
Abstract
Neuromuscular junctions (NMJs) are specialized synapses that connect motor neurons to skeletal muscle fibers and orchestrate proper signal transmission from the nervous system to muscles. The efficient formation and maintenance of the postsynaptic machinery that contains acetylcholine receptors (AChR) are indispensable for proper NMJ function. Abnormalities in the organization of synaptic components often cause severe neuromuscular disorders, such as muscular dystrophy. The dystrophin-associated glycoprotein complex (DGC) was shown to play an important role in NMJ development. We recently identified liprin-α-1 as a novel binding partner for one of the cytoplasmic DGC components, α-dystrobrevin-1. In the present study, we performed a detailed analysis of localization and function of liprin-α-1 at the murine NMJ. We showed that liprin-α-1 localizes to both pre- and postsynaptic compartments at the NMJ, and its synaptic enrichment depends on the presence of the nerve. Using cultured muscle cells, we found that liprin-α-1 plays an important role in AChR clustering and the organization of cortical microtubules. Our studies provide novel insights into the function of liprin-α-1 at vertebrate neuromuscular synapses.
Similar content being viewed by others


Introduction
The function of neuromuscular connections depends on the ability of muscle cells to maintain a high density of postsynaptic components, including acetylcholine receptors (AChR), at the cell surface1,2,3. Abnormalities in the clustering, stabilization, and turnover of AChR are often associated with severe neuromuscular disorders, such as myasthenia gravis4. There are an estimated 300 diseases of the neuromuscular system, and approximately 50% of them have an unknown etiology. The dystrophin-associated glycoprotein complex (DGC) is a multiprotein transmembrane complex. In muscles, it serves as a major laminin receptor that plays an important role in muscle integrity5, 6. At the neuromuscular junction (NMJ), the DGC is involved in stabilizing postsynaptic machinery by linking it to the extracellular matrix and actin-cytoskeleton5, 6. Mutations in several components of the DGC are known to cause muscular dystrophies that are often fatal4, 7. Apart from its function in skeletal muscles, the DGC was also shown to regulate many important processes, including cell migration, tumor progression, blood-brain barrier formation, and synaptic plasticity in the central nervous system (CNS)6, 8,9,10. Although the molecular mechanism that underlies the DGC-mediated stabilization of postsynaptic components is not fully understood, the cytoplasmic protein α-dystrobrevin-1 (αDB1) has been shown to play an important role in this process. αDB1 binds to dystrophin, utrophin, and syntrophin and regulates the dynamics of AChR at the NMJ11,12,13,14. To gain further insights into the molecular function of αDB1, we recently performed a biochemical screen for αDB1-interacting partners and identified liprin-α-112 as one of these.
Liprin-α-1 belongs to the liprin family of cytosolic scaffold proteins, which consists of four liprin-α and two liprin-β isoforms in mammals15. α-Liprins form homodimers and heterodimers with β-liprins15, 16. Lyssand et al. reported that liprin-α-1 in HEK-293 cells interacts with α-catulin, another component of the DGC that also plays a crucial role in the organization of AChR clustering12, 17. Liprin-α-1 was additionally shown to interact with the LL5β-ELKS-CLASP complex that forms a specialized plasma membrane platform that is involved in regulating focal adhesion turnover, cortical microtubules, cell migration, and tumor cell invasion18,19,20,21. LL5β, ELKS, and CLASP are also associated with the local exocytosis and organization of intracellular vesicle transport22, 23. LL5β and CLASP were shown to be involved in the organization of AChR clusters in cultured myotubes, AChR exocytosis, and microtubule attachment at the postsynaptic specialisation24,25,26,27,28.
Different liprin-α isoforms are differentially expressed in various brain regions, suggesting their specialized functions. In CNS neurons, liprin-α proteins were found at both presynaptic and postsynaptic compartments where they act as a molecular anchor for receptors and other proteins during synaptic assembly29,30,31. The function of liprins in the nervous system was also studied in invertebrates. A mutation of liprin-α homolog that is present in Drosophila (dliprin-α) displayed impairments in the accumulation of synaptic vesicles along the axon, in addition to fewer boutons and a reduction of the synaptic complexity of larval NMJs32,33,34. Similarly, a mutation of the C. elegans homolog of liprin-α, SYD-2, was associated with diffused localization of synaptic vesicles in motor neurons and defects in the assembly of active zones. Consequently, SYD-2 mutant worms are characterized by uncoordinated movements and defects in egg-laying behaviour as a result of impairments in synaptic transmission35,36,37,38,39.
Despite the well-documented function of liprin-α in the nervous system, the localization and function of this protein in vertebrate NMJs has not been reported. In the present study, we characterized the localization of liprin-α-1 at the murine NMJ and found evidence that this protein is implicated in the organization of postsynaptic machinery.
Results
Liprin-α-1 is localized to contractile machinery and concentrates at the NMJ
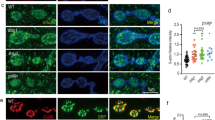
Previous studies highlighted a critical role for the DGC in muscle fiber integrity and in the development of the postsynaptic machinery6. Using a biochemical screen, we recently identified liprin-α-1 as a binding partner for αDB1, a cytoplasmic component of the DGC12. Liprin-α-1 has not been reported at the vertebrate NMJ. Therefore, we performed a histochemical analysis of its localization in mouse skeletal muscles. Liprin-α-1 immunoreactivity was detected in a striated pattern along the muscle fiber, which could be attributable to its association with a specific domain of contractile machinery. We performed co-localization experiments using various markers of contractile machinery, including α-actinin (an actin-binding protein that anchors myofibrillar actin thin filaments and titin to Z-discs; Fig. 1a,b), ryanodine receptor Ca2+ channels (RyRs) that are located in sarcoplasmic reticulum abutting Z-discs (Fig. 1c), and myomesin (a myosin crosslinking protein that is mainly present at the M-line; Fig. 1d)40,41,42. Immunohistochemical analyses of liprin-α-1 on fibers that were isolated from tibialis anterior muscles showed that it is predominantly associated with Z-discs that are enriched in α-actinin (Fig. 1a,b).

Liprin-α-1 localizes to Z-discs and NMJs in skeletal muscles. (a–d) Liprin-α-1 localized to the Z-discs of contractile machinery. Tibialis anterior muscle fibers from P30 mice were stained with antibody against liprin-α-1 protein (green) and indicated markers for contractile machinery (red). (b) Shows a higher magnification of an area in (a). (e) Liprin-α-1 (green) was enriched mainly at the postsynaptic machinery of the NMJ. Fluorescently labeled BTX was used to visualize AChR (red), and anti-synaptophysin and anti-neurofilament were used to visualize nerve terminals (blue). (f) Enrichment of liprin-α-1 at the postsynaptic machinery. Fluorescence intensity for liprin-α-1 immunochemistry (green) and AChR (BTX, red) along the line shown in (e) was plotted using RGB profiler plugin to the ImageJ/Fiji software. (g) Liprin-α-1-GFP fusion protein localized to the NMJ on transfected fiber. AChR (red); liprin-α-1-GFP (green). Scale bar 10 μm.
Strong liprin-α-1 immunoreactivity was also detected at neuromuscular synapses, visualized by fluorescently-labelled bungarotoxin (BTX) that specifically binds to the postsynaptic AChR (Fig. 1e,f). Optical cross-sections of confocal Z-stack images showed that liprin-α-1 was predominantly concentrated at the postsynaptic machinery (below or at the same level as AChR) but was also present at the presynaptic compartment where it co-localized with synaptophysin and neurofilament (Fig. 1e). We confirmed this observation in an independent experiment in which we ectopically expressed liprin-α-1-green fluorescent protein (GFP) fusion protein in muscle fibers and confirmed that it localized to the postsynaptic machinery (Fig. 1g). Similarly, overexpressed liprin-α-1-GFP was enriched at the AChR clusters in myotubes derived from C2C12 myoblasts or primary myoblasts (Supplementary Fig. 2a,b).
Liprin-α-1 NMJ localization during development and in various types of muscles
Fibers in different muscles vary in their molecular composition and physiological properties43, 44. They can be divided into groups of either fast or slow contraction, based on the types of myosin heavy chain isoforms that are expressed. Tibialis anterior, triangularis sterni, and gastrocnemius muscles comprise predominantly fast fibers, and soleus muscles consist of predominantly slow fibers43, 44. Muscle fibers in different groups of muscles can also vary in their synaptic organization, reflected by the overall size and shape of NMJs45. Importantly, Chakkalakal et al. also found molecular differences between NMJs on fast and slow fibers46. Thus, liprin-α-1 expression and localization in various muscle types need to be investigated. We found that anti-liprin-α-1 antibody immunoreactivity was similar in tibialis anterior, triangularis sterni, gastrocnemius, and soleus muscles and presented a striated pattern along the fibers and strong enrichment at the NMJ (Fig. 2a).

Localization of liprin-α-1 is similar in various muscles and maintained throughout the development. (a) Liprin-α-1 immunoreactivity in different muscles. TS, triangularis sterni; TA, tibialis anterior; SOL, soleus; GAS, gastrocnemius. Muscles were collected on P30. (b) Immunohistochemical analysis of TA muscle fibers on the indicated embryonic (E) and postnatal (P) days. Anti-liprin-α-1 antibody (green); AChR (red). Scale bar 10 μm.
During the early postnatal period, NMJs undergo intensive remodelling. Until postnatal day 7 (P7), clusters of postsynaptic machinery mostly grew in size, and the transformation of juvenile oval synapses into topologically complex structures occurred between P7 and P30 (Fig. 2b). Specific synaptic components could be recruited to the synapse at particular time points of development and maturation12, 26, 47. To study the presence of liprin-α-1 at the NMJ during these transitional stages, we performed immunohistochemical analyses of tibialis anterior muscles from mice at embryonic day 16 (E16) and at different postnatal days: P9, P14, P22, and P30. We observed a stable and constant level of liprin-α-1 during all stages of development. Liprin-α-1 was present at juvenile NMJs before their perforation, and similar localization was apparent at later stages (Fig. 2b).
Localization of liprin-α-1 is nerve-dependent
When NMJ maturation is accomplished, postsynaptic machinery still undergoes remodelling, including the continuous endocytosis and exocytosis of synaptic components. The instability of postsynaptic machinery can be induced after nerve injury, which leads to the enhanced internalization of AChR. To study potential correlations between increases in NMJ plasticity and the localization of liprin-α-1, we performed a nerve cut procedure that leads to permanent degeneration of the nerve terminal48, 49. We carried out immunohistochemical analyses of teased fibers that were isolated from the tibialis anterior muscle after sciatic nerve cut 3, 7, and 14 days after surgery (Fig. 3a). In uninjured, control tissues, liprin-α-1 was present at both pre- and postsynaptic sites and also at sarcomeres. Three days after surgery, the nerve terminal began to withdraw, and AChR became more diffused at the postsynaptic membrane. Interestingly, 7 days after surgery, we observed the mislocalization of liprin-α-1 from the postsynaptic specialization, which became more apparent 7 days later. A similarly striated pattern of liprin-α-1 immunoreactivity along the fibers gradually disappeared following nerve degradation (Fig. 3a). Interestingly, 14 days after denervation αDB1 immunoreactivity was still detected at the postsynaptic machinery, although it appeared slightly more diffuse (Fig. 3b). Although liprin-α-1 interacts with αDB1, its recruitment to the postsynaptic machinery appears to be independent of αDB1, since liprin-α-1 colocalized with AChRs in αDB KO muscles (Supplementary Fig. 2b,c). Collectively, these observations indicate that liprin-α-1 localization in skeletal muscle fibers depends on innervation but not the presence of αDB1 protein.

Liprin-α-1 NMJ localization is nerve-dependent. Fibers were isolated from the tibialis anterior muscle from P30 mice on the indicated days after the nerve cut procedure. (a) Liprin-α-1 (green) localisation at different days after nerve cut. The bottom panel represents higher magnification of the box in the upper panel (D14). (b) α-dystrobrevin-1 immunostaining (green) 14 days after nerve cut. AChR are visualized with BTX in red and anti-synaptophysin/anti-neurofilament immunostaining is shown in blue. All of the images were collected using the same exposure and imaging parameters. Scale bar 10 μm.
Liprin-α-1 is required for AChR cluster formation
To determine whether liprin-α-1 plays a functional role in AChR cluster organization, we performed RNA interference (RNAi) knockdown experiments with myotubes that were derived from C2C12 myoblasts using three synthetic small-interfering RNAs (siRNAs) that were validated with regard to knockdown efficiency (Fig. 4a,b). Myotubes that were cultured on laminin-coated surfaces and transfected with non-targeting siRNA (control) formed numerous clusters of AChR that often acquired a complex topology (Fig. 4c), similar to postsynaptic machinery at the NMJ. In contrast, myotubes that were transfected with liprin-α-1-targeting siRNAs presented significantly fewer AChR clusters and fewer topologically complex assemblies (Fig. 4c–e). Liprin knockdown had similar effects on the formation of agrin-induced AChR clusters. The addition of this nerve-derived glycoprotein to myotubes transfected with non-targeting siRNA (control) induced the formation of small AChR clusters scattered at the myotube surface (Fig. 4f). The inhibition of liprin-α-1 expression significantly affected the formation of agrin-induced clusters (Fig. 4f,g).

Liprin-α-1 is required for the formation of AChR clusters. (a) Validation of liprin-α-1 knockdown efficiency by Western blot. Hek-293 cells were co-transfected with liprin-α-1-GFP and the indicated siRNAs that targeted liprin-α-1 or indicated control siRNAs. Western blot for tubulin was used as a loading control. (b) Validation of liprin-α-1 knockdown efficiency by qRT-PCR. C2C12 differentiated myotubes were transfected with indicated siRNAs and 2 days later the total RNA was isolated from cell extract followed by qRT-PCR analysis of liprin-α-1 expression. (c) C2C12 myotubes cultured on laminin-coated surfaces were transfected with siRNAs that targeted liprin-α-1 and AChR clusters were visualised with BTX. (d) Quantification of the total number of clusters. (e) Quantification of topologically complex (perforated) clusters. (f,g) Liprin-α-1 was essential for the formation of agrin-induced AChR clusters. C2C12 myotubes were cultured on gelatin-coated surfaces, transfected with siRNAs that targeted liprin-α-1, and stimulated with soluble agrin (Z-fragment) to form AChR clusters. (g) Quantification of AChR clusters in (f). (h) Western Blot analysis of AChR and rapsyn expression levels after liprin-α-1 knockdown. Lysates from C2C12 myotubes transfected with liprin-α-1 targeting siRNAs or non-targeting RNAs were probed with antibodies against specified proteins. Levels of tubulin were used as a loading control. Negative control -non-targeting siRNA; positive control - muscle-specific kinase (MuSK) siRNA. Statistical significance was analysed using 2-tailed t-test. *p < 0.05; **p < 0.01; ****p < 0.0001. Error bars represent standard error of the mean (SEM). Scale bar 20 μm.
Liprin-α-1 regulates AChR clustering by organizing cortical microtubules
Defects in AChR cluster formation observed in liprin-α-1-depleted myotubes could be due to the lack of expression of synaptic proteins, impaired cell surface delivery, or impaired assembly of the receptors into postsynaptic structures. Neither levels of AChR receptor subunit α, nor levels of rapsyn, a scaffold protein required for AChR clustering, were reduced in siRNA-transfected myotubes (Fig. 4h). We then checked if the liprin siRNA-mediated reduction of the number of AChR clusters could be due to the defective transport of AChR to the cell surface. For this, we visualized surface AChRs by labelling live cells with Alexa-488-tagged BTX shortly prior to cell fixation. Fixed myotubes were permeabilized and additionally stained with Alexa-555-tagged BTX to label internal AChRs. Liprin-α-1-depleted myotubes failed to assemble AChR clusters observed in control cells, however significant amount of fluorescence was detected at the myotubes surfaces, suggesting that high amounts of dispersed AChRs were still present (Fig. 5a,b). The level of internal AChRs detected with Alexa-555-tagged BTX was unchanged upon liprin-α-1 depletion (Fig. 5a). To eliminate the possibility that Alexa-488 signal (surface AChRs) came from the autofluorescence of the fixed cells we performed a control experiment in which we masked surface receptors by incubation with saturating dose of unlabelled BTX followed by incubation with Alexa-488-tagged BTX. As expected, pre-masking of surface receptors completely blocked the Alexa-488 signal, which proves that our detection method for surface AChRs is specific (Fig. 5a,b). To independently determine the amount of surface AChRs in control and liprin-α-1 knockdown cells, we labeled AChRs with biotin-conjugated BTX either in live cells (to label surface receptors) or in cell lysates (to label total cellular AChR pool) and precipitated both pools of AChR with streptavidin-coated beads. As shown in Fig. 5c, myotubes depleted of liprin-α-1 had significant amounts of AChRs at the plasma membrane.

Liprin-α-1 regulates clustering of AChRs by organizing microtubules. (a–c) C2C12 myotubes depleted of liprin-α-1 have substantial amounts of surface AChR. (a) Surface AChR (green) were stained with Alexa-488-BTX and internal AChR were labelled with Alexa-555-BTX. Lower panel shows control experiment for labelling specificity of the Alexa-488-BTX staining shown in the middle panel. Unlabelled BTX was applied to live cells prior to Alexa-488-BTX labelling to mask BTX binding sites on AChR. (b) Fluorescence intensity quantification of surface AChR labelling, values normalized to control. (c) Analysis of surface AChRs in liprin-α-1-depleted myotubes by AChR precipitation and Western blotting. S-surface and T-total AChR precipitates. (d,e) Liprin-α-1 knockdown reduces the number of plasma membrane-attached microtubules in myoblasts. (d) Cells were transfected with indicated siRNAs and microtubules were visualized with anti-α-tubulin antibody. Lower panel represents higher magnifications of areas in the upper panels. Arrows indicate examples of microtubules in contact with cell surface. (e) Quantification of cell surface-attached microtubules per micrometre of the cell edge from 60 cells in three independent experiments. (f,g) Liprin-α-1 knockdown reduces the number of EB1 foci at the AChR clusters in myotubes. Microtubule tips were visualised with anti-EB1 antibody. Columns at the right represent higher magnifications of boxed areas. Asterisks mark the nucleous of an undifferentiated myoblast. (f) Quantification of EB1 foci at from 20 AChR clusters from three independent experiments. (h,i) Microtubules are dispensable for maintenance of AChR assemblies but are required for the cluster expansion. (h) Pre-existing AChR were labelled in live cells with Alexa-555-BTX (Old AChR) and nocodazole or DMSO (control) was added to the cells for 6 h and live cells were labelled again with Alexa-488-BTX (New AChR). (i) Fluoresence intensity quantification of surface AChRs. (j) Nocodazole treatment does not significantly affect AChR exocytosis. Western blot analysis of surface and total AChR in cell lysates (Lys), flow-through (FL) and elution (EL) fractions from control (DMSO) and nocodazole experiments. Scale bar 10 μm. Statistical significance was analysed using 2-tailed t-test. ****p < 0.0001. Error bars represent SEM.
These experiments suggest that defects in the formation of AChR assemblies upon liprin-α-1 knockdown were not due to the impaired synthesis of AChRs or their insufficient amount at the plasma membrane.
Previous studies by van der Vaart et al., suggested that liprin-α-1 plays a role in the anchoring of microtubule ends at the cell cortex in HeLa cells21. We therefore examined if liprin-α-1 may have similar function in cultured muscle cells. We first performed microtubule cytoskeleton analysis in myoblasts that were transfected with liprin-α-1 targeting siRNAs or control, non-specific siRNA. Both control and liprin-α-1 depleted myoblasts assembled a dense microtubule network, however, liprin-α-1 knockdown significantly reduced the number of microtubule filaments attached to the cell cortex (Fig. 5d,e). Analysis of microtubule attachment to the plasma membrane in differentiated myotubes is complicated because myotubes make a very dense microtubule network making the distinction of individual filaments and their tips very difficult (see Supplementary Fig. 1). In order to distinguish microtubule tips we stained myotubes for end-binding protein 1 (EB1), a molecular scaffold that is present at the plus-end of microtubules and analysed EB1 density on single plane images of plasma membrane collected with a confocal microscope, as had been done previously25. We observed that liprin-α-1 depletion led to significant reduction of EB1 foci at the AChR clusters (Fig. 5f,g).
Interestingly, enrichment of microtubule–plasma membrane contacts specifically at the postsynaptic specialization was shown previously to be crucial for AChR clustering, suggesting that microtubule cytoskeleton plays important role in the receptor aggregation apart from its role in the intracellular trafficking25. Moreover, the impaired microtubule attachment at the AChR clusters did not disassemble pre-existing clusters but significantly inhibited formation of new assemblies25. These observations prompted us to study if the abrupt disruption of microtubule cytoskeleton with nocodazole treatment has similar effect on AChR organization. For this purpose we labelled pre-existing surface AChRs with Alexa-555-tagged BTX in live cells and applied nocodazole for the next six hours. After this chase period we performed a second labelling of surface AChR with BTX conjugated to Alexa-488, to visualize newly synthesized AChR. In control myotubes (h), newly synthesized AChR were circumferentially incorporated into the postsynaptic assembly surrounding centrally located pre-existing receptors, as published previously50. Nocodazole treatment did not affect the organization of the pre-existing AChR clusters, however, it abolished incorporation of the new AChR into the assemblies (Fig. 5h,i). To check if nocodazole treatment abolishes AChR exocytosis we performed precipitation of surface receptors labelled with biotin-conjugated BTX. As shown in Fig. 5j the amounts of surface AChRs were not significantly altered after nocodazole treatment. These results suggest that in cultured myotubes microtubules play an important role in the clustering of AChRs and play a minor role in AChR cell surface delivery.
Discussion
In vertebrates, liprin-α-1 has been shown to play a role in the organization of synapses in the CNS, but has not been studied at the NMJ. We employed several approaches to characterize the localization of liprin-α-1 at the murine NMJ and its involvement in the formation of AChR clusters in cultured myotubes. Our immunohistochemical analysis and the ectopic expression of GFP-tagged proteins revealed that liprin-α-1 was localized to muscle contractile machinery where it concentrated at Z-discs and co-localized with α-actinin. Additionally, liprin-α-1 was strongly enriched at the NMJ where it was present at both pre- and postsynaptic compartments in different types of muscles. The amount of liprin-α-1 protein at the synapse appeared to be unchanged during development, but its localization was affected by nerve injury. Liprin-α-1 recruitment to the postsynaptic machinery appeared to be independent of interaction with αDB1. This suggests that liprin-α-1 could bind more than one postsynaptic protein. One candidate could be catulin-α-1, which binds to several DGC components17, and similarly to liprin-α-1 appeared to localize to the NMJ in the absence of αDB112, 51. Our functional studies revealed that liprin-α-1 is involved in the organization of postsynaptic machinery because it was required for the formation of laminin- and agrin-induced AChR clusters. The defects in the formation of AChR clusters observed with liprin-α-1 knockdown were more severe than those observed in αDB knockout cells12, 52. This suggests that liprin-a1 has additional functions at the NMJ, independent from αDB1. Our and previous studies19, 21 demonstrated that it is involved in the organization of plasma membrane-attached microtubules. Basu et al., have shown that LL5β-dependent attachment of microtubule tips to the postsynaptic membrane plays a crucial role in the AChR clustering25. Importantly, LL5β knockdown appeared to have no effect on the pre-existing AChR clusters, but inhibited aggregation of the newly synthesized AChR24, 25. In agreement with these observations, our experiments on acute disruption of microtubule cytoskeleton with nocodazole showed that the microtubules are dispensable for the maintenance of the pre-existing AChR clusters but are required for the cluster expansion in C2C12 myotubes (Fig. 5h–j). Microtubules have an important function in intracellular vesicular trafficking, and an obvious possibility would be that microtubule disassembly blocks biosynthetic transport to the plasma membrane. Connolly J.A., however, demonstrated that disruption of microtubules with nocodazole in cultured chick myotubes had a minor effect on the AChR transport to the cell surface and newly synthesized AChR were efficiently exocytosed53. This is in agreement with our observation that disassembly of the microtubule cytoskeleton with nocodazole does not substantially alter AChR delivery to the plasma membrane (Fig. 5j). Similarly, liprin-α-1 depleted myotubes appeared to have significant amount of AChR at the cell surface that failed to aggregate (Fig. 5a–c). Also LL5β knockdown, which blocks clustering of AChR on myotubes surfaces24 was proposed to reduce the density specifically of AChR-associated cortical microtubules25. These observations argue that microtubules attached at the postsynaptic machinery could have important functions in the clustering of the postsynaptic components apart from their role in exocytosis. It is believed that microtubules are utilized for vesicular transport over longer distances and actin filaments play a more important role in short-distance delivery54, 55. At the neuromuscular junctions, as well as at the laminin-cultured myotubes synaptic genes are expressed predominantly from specialized postsynaptic nuclei that are clustered directly underneath the postsynaptic membrane50. Such compartmentalization together with local synthesis leads to the enrichment of synaptic mRNAs and proteins at the synapse, thus reducing the need for long-distance transport. Therefore, vesicular transport to the muscle postsynaptic specialization could rely more on the actin cytoskeleton. Microtubules could have either mechanical or signalling role, which may influence clustering of AChR, but the underlying mechanism is not clear. It could be possible that local exocytosis directed specifically to the AChR cluster25 could be crucial for AChR incorporation into the assembly. Recent studies utilizing quantum dots, however, provided evidence supporting the diffusion-trapping hypothesis for incorporation of AChR into the postsynaptic membrane56. If this is the case, one would expect circumferential localization of newly incorporated receptors as shown in Fig. 5 and reported previously for AChR clusters formed by laminin-cultured myotubes and postsynaptic machinery in vivo 50.
The role of liprin-α-1 at the postsynaptic membrane may not be restricted to the regulation of cortical microtubules. We have recently demonstrated that liprin-α-1 binds to αDB1 and Lyssand et al. showed that it also interacts with catulin-α-1 in HEK-293 cells17. These two cytoplasmic DGC-associated proteins play an important role in the organization of the postsynaptic apparatus12, 13. Liprin-α-1 may also function as a scaffold, recruiting signalling proteins such as: LAR transmembrane protein-tyrosine phosphatase (which controls the development of invertebrate NMJs)57, MAGUK (a kinase that is involved in the muscle-specific kinase [MuSK] signaling pathway at the vertebrate NMJ)58, and CamKII (which is critical for AChR recycling)59. de Curtis and colleagues proposed that the liprin-α-1 and its binding partners function as a hub for the recruitment of additional effector proteins that form dynamic supramolecular assemblies at specific membrane locations, such as presynaptic nerve terminals, the leading edge of migrating cells or invasive protrusions called podosomes18, 19. Thus, additional studies should be conducted to shed more light in the function of this important synaptic protein.
Methods
Cell culture
C2C12 cells were purchased from the American Type Culture Collection (CRL1772) and cultured for up to five passages in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 20% fetal bovine serum (FBS), 4.5 g/L glucose, L-glutamine, penicillin, streptomycin, and fungizone on 0.2% gelatin-coated plastic dishes. Prior to the experiment, the cells were trypsinized and plated on Permanox slides in eight-well Flexiperm chambers (catalog no. 177445, Sigma-Aldrich). The slides were coated with laminin 111 (catalog no. 23017-015, Invitrogen) in DMEM. To induce cell fusion, the media were replaced with DMEM that contained 2% horse serum, 4.5 g/L glucose, L-glutamine, penicillin, streptomycin, and fungizone.
For the agrin experiments, slides were coated with 0.2% gelatin and myotubes were stimulated with 10 nM agrin (catalog no. 550-AG-100/CF, Biokom). Hek293 cells were from the American Type Culture Collection (CRL-1573) and maintained in DMEM with 10% FBS, 4.5 g/L glucose, L-glutamine, penicillin, streptomycin, and fungizone.
Primary myoblast cultures were performed as described previously60, 61. Briefly, tibialis anterior and extensor digitorum longus muscles were digested with 0.2% collagenase in DMEM for 1, 5 h. Next, muscles were transferred to DMEM containing 10% horse serum, 0.5% chicken embryo extract and penicillin and streptomycin and single fibers were isolated from dissociated muscles. Fibers were stripped using syringe with 21-gauge needle. Cells were filtered through 70 µm nylon cell strainer and plated on Flexiperm chamber slides covered with laminin and agrin28. Myoblasts were cultured in DMEM containing 20% FBS, 10% horse serum, 0.5% chicken embryo extract, penicillin and streptomycin. After 4 days of culture myoblasts were transfected with liprin-α1-GFP plasmid using Lipofectamine 2000 (catalog no. 11668-030, ThermoFisher) according to manufacturer’s instructions. Six days after transfection cells were fixed in 4% PFA and labeled with AlexaFluor-555-conjugated BTX.
siRNA transfection
Differentiated C2C12 myotubes were transfected with 20 nM siRNAs (Qiagen) that targeted liprin-α-1 (catalog no. and targeted sequences: siRNA1 [SI04927860, CTGCTTGACGGAAACCATGAA], siRNA2 [SI04927867, CACGTCTGTGCATGACCTCAA], siRNA3 [SI04927874, ATCCTGTCGATTGGCCTTAAA]) using Lipofectamine RNAiMAX (catalog no. 13778075, Life Technologies) at 48–72 h after fusion according to the manufacturer’s instructions. The cells were fixed 2–3 days later and examined by confocal microscopy. Transfections of HEK293 cells were performed with Lipofectamine 2000 Transfection Reagent.
Cell staining
The cells were fixed in 4% paraformaldehyde (PFA) and stained using AlexaFluor-555-bugarotoxin (BTX) (catalog no. B35451, Life Technologies) and Alexa-488-taged BTX (catalog no. B13422, Life Technologies). For labelling of AChRs in live cells BTX conjugates were added to the culture media for 5–7 min; internal AChR staining was performed after cell membrane permabilization with 0.5% Triton X-100 for 1 h. For masking of BTX binding sites on surface AChRs unlabelled BTX (catalog no. B1601, Life Technologies) was added to the culture media for 20 minutes. For microtubule disruption 10 µg/ml nocodazole (catalog no. M1404, Sigma) or DMSO (control) was added to the media for 6 hours. Microtubules were visualised with anti-tubulin (catalog no. ab18251, Abcam) and EB1 with anti-EB1 (catalog no. 610534, BD Biosciences). F-actin was stained with Acti-stain-670 (catalog no. PHDN1, Cytoskeleton).
Microscopy and image analysis
Microscopic analysis was performed at the Confocal Microscopy Facility, Nencki Institute, using a Zeiss Spinning Disc confocal microscope or Leica TCS SP8 scanning confocal microscope that were equipped with diode or white light lasers, respectively. Images were collected using ZEN software (ZEISS International) and analysed using ImageJ/Fiji software. Enrichment of liprin-α-1 at the NMJ postsynaptic machinery was analysed using RGB profiler plugin (https://imagej.nih.gov/ij/plugins/rgb-profiler.html) at the ImageJ/Fiji software. Quantitative image analysis was performed using ZEN Lite software (ZEISS International). For the quantification of surface AChRs, mean fluorescence counts from whole images was collected. Values obtained for liprin-α-1 knockdown and masking experiments were calculated as percentages of the mean fluorescence from the negative control.
Western blotting and AChR precipitation
The cells were lysed in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Nonidet-P40, 0.5% sodium dodecyl sulfate [SDS], 10% glycerol, 1 mM dithiothreitol [DTT], 1 mM NaF, and ethylenediaminetetraacetic acid-free mini protease inhibitor cocktail [catalog no. 11873580001, Roche], pH 8.0). The cells were scraped off the dish, incubated briefly on ice, passed three times through a 25-gauge needle, incubated on ice for 15 min; and centrifuged at 20,000 × g for 30 min, at 4 °C. For Western blot, the supernatant samples were mixed with sample buffer and boiled for 5 min. The proteins were then resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (catalog no. 66485, Pall Corporation) using Trans Blot Turbo (catalog no. 1704270, Bio-Rad). Membranes were blocked with 5% milk in TBST (20 mM Tris-HCl, 150 mM NaCl [pH 7.6], and 0.1% Tween20) for 1 h at room temperature and probed with primary antibodies that were diluted in 5% milk in TBST at 4 °C overnight. After washing with TBST buffer, the membranes were incubated with appropriate secondary antibodies conjugated to horseradish peroxidase. For protein detection, we used Clarity chemiluminescent substrate (catalog no. 1705060, Bio-Rad). The primary antibodies were anti-GFP (catalog no. ab32146, Abcam), anti-AChR-α1 (catalog no. 10613-1-AP, Proteintech), anti-rapsyn (catalog no. ab156002, Abcam) and anti-tubulin (catalog no. ab18251, Abcam).
For precipitation of surface AChR62, BTX-biotin (catalog no. B1196, Invitrogen) was added to the media for 5 minutes at 37 °C, washed with PBS, followed by cell lysis. For precipitation of total AChR, cells were lysed and incubated with BTX-biotin for 30 minutes at 4 °C. Lysates were incubated with NeutrAvidin beads (catalog no. 29200, Thermo Scientific) overnight in 4 °C. Beads were collected by centrifugation (2500x g), washed three times with wash buffer (100 mM Hepes pH 7.4, 150 mM NaCl, 1 mM PMSF, 0.5% NP-40), resuspended in 2x sample buffer and boiled. Rat polyclonal antibody (catalog no. MRT-609R, BioLegend) was used to detect precipitated AChR.
Sciatic nerve cut and plasmid electroporation
P30 mice were anesthetized with a ketamine (150 mg/kg)/xylazine (10 mg/kg) cocktail, and the surgery area was shaved and disinfected with ethanol. A 1 cm incision was made in the longitudinal direction proximal to the knee and a 5 mm fragment of the sciatic nerve was removed with sterile scissors. The skin was sutured and Lidocaine was applied to the wound, and analgesics were administered.
DNA electroporation was performed under general anesthesia (see above), and 25 μg DNA (1 μg/μl) was injected into the tibialis anterior muscle using a Hamilton syringe. The muscle was electroporated with 10 pulses of electrical current (180 V/cm) of 20 ms each at 1 s intervals using an ECM-830 electroporator (catalog no. 45-0052INT, BTX Harvard Apparatus). Lidocaine was applied to the wound and analgesics were administered. The animals were sacrificed 14 days after surgery.
αDB KO mice63 were a kind gift from Joshua R. Sanes (Harvard University, USA). C57BL6 mice were obtained from Jackson Laboratories and maintained at the Nencki Institute of Experimental Biology in the animal facility equipped with a selected pathogen free (SPF) barrier (standard). For the animal experiments, we obtained approval from the First Warsaw Local Ethics Committee for Animal Experimentation, and all of the experiments were conducted in accordance with all applicable laws and regulations. The health of the animals was monitored by veterinarian on-staff.
Muscle tissue preparation
The mice were sacrificed by a lethal injection of pentobarbital (200 mg/kg). Mouse embryos were extracted at E16. The tibialis anterior, triangularis sterni, soleus, and gastrocnemius muscles were excised and fixed in 4% PFA or cold methanol. Muscles were stripped of connective tissues and either used for the preparation of teased fibers or stained in a whole-mount procedure. Nonspecific staining was blocked with 2% bovine serum albumin and 2% goat serum in PBS supplemented with 0.5% Triton-X100 before overnight incubation with primary antibodies. The muscles were stained using fluorescently labeled BTX (catalog no. B35451, Life Technologies) and primary antibodies against anti-liprin-α-1 (catalog no. 115098, GeneTex), anti-α-actinin (catalog no. sc-17829, Santa Cruz Biotechnology), anti-ryr (catalog no. ab2868, Abcam), anti-myomesin (catalog no. ab111433, Abcam), anti-α-dystrobrevin-1 (catalog no. 610766, BD Transduction Laboratories), anti-neurofilament (catalog no. 2H3, Developmental Studies Hybridoma Bank), and anti-synaptophysin (catalog no. 101 004, Synaptic Systems). For cryosectioning tibialis anterior muscles were flash-frozen in isopentane cooled with liquid nitrogen and sectioned at 10–20 µm in a cryostat. Cryosections were fixed and stained as described above.
DNA constructs, RNA isolation and quantitative PCR analysis
The liprin-α-1-GFP (human) plasmid was a kind gift from Ivan de Curtis (San Raffaele Scientific Institute, Milano, Italy)20. The liprin-α-1-GFP (mouse) plasmid was constructed by amplification of the liprin-α-1 coding sequence from pLiprin plasmid (catalog no. IRCKp5014O0117Q, Source BioScience) using GATCGCTAGCATGATGTGCGAGGTGATGCCGACCATTAGT and CATTGGTACCAGCAGGAGTAAGTCCTGACTGTGGCAGAGTC primers and subcloned into the pEGFP-N2 vector.
RNA was isolated with Trisure Reagent (catalog no. BIO-38032, Bioline) according to manufacturer’s instructions. cDNA was generated from 1 µg of templates using the High Capacity cDNA Reverse Transcription Kit (catalog no. 4374966, Applied Biosystems). RT-qPCR was performed in triplicates for RNAs obtained from two independent experiments using TaqMan Fast Universal PCR Master Mix (catalog no. 4352042, Applied Biosystems) with predesigned liprin-α1 TaqMan probe (catalog no. 4448892, ThermoFisher) on Step One Plus instrument (Applied Biosystems). Results were analyzed with Step One software v2.3 using the standard curve method and standardized to 18 S rRNA (catalog no. 4333760, ThermoFisher).
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Sanes, J. R. & Lichtman, J. W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2, 791–805, doi:10.1038/35097557 (2001).
Strack, S., Khan, M. M., Wild, F., Rall, A. & Rudolf, R. Turnover of acetylcholine receptors at the endplate revisited: novel insights into nerve-dependent behavior. J Muscle Res Cell Motil 36, 517–524, doi:10.1007/s10974-015-9418-0 (2015).
Tintignac, L. A., Brenner, H. R. & Ruegg, M. A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiological reviews 95, 809–852, doi:10.1152/physrev.00033.2014 (2015).
Verschuuren, J., Strijbos, E. & Vincent, A. Neuromuscular junction disorders. Handb Clin Neurol 133, 447–466, doi:10.1016/B978-0-444-63432-0.00024-4 (2016).
Ervasti, J. M. & Campbell, K. P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 122, 809–823 (1993).
Pilgram, G. S., Potikanond, S., Baines, R. A., Fradkin, L. G. & Noordermeer, J. N. The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol Neurobiol 41, 1–21, doi:10.1007/s12035-009-8089-5 (2010).
Shin, J., Tajrishi, M. M., Ogura, Y. & Kumar, A. Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol 45, 2266–2279, doi:10.1016/j.biocel.2013.05.001 (2013).
Bao, X. et al. Tumor suppressor function of laminin-binding alpha-dystroglycan requires a distinct beta3-N-acetylglucosaminyltransferase. Proc Natl Acad Sci USA 106, 12109–12114, doi:10.1073/pnas.0904515106 (2009).
Gong, Y. et al. Alpha-dystroglycan is involved in positive selection of thymocytes by participating in immunological synapse formation. FASEB J 22, 1426–1439, doi:10.1096/fj.07-9264com (2008).
Lien, C. F. et al. Absence of glial alpha-dystrobrevin causes abnormalities of the blood-brain barrier and progressive brain edema. J Biol Chem 287, 41374–41385, doi:10.1074/jbc.M112.400044 (2012).
Peters, M. F. et al. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J Cell Biol 142, 1269–1278 (1998).
Gingras, J. et al. Alpha-Dystrobrevin-1 recruits Grb2 and alpha-catulin to organize neurotransmitter receptors at the neuromuscular junction. J Cell Sci 129, 898–911, doi:10.1242/jcs.181180 (2016).
Grady, R. M. et al. Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: roles in skeletal muscle and its neuromuscular and myotendinous junctions. J Cell Biol 160, 741–752, doi:10.1083/jcb.200209045 (2003).
Aittaleb, M., Martinez-Pena, Y. V. I. & Akaaboune, M. Spatial distribution and molecular dynamics of dystrophin glycoprotein components at the neuromuscular junction in vivo. Journal of cell science, doi:10.1242/jcs.198358 (2017).
Serra-Pages, C., Medley, Q. G., Tang, M., Hart, A. & Streuli, M. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem 273, 15611–15620 (1998).
Kriajevska, M. et al. Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1). J Biol Chem 277, 5229–5235, doi:10.1074/jbc.M110976200 (2002).
Lyssand, J. S. et al. Alpha-dystrobrevin-1 recruits alpha-catulin to the alpha1D-adrenergic receptor/dystrophin-associated protein complex signalosome. Proc Natl Acad Sci USA 107, 21854–21859, doi:10.1073/pnas.1010819107 (2010).
Astro, V., Chiaretti, S., Magistrati, E., Fivaz, M. & de Curtis, I. Liprin-alpha1, ERC1 and LL5 define polarized and dynamic structures that are implicated in cell migration. J Cell Sci 127, 3862–3876, doi:10.1242/jcs.155663 (2014).
Astro, V. & de Curtis, I. Plasma membrane-associated platforms: dynamic scaffolds that organize membrane-associated events. Sci Signal 8, re1, doi:10.1126/scisignal.aaa3312 (2015).
Asperti, C., Astro, V., Totaro, A., Paris, S. & de Curtis, I. Liprin-alpha1 promotes cell spreading on the extracellular matrix by affecting the distribution of activated integrins. Journal of cell science 122, 3225–3232, doi:10.1242/jcs.054155 (2009).
van der Vaart, B. et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell 27, 145–160, doi:10.1016/j.devcel.2013.09.010 (2013).
Stehbens, S. J. et al. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat Cell Biol 16, 561–573, doi:10.1038/ncb2975 (2014).
Lansbergen, G. et al. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev Cell 11, 21–32, doi:10.1016/j.devcel.2006.05.012 (2006).
Kishi, M., Kummer, T. T., Eglen, S. J. & Sanes, J. R. LL5beta: a regulator of postsynaptic differentiation identified in a screen for synaptically enriched transcripts at the neuromuscular junction. J Cell Biol 169, 355–366, doi:10.1083/jcb.200411012 (2005).
Basu, S. et al. CLASP2-dependent microtubule capture at the neuromuscular junction membrane requires LL5beta and actin for focal delivery of acetylcholine receptor vesicles. Mol Biol Cell 26, 938–951, doi:10.1091/mbc.E14-06-1158 (2015).
Proszynski, T. J. & Sanes, J. R. Amotl2 interacts with LL5beta, localizes to podosomes and regulates postsynaptic differentiation in muscle. J Cell Sci 126, 2225–2235, doi:10.1242/jcs.121327 (2013).
Basu, S. et al. Acetylcholine receptor (AChR) clustering is regulated both by glycogen synthase kinase 3beta (GSK3beta)-dependent phosphorylation and the level of CLIP-associated protein 2 (CLASP2) mediating the capture of microtubule plus-ends. The Journal of biological chemistry 289, 30857–30867, doi:10.1074/jbc.M114.589457 (2014).
Schmidt, N. et al. Agrin regulates CLASP2-mediated capture of microtubules at the neuromuscular junction synaptic membrane. The Journal of cell biology 198, 421–437, doi:10.1083/jcb.201111130 (2012).
Spangler, S. A. & Hoogenraad, C. C. Liprin-alpha proteins: scaffold molecules for synapse maturation. Biochem Soc Trans 35, 1278–1282, doi:10.1042/BST0351278 (2007).
Spangler, S. A. et al. Differential expression of liprin-alpha family proteins in the brain suggests functional diversification. J Comp Neurol 519, 3040–3060, doi:10.1002/cne.22665 (2011).
Zurner, M., Mittelstaedt, T., tom Dieck, S., Becker, A. & Schoch, S. Analyses of the spatiotemporal expression and subcellular localization of liprin-alpha proteins. J Comp Neurol 519, 3019–3039, doi:10.1002/cne.22664 (2011).
Kaufmann, N., DeProto, J., Ranjan, R., Wan, H. & Van Vactor, D. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron 34, 27–38 (2002).
Choe, K. M., Prakash, S., Bright, A. & Clandinin, T. R. Liprin-alpha is required for photoreceptor target selection in Drosophila. Proc Natl Acad Sci USA 103, 11601–11606, doi:10.1073/pnas.0601185103 (2006).
Astigarraga, S., Hofmeyer, K., Farajian, R. & Treisman, J. E. Three Drosophila liprins interact to control synapse formation. J Neurosci 30, 15358–15368, doi:10.1523/JNEUROSCI.1862-10.2010 (2010).
Dai, Y. et al. SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat Neurosci 9, 1479–1487, doi:10.1038/nn1808 (2006).
Ko, J., Na, M., Kim, S., Lee, J. R. & Kim, E. Interaction of the ERC family of RIM-binding proteins with the liprin-alpha family of multidomain proteins. J Biol Chem 278, 42377–42385, doi:10.1074/jbc.M307561200 (2003).
Kowalski, J. R. et al. The Anaphase-Promoting Complex (APC) ubiquitin ligase regulates GABA transmission at the C. elegans neuromuscular junction. Mol Cell Neurosci 58, 62–75, doi:10.1016/j.mcn.2013.12.001 (2014).
Patel, M. R. et al. Hierarchical assembly of presynaptic components in defined C. elegans synapses. Nat Neurosci 9, 1488–1498, doi:10.1038/nn1806 (2006).
Zhen, M. & Jin, Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 401, 371–375, doi:10.1038/43886 (1999).
Sjoblom, B., Salmazo, A. & Djinovic-Carugo, K. Alpha-actinin structure and regulation. Cell Mol Life Sci 65, 2688–2701, doi:10.1007/s00018-008-8080-8 (2008).
Lanner, J. T., Georgiou, D. K., Joshi, A. D. & Hamilton, S. L. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2, a003996, doi:10.1101/cshperspect.a003996 (2010).
Pinotsis, N. et al. Superhelical architecture of the myosin filament-linking protein myomesin with unusual elastic properties. PLoS Biol 10, e1001261, doi:10.1371/journal.pbio.1001261 (2012).
Penney, R. K., Prentis, P. F., Marshall, P. A. & Goldspink, G. Differentiation of muscle and the determination of ultimate tissue size. Cell Tissue Res 228, 375–388 (1983).
Sher, J. & Cardasis, C. Skeletal muscle fiber types in the adult mouse. Acta Neurol Scand 54, 45–56 (1976).
Jones, R. A. et al. NMJ-morph reveals principal components of synaptic morphology influencing structure-function relationships at the neuromuscular junction. Open Biol 6, doi:10.1098/rsob.160240 (2016).
Chakkalakal, J. V., Nishimune, H., Ruas, J. L., Spiegelman, B. M. & Sanes, J. R. Retrograde influence of muscle fibers on their innervation revealed by a novel marker for slow motoneurons. Development 137, 3489–3499, doi:10.1242/dev.053348 (2010).
Bernadzki, K. M., Rojek, K. O. & Proszynski, T. J. Podosomes in muscle cells and their role in the remodeling of neuromuscular postsynaptic machinery. Eur J Cell Biol 93, 478–485, doi:10.1016/j.ejcb.2014.06.002 (2014).
Levitt, T. A., Loring, R. H. & Salpeter, M. M. Neuronal control of acetylcholine receptor turnover rate at a vertebrate neuromuscular junction. Science 210, 550–551 (1980).
Matsuda, Y. et al. Scanning electron microscopic study of denervated and reinnervated neuromuscular junction. Muscle Nerve 11, 1266–1271, doi:10.1002/mus.880111211 (1988).
Kummer, T. T., Misgeld, T., Lichtman, J. W. & Sanes, J. R. Nerve-independent formation of a topologically complex postsynaptic apparatus. J Cell Biol 164, 1077–1087, doi:10.1083/jcb.200401115 (2004).
Oh, H. J. et al. Interaction of alpha-catulin with dystrobrevin contributes to integrity of dystrophin complex in muscle. The Journal of biological chemistry 287, 21717–21728, doi:10.1074/jbc.M112.369496 (2012).
Pawlikowski, B. T. & Maimone, M. M. Formation of complex AChR aggregates in vitro requires alpha-dystrobrevin. Developmental neurobiology 69, 326–338, doi:10.1002/dneu.20703 (2009).
Connolly, J. A. Microtubules, microfilaments and the transport of acetylcholine receptors in embryonic myotubes. Exp Cell Res 159, 430–440 (1985).
Vale, R. D. Intracellular transport using microtubule-based motors. Annu Rev Cell Biol 3, 347–378, doi:10.1146/annurev.cb.03.110187.002023 (1987).
Woehlke, G. & Schliwa, M. Walking on two heads: the many talents of kinesin. Nat Rev Mol Cell Biol 1, 50–58, doi:10.1038/35036069 (2000).
Geng, L., Zhang, H. L. & Peng, H. B. The formation of acetylcholine receptor clusters visualized with quantum dots. BMC neuroscience 10, 80, doi:10.1186/1471-2202-10-80 (2009).
Johnson, K. G. et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron 49, 517–531, doi:10.1016/j.neuron.2006.01.026 (2006).
Strochlic, L. et al. MAGI-1c: a synaptic MAGUK interacting with muSK at the vertebrate neuromuscular junction. J Cell Biol 153, 1127–1132 (2001).
Martinez-Pena y Valenzuela, I., Mouslim, C. & Akaaboune, M. Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J Neurosci 30, 12455–12465, doi:10.1523/JNEUROSCI.3309-10.2010 (2010).
Rosenblatt, J. D., Lunt, A. I., Parry, D. J. & Partridge, T. A. Culturing satellite cells from living single muscle fiber explants. In vitro cellular & developmental biology. Animal 31, 773–779, doi:10.1007/BF02634119 (1995).
Pasut, A., Jones, A. E. & Rudnicki, M. A. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. Journal of visualized experiments: JoVE, e50074, doi:10.3791/50074 (2013).
Mouslim, C., Aittaleb, M., Hume, R. I. & Akaaboune, M. A role for the calmodulin kinase II-related anchoring protein (alphakap) in maintaining the stability of nicotinic acetylcholine receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience 32, 5177–5185, doi:10.1523/JNEUROSCI.6477-11.2012 (2012).
Grady, R. M. et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nature cell biology 1, 215–220, doi:10.1038/12034 (1999).
Acknowledgements
This work was supported by Polish National Science Center Opus 5 grant no. 2013/09/B/NZ3/03524 (awarded to T.J.P.) and Preludium 10 grant no. 2015/19/N/NZ5/02268 (awarded to K.M.B.). M.P. was also supported by the European Union’s Horizon 2020 research and innovation program under Marie Sklodowska-Curie grant no. 665735 (Bio4Med) and the Polish Ministry of Science and Higher Education within 2016–2020 funds for the implementation of international projects (agreement no. 3548/H2020/COFUND/2016/2). The project was performed using the CePT infrastructure financed by the European Union, European Regional Development Fund, within the Operational Program “Innovative Economy” for 2007–2013.
Author information
Authors and Affiliations
Contributions
K.M.B., M.G., M.P, and P.M. performed the experiments and analyzed data. M.J.R. and P.N. provided reagents and suggestions for the experiments. K.M.B., M.P and T.J.P wrote the manuscript. All authors planned the experiments, analyzed data and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bernadzki, K.M., Gawor, M., Pęziński, M. et al. Liprin-α-1 is a novel component of the murine neuromuscular junction and is involved in the organization of the postsynaptic machinery. Sci Rep 7, 9116 (2017). https://doi.org/10.1038/s41598-017-09590-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09590-7
This article is cited by
-
Rare disease research workflow using multilayer networks elucidates the molecular determinants of severity in Congenital Myasthenic Syndromes
Nature Communications (2024)
-
Optical recognition of constructs using hyperspectral imaging and detection (ORCHID)
Scientific Reports (2022)
-
Liprins in oncogenic signaling and cancer cell adhesion
Oncogene (2021)
-
The ERC1 scaffold protein implicated in cell motility drives the assembly of a liquid phase
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
