSO and kept for up to 1 week stored at −20 °C. The final DMSO concentration did not exceed 0.5%. Coverslips containing VSNs were placed in a laminar-flow chamber (Luigs and Neumann) and constantly perfused with extracellular Hepes-buffered solution. We used an upright scanning confocal microscope (Zeiss LSM 880 Indimo) equipped with a standard Argon laser for excitation at wavelength of 488 nm (fluo-4 excitation) and a UV laser (Coherent) emitting 355 nm (InsP3 uncaging). Images were acquired at 0.5 Hz. Emitted fluorescence was collected between 500 and 560 nm. All scanning head settings were kept constant during each experiment. The UV laser light was coupled to the confocal microscope and focused onto the image plane through a 20 × 1.0 NA Plan-Apochromat water immersion objective (Zeiss). The depth of focus was 16 µm which ensured, together with the region of interest (ROI) diameter, illumination of individual cells. Before starting photolysis of caged InsP3, UV laser light was optimally focused using 18 µm thick brain tissue sections loaded with Hoechst 33342 (1:10000; ThermoFisher) and the semi-automated correction tool of the Zen software (Zeiss). Photolysis of caged InsP3 was achieved by directing UV laser light (1.036 mW) on preselected ROIs (spot diameter ~10 µm) using the Zen software (Zeiss) before reverting back to the visible wavelength laser to resume monitoring of fluo-4 fluorescence. Photorelease of caged InsP3 with ROI spot illumination was performed on single cells previously identified to respond to HMW or E1050. In some experiments, we used UV whole-field illumination (ROI area, 425 × 425 µm) to photorelease InsP3 in a larger area containing multiple fluo-4 loaded cells, in order to record also from IP3R3-deficient cells that did not respond to HMW or E1050 but were potentially sensitive to InsP3 (such as non-VSN cell types present in the VNO and expressing IP3R1 or IP3R2, serving as positive controls). The estimated intracellular InsP3 concentration after photolysis was expected to be in the 0.1–5 μM range56. Ca2+ changes were generally expressed as relative fluorescence changes, i.e. ΔF/F0 (F0 was the average of the fluorescence values of 5–10 frames before stimulation). Images were acquired at 0.5 Hz and analysed using ImageJ (NIH). Peak Ca2+ signals evoked by photoreleased InsP3 were calculated from the temporal profiles of ROI values during the first 10–20 s after a UV stimulus.
Virus production
Virus production, packaging of herpes simplex virus type 1 (HSV-1) vectors and VSN infection was performed as described44. This virus-based amplicon delivery system was initially developed to overexpress vomeronasal receptors of the V1r, V2r, and Fpr families in VSNs44. To produce viral vectors, a HindIII/EcoRI fragment containing the erGAP2 cDNA was cloned into the herpes simplex virus plasmid pHSVpUC. Packaging and titration of virus particles were performed as reported earlier75. VSN cultures were infected with a multiplicity of infection (moi) ranging between 0.01 and 0.1 one day before use.
Immunohistochemistry
Mice were deeply anesthetized with CO2 prior to decapitation and VNOs were removed, fixed for 3 h in 4% paraformaldehyde, equilibrated overnight in PBS containing 30% sucrose, embedded in OCT (Tissue-Tek), and snap-frozen in a dry ice/2-methylbutane bath. Frozen tissue sections (16 µm) were collected on glass slides (Superfrost Plus, Polysciences) and stored at −80 °C until use. Sections were post-fixed 15 min in 4% paraformaldehyde, washed 3 times in PBS (10 min each), incubated in blocking solution (0.5% Triton X-100, 3% horse serum, in PBS) for 3 h, and incubated overnight at 4 °C in blocking solution containing anti-InsP3R3 primary antibody (1:500, Millipore). The tissue was then washed 3 times in PBS (10 min each), incubated in AlexaFluor 488 goat anti-rabbit secondary antibody (1:500, Vector) 1 h at room temperature, and in Hoechst (10 µg/ml, Life Technologies) 5 min at RT. Sections were mounted using Vectashield Mounting Medium (Vector). Fluorescent images were acquired on a Nikon 80i microscope.
Single-cell RT-PCR
Single-cell cDNA libraries were prepared as described10, 44. VSNs were collected using a glass capillary (~10 m tip size) in 1 µl of extracellular solution. Single cells were then transferred to a PCR tube containing 1 µl of diethylpyrocarbonate (DEPC)-treated water. As control, 1 µl of extracellular solution containing no cells was collected with the glass capillary. Samples were immediately frozen on dry ice and kept at −80 °C until use. Second rounds of PCR were performed using the following primers: Omp: GCACAGTTAGCAGGTTCAGCT and GGTTTGCAGTCCTGGCAGC; Itpr1: CGAGGCTGGAAATGAAGGGT and CCACTGAGGGCTGAAACTCC; Itpr2: GGCTGCAAAGAGGTGAATGC and GACGCGATGTCATTTCCGTG; Itpr3: TGCCATGTCCCTGGTGAGC and GACCTGAAGGAAGGGCAGTG. PCR products were sequenced to exclude unspecific amplification or false positives. Total VNO mRNA was obtained from pooled VNO tissue of C57BL/6 adult mice (both genders) using PureLink RNA Mini Kit (Ambion) according to the manufacturer’s instructions. Traces of genomic DNA were digested by incubation with 30 U of DNase I (Fermentas).
Electrophysiology
Whole-cell voltage-clamp or loose-patch recordings from optically identified VSNs were performed in acute VNO tissue slices5, 32, 57. The recording chamber was perfused at a rate of ~1 ml/min with bicarbonate-buffered oxygenated (95% O2/5% CO2) extracellular solution (ACSF) containing (in mM) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 10 glucose; pH, 7.3; 300 mOsm. The intracellular solution contained, in mM: KCl 140, EGTA 1, Hepes 10, Mg-ATP 2, Na-GTP 1, pH, 7.1; 290 mOsm for urine-evoked responses and: CsCl 140, EGTA 1, Hepes 10, Mg-ATP 2, Na-GTP 1, pH, 7.1; 290 mM for some E1050-evoked currents. Recordings were performed at room temperature using an EPC-9 patch clamp amplifier (HEKA Elektronik, Lambrecht, Germany) and Pulse 8.80 software. In voltage-clamp experiments the membrane potential was clamped to −70 mV, if not otherwise noted. For urine-evoked responses, we chose VSNs from both apical and basal layers. For E1050-evoked responses we focused on VSNs located in the apical layer. In some experiments, we elicited voltage ramps (duration, 60 ms; slope of −3.3 mV/ms; from 80 mV to −120 mV) at the peak of sensory currents. Urine samples were freshly collected from mature C57/BL6 mice (either sex) and frozen for up to three months at −80 °C. Chemostimuli were delivered for 1 or 5 s via multi-barrel pipettes using a pressurized perfusion system (Picospritzer II, General Valve Corp.). Extracellular loose-patch recordings were performed as described52, 58. Local field potentials from intact VNO (fluid phase) were recorded as described previously32, 47, 52, 58, 63.
Statistics
Student’s t test and Mann-Whitney U test were used for measuring the significance of difference between two independent distributions, Wilcoxon signed-rank test was used when comparing two related samples, and Kruskal-Wallis test for three or more independent samples. Analyses were performed using Origin8.6 (OriginLab) or Igor Pro (WaveMetrics) software. Unless otherwise stated, results are presented as means ± SEM. Box-whisker plots show median values, mininimum-maximum outliers, and interquartile (25–75%) ranges.
Data availability statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Munger, S. D., Leinders-Zufall, T. & Zufall, F. Subsystem organization of the mammalian sense of smell. Annu. Rev. Physiol. 71, 115–140 (2009).
Kaupp, U. B. Olfactory signalling in vertebrates and insects: differences and commonalities. Nat. Rev. Neurosci. 11, 188–200 (2010).
Leypold, B. G. et al. Altered sexual and social behaviors in trp2 mutant mice. Proc. Natl. Acad. Sci. USA 99, 6376–6381 (2002).
Stowers, L., Holy, T. E., Meister, M., Dulac, C. & Koentges, G. Loss of sex discrimination and male-male aggression in mice deficient for TRP2. Science 295, 1493–1500 (2002).
Lucas, P., Ukhanov, K., Leinders-Zufall, T. & Zufall, F. A diacylglycerol-gated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: mechanism of pheromone transduction. Neuron 40, 551–561 (2003).
Tirindelli, R., Dibattista, M., Pifferi, S. & Menini, A. From pheromones to behavior. Physiol. Rev. 89, 921–956 (2009).
Chamero, P., Leinders-Zufall, T. & Zufall, F. From genes to social communication: molecular sensing by the vomeronasal organ. Trends Neurosci. 35, 597–606 (2012).
Liberles, S. D. Mammalian pheromones. Annu. Rev. Physiol. 76, 151–175 (2014).
Omura, M. & Mombaerts, P. Trpc2-expressing sensory neurons in the main olfactory epithelium of the mouse. Cell Rep. 8, 583–595 (2014).
Omura, M. & Mombaerts, P. Trpc2-expressing sensory neurons in the mouse main olfactory epithelium of type B express the soluble guanylate cyclase Gucy1b2. Mol. Cell. Neurosci. 65, 114–124 (2015).
Bleymehl, K. et al. A sensor for low environmental oxygen in the mouse main olfactory epithelium. Neuron 92, 1196–1203 (2016).
Liman, E. R. & Dulac, C. TRPC2 and the molecular biology of pheromone detection in mammals in TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades (eds Liedtke, W. B. & Heller, S.) Ch. 3 (CRC Press/Taylor & Francis, 2007).
Yildirim, E. & Birnbaumer, L. TRPC2: molecular biology and functional importance in Handbook of Experimental Pharmacology (eds Flockerzi, V. & Nilius, B.) 53–75 (Springer, 2007).
Miller, B. A. Trpc2 in Handbook of Experimental Pharmacology (eds Nilius, B. & Flockerzi, V.) 53–65 (Springer, 2014).
Spehr, M. Vomeronasal transduction and cell signaling in Chemosensory Transduction: The Detection of Odors, Tastes, and Other Chemostimuli (eds Zufall, F. & Munger, S. D.) 191–223 (Academic Press/Elsevier, 2016).
Yu, C. R. TRICK or TRP? What Trpc2−/− mice tell us about vomeronasal organ mediated innate behaviors. Front. Neurosci. 9, 221 (2015).
Holy, T. E., Dulac, C. & Meister, M. Responses of vomeronasal neurons to natural stimuli. Science 289, 1569–1572 (2000).
Zufall, F., Ukhanov, K., Lucas, P., Liman, E. R. & Leinders-Zufall, T. Neurobiology of TRPC2: from gene to behavior. Pflügers Arch. 451, 61–71 (2005).
Vannier, B. et al. Mouse trp2, the homologue of the human trpc2 pseudogene, encodes mTrp2, a store depletion-activated capacitative Ca2+ entry channel. Proc. Natl. Acad. Sci. USA 96, 2060–2064 (1999).
Gailly, P. & Colson-Van Schoor, M. Involvement of trp-2 protein in store-operated influx of calcium in fibroblasts. Cell Calcium 30, 157–165 (2001).
Tong, Q. et al. Erythropoietin-modulated calcium influx through TRPC2 is mediated by phospholipase Cγ and IP3R. Am. J. Physiol. Cell Physiol. 287, C1667–1678 (2004).
Inamura, K., Kashiwayanagi, M. & Kurihara, K. Inositol-1,4,5-trisphosphate induces responses in receptor neurons in rat vomeronasal sensory slices. Chem. Senses 22, 93–103 (1997).
Wekesa, K. S. & Anholt, R. R. Pheromone regulated production of inositol-(1,4,5)-trisphosphate in the mammalian vomeronasal organ. Endocrinology 138, 3497–3504 (1997).
Sasaki, K., Okamoto, K., Inamura, K., Tokumitsu, Y. & Kashiwayanagi, M. Inositol-1,4,5-trisphosphate accumulation induced by urinary pheromones in female rat vomeronasal epithelium. Brain Res. 823, 161–168 (1999).
Taniguchi, M., Wang, D. & Halpern, M. Chemosensitive conductance and inositol 1,4,5-trisphosphate-induced conductance in snake vomeronasal receptor neurons. Chem. Senses 25, 67–76 (2000).
Cinelli, A. R., Wang, D., Chen, P., Liu, W. & Halpern, M. Calcium transients in the garter snake vomeronasal organ. J. Neurophysiol. 87, 1449–1472 (2002).
Gjerstad, J., Valen, E. C., Trotier, D. & Doving, K. Photolysis of caged inositol 1,4,5-trisphosphate induces action potentials in frog vomeronasal microvillar receptor neurones. Neuroscience 119, 193–200 (2003).
Brann, J. H., Dennis, J. C., Morrison, E. E. & Fadool, D. A. Type-specific inositol 1,4,5-trisphosphate receptor localization in the vomeronasal organ and its interaction with a transient receptor potential channel, TRPC2. J. Neurochem. 83, 1452–1460 (2002).
Mast, T. G., Brann, J. H. & Fadool, D. A. The TRPC2 channel forms protein-protein interactions with Homer and RTP in the rat vomeronasal organ. BMC Neurosci. 11, 61 (2010).
Liman, E. R., Corey, D. P. & Dulac, C. TRP2: a candidate transduction channel for mammalian pheromone sensory signaling. Proc. Natl. Acad. Sci. USA 96, 5791–5796 (1999).
Menco, B. P., Carr, V. M., Ezeh, P. I., Liman, E. R. & Yankova, M. P. Ultrastructural localization of G-proteins and the channel protein TRP2 to microvilli of rat vomeronasal receptor cells. J. Comp. Neurol. 438, 468–489 (2001).
Spehr, J. et al. Ca2+-calmodulin feedback mediates sensory adaptation and inhibits pheromone-sensitive ion channels in the vomeronasal organ. J. Neurosci. 29, 2125–2135 (2009).
Yang, C. & Delay, R. J. Calcium-activated chloride current amplifies the response to urine in mouse vomeronasal sensory neurons. J. Gen. Physiol. 135, 3–13 (2010).
Billig, G. M., Pal, B., Fidzinski, P. & Jentsch, T. J. Ca2+-activated Cl− currents are dispensable for olfaction. Nat. Neurosci. 14, 763–769 (2011).
Kim, S., Ma, L. & Yu, C. R. Requirement of calcium-activated chloride channels in the activation of mouse vomeronasal neurons. Nat. Commun. 2, 365 (2011).
Dibattista, M. et al. Calcium-activated chloride channels in the apical region of mouse vomeronasal sensory neurons. J. Gen. Physiol. 140, 3–15 (2012).
Amjad, A. et al. Conditional knockout of TMEM16A/anoctamin1 abolishes the calcium-activated chloride current in mouse vomeronasal sensory neurons. J. Gen. Physiol. 145, 285–301 (2015).
Kim, S. et al. Paradoxical contribution of SK3 and GIRK channels to the activation of mouse vomeronasal organ. Nat. Neurosci. 15, 1236–1244 (2012).
Liman, E. R. Regulation by voltage and adenine nucleotides of a Ca2+-activated cation channel from hamster vomeronasal sensory neurons. J. Physiol. 548, 777–787 (2003).
Futatsugi, A. et al. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309, 2232–2234 (2005).
Hisatsune, C. et al. Abnormal taste perception in mice lacking the type 3 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 282, 37225–37231 (2007).
Navas-Navarro, P. et al. GFP-aequorin protein sensor for ex vivo and in vivo imaging of Ca2+ dynamics in high-Ca2+ organelles. Cell Chem. Biol. 23, 738–745 (2016).
Rodriguez-Garcia, A. et al. GAP, an aequorin-based fluorescent indicator for imaging Ca2+ in organelles. Proc. Natl. Acad. Sci. USA 111, 2584–2589 (2014).
Stein, B., Alonso, M. T., Zufall, F., Leinders-Zufall, T. & Chamero, P. Functional overexpression of vomeronasal receptors using a herpes simplex virus type 1 (HSV-1)-derived amplicon. PLoS One 11, e0156092 (2016).
Chamero, P. et al. Identification of protein pheromones that promote aggressive behaviour. Nature 450, 899–902 (2007).
Nodari, F. et al. Sulfated steroids as natural ligands of mouse pheromone-sensing neurons. J. Neurosci. 28, 6407–6418 (2008).
Chamero, P. et al. G protein Gαo is essential for vomeronasal function and aggressive behavior in mice. Proc. Natl. Acad. Sci. USA 108, 12898–12903 (2011).
Celsi, F., D’Errico, A. & Menini, A. Responses to sulfated steroids of female mouse vomeronasal sensory neurons. Chem. Senses 37, 849–858 (2012).
Haga-Yamanaka, S., Ma, L. & Yu, C. R. Tuning properties and dynamic range of type 1 vomeronasal receptors. Front. Neurosci. 9, 244 (2015).
McPherson, P. S. et al. The brain ryanodine receptor: a caffeine-sensitive calcium release channel. Neuron 7, 17–25 (1991).
Pérez-Gómez, A. et al. Innate predator odor aversion driven by parallel olfactory subsystems that converge in the ventromedial hypothalamus. Curr. Biol. 25, 1340–1346 (2015).
Leinders-Zufall, T. et al. Ultrasensitive pheromone detection by mammalian vomeronasal neurons. Nature 405, 792–796 (2000).
Ibarra-Soria, X., Levitin, M. O., Saraiva, L. R. & Logan, D. W. The olfactory transcriptomes of mice. PLoS Genet. 10, e1004593 (2014).
Potter, S. M. et al. Structure and emergence of specific olfactory glomeruli in the mouse. J. Neurosci. 21, 9713–9723 (2001).
Li, W., Llopis, J., Whitney, M., Zlokarnik, G. & Tsien, R. Y. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 392, 936–941 (1998).
Decrock, E. et al. Flash photolysis of caged IP3 to trigger intercellular Ca2+ waves. Cold Spring Harb. Protoc. 2015, 289–292 (2015).
Ukhanov, K., Leinders-Zufall, T. & Zufall, F. Patch-clamp analysis of gene-targeted vomeronasal neurons expressing a defined V1r or V2r receptor: ionic mechanisms underlying persistent firing. J. Neurophysiol. 98, 2357–2369 (2007).
Leinders-Zufall, T. et al. MHC class I peptides as chemosensory signals in the vomeronasal organ. Science 306, 1033–1037 (2004).
Leinders-Zufall, T., Ishii, T., Mombaerts, P., Zufall, F. & Boehm, T. Structural requirements for the activation of vomeronasal sensory neurons by MHC peptides. Nat. Neurosci. 12, 1551–1558 (2009).
Yildirim, E., Dietrich, A. & Birnbaumer, L. The mouse C-type transient receptor potential 2 (TRPC2) channel: alternative splicing and calmodulin binding to its N terminus. Proc. Natl. Acad. Sci. USA 100, 2220–2225 (2003).
Zhang, Z. et al. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. USA 98, 3168–3173 (2001).
Del Punta, K. et al. Deficient pheromone responses in mice lacking a cluster of vomeronasal receptor genes. Nature 419, 70–74 (2002).
Leinders-Zufall, T. & Zufall, F. The electrovomeronasogram: field potential recordings in the mouse vomeronasal organ. Methods Mol. Biol. 1068, 221–236 (2013).
Kelliher, K. R., Spehr, M., Li, X. H., Zufall, F. & Leinders-Zufall, T. Pheromonal recognition memory induced by TRPC2-independent vomeronasal sensing. Eur. J. Neurosci. 23, 3385–3390 (2006).
Brann, J. H. & Fadool, D. A. Vomeronasal sensory neurons from Sternotherus odoratus (stinkpot/musk turtle) respond to chemosignals via the phospholipase C system. J. Exp. Biol. 209, 1914–1927 (2006).
Rodriguez, I. Vomeronasal receptors: V1Rs, V2Rs, and FRPs in Chemosensory Transduction: The Detection of Odors, Tastes, and Other Chemostimuli (eds Zufall, F. & Munger, S. D.) 175–190 (Academic Press/Elsevier, 2016).
Nakahara, T. S. et al. Detection of pup odors by non-canonical adult vomeronasal neurons expressing an odorant receptor gene is influenced by sex and parenting status. BMC Biol. 14, 12 (2016).
Nilius, B. & Flockerzi, V. Mammalian Transient Receptor Potential (TRP) Cation Channels. Vol. 223 (Springer, 2014).
Hofmann, T. et al. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259–263 (1999).
Kim, S., Ma, L., Unruh, J., McKinney, S. & Yu, C. R. Intracellular chloride concentration of the mouse vomeronasal neuron. BMC Neurosci. 16, 90 (2015).
Untiet, V. et al. Elevated cytosolic Cl− concentrations in dendritic knobs of mouse vomeronasal sensory neurons. Chem. Senses 41, 669–676 (2016).
Jin, X. et al. Activation of the Cl− channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci. Signal. 6, ra73 (2013).
Haga-Yamanaka, S. et al. Integrated action of pheromone signals in promoting courtship behavior in male mice. eLife 3, e03025 (2014).
Xu, P. S., Lee, D. & Holy, T. E. Experience-dependent plasticity drives individual differences in pheromone-sensing neurons. Neuron 91, 878–892 (2016).
Alonso, M. T. et al. Functional measurements of [Ca2+] in the endoplasmic reticulum using a herpes virus to deliver targeted aequorin. Cell Calcium 24, 87–96 (1998).
Acknowledgements
We thank Hajime Hirase for valuable help in getting this project started, Benjamin Stein for experimental assistance, and Roberto Tirindelli for advice on InsP3R3 antibodies. This work was supported by Deutsche Forschungsgemeinschaft (DFG) grants CH 920/2-1 (P.C.), Sonderforschungsbereich 894 project A17 (F.Z.), Transregio SFB 152 projects P09 (T.L.-Z.) and P10 (F.Z.) and INST 256/427-1 FUGB (T.L.-Z.), a Saarland University HOMFORexcellent grant (P.C.), the Volkswagen Foundation (T.L.-Z.), and the Spanish Ministerio de Economía y Competitividad (BFU2014-53469P (M.T.A.). T.L.-Z. is a Lichtenberg Professor of the Volkswagen Foundation.
Author information
Authors and Affiliations
Contributions
P.C., T.L.-Z. and F.Z. conceived the study. P.C., J.W. and T.L.-Z. performed research and analysed data. M.T.A., M.R.P., C.H. and K.M. contributed key reagents. P.C. and F.Z. wrote the manuscript. All authors contributed to editing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chamero, P., Weiss, J., Alonso, M.T. et al. Type 3 inositol 1,4,5-trisphosphate receptor is dispensable for sensory activation of the mammalian vomeronasal organ. Sci Rep 7, 10260 (2017). https://doi.org/10.1038/s41598-017-09638-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09638-8
This article is cited by
-
Natural and Pathological Aging Distinctively Impacts the Pheromone Detection System and Social Behavior
Molecular Neurobiology (2023)
-
In-vivo activation of vomeronasal neurons shows adaptive responses to pheromonal stimuli
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
