
Abstract
The human cytomegalovirus (HCMV) terminase complex consists of several components acting together to cleave viral DNA into unit length genomes and translocate them into capsids, a critical process in the production of infectious virions subsequent to DNA replication. Previous studies suggest that the carboxyl-terminal portion of the pUL56 subunit interacts with the pUL89 subunit. However, the specific interacting residues of pUL56 remain unknown. We identified a conserved sequence in the C-terminal moiety of pUL56 (671WMVVKYMGFF680). Overrepresentation of conserved aromatic amino acids through 20 herpesviruses homologues of pUL56 suggests an involvement of this short peptide into the interaction between the larger pUL56 terminase subunit and the smaller pUL89 subunit. Use of Alpha technology highlighted an interaction between pUL56 and pUL89 driven through the peptide 671WMVVKYMGFF680. A deletion of these residues blocks viral replication. We hypothesize that it is the consequence of the disruption of the pUL56-pUL89 interaction. These results show that this motif is essential for HCMV replication and could be a target for development of new small antiviral drugs or peptidomimetics.
Similar content being viewed by others


Introduction
Human cytomegalovirus (HCMV), a beta herpesvirus, can cause serious diseases in immunocompromised patients. Current antiviral inhibitors (ganciclovir, cidofovir and foscarnet) all target the viral DNA polymerase. They have adverse effects and prolonged treatment can select for drug resistance mutations either in the viral polymerase pUL54, the kinase pUL97 or both of them1, 2. Thus, we need new drugs targeting others stages of replication. The terminase complex is highly specific for HCMV, has no counterpart in the human organism, and thus represents a target of choice for new antivirals development. This has been confirmed by the recent development of letermovir in the transplant setting3, 4.
DNA packaging process requires several proteins such as pUL56 and pUL89, the large and small terminase subunits, respectively. Recently, four additional proteins were shown to be also implicated in this process, namely, pUL51, pUL52, pUL77, pUL935,6,7,8,9,10. This process is driven by specific interactions of protein-DNA and protein-protein to cleave and package unit length genomic DNA into an empty capsid.
Evidence suggests that the large subunit pUL56 has a crucial role in DNA cleavage/packaging, containing many of the functional sites required for this process like interaction with the portal protein pUL104, endonuclease activity, and more interestingly an ATP-binding site (amino acids 709 to 723)11. Although the association between pUL56 and pUL89 has already been reported, the residues of pUL56 involved in the terminase complex integrity are still unknown12, 13. Nevertheless, co-immunoprecipitation experiments showing an interaction between the C-terminal half of pUL56 (pUL56-Cter) and pUL89 were confirmed by other results12, 13.
Because knowledge of terminase functional and interaction domains is important both for the development of drugs targeting the DNA packaging stage and for the improvement of existing ones such as letermovir, the aim of the current study is to identify a minimum peptide of pUL56 with a putative key role in its interaction with pUL89. Sequence alignments encouraged us to focus on the putative involvement of one part of the pUL56 sequence into its interface with pUL89. BAC mutagenesis and Alpha technology using purified proteins subsequently validated that the aromatic rich peptide 671WMVVKYMGFF680 pUL56(671-680) in the C-terminal of pUL56 is involved in interaction with pUL89. These results could contribute for development of new antiviral drugs, peptides or antibodies against HCMV.
Results
A putative conserved protein interface in pUL56 subunit
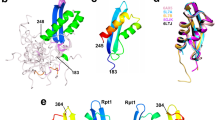
Selection of a potent pUL56 fragment for pUL89 interaction was supported by three hints. First, based on the sequences alignment of pUL56 with 20 herpesviruses homologues, the peptide 671WMVVKYMGFF680 pUL56(671-680) seems to be broadly conserved in betaherpesviruses proteins, which supported a major role either in function or structure of pUL56. Secondly, as shown in Fig. 1, its secondary structure is predicted as an alpha helix. Previous studies demonstrated that the peptide pUL89(580-600) implicated in the pUL56-pUL89 interface13 adopts an alpha helix secondary structure14, 15. Moreover, wide protein-protein interfaces analyses revealed a preferential interaction of an helix of one protein with one of its counterpart16, 17.
Thirdly, pUL56(671-680) is within the C-terminal part previously described to be sufficient for interaction with pUL8912. Interestingly, this motif belongs to the pUL56 region carrying the ATP binding site. As a parallel, pUL89(580-600) is enclosed into the endonuclease domain of pUL8915. Both activities, ATPase of pUL56 on the one hand and nuclease of pUL89 on the other hand are dependent on the association between the two terminase subunits12, 18. Taken together, these observations make pUL56(671-680) a good candidate to interact with pUL89.

(A) Structure of HCMV terminase subunit pUL56 with a putative leucine zipper pattern annotated as pUL56-LZ22. (B) Sequences alignment of conserved regions from 21 herpesviruses. Sequence numbering is consistent with that of AD169 residues. Key residues are highlighted as white letters on a black background. SSP: Secondary structure prediction of pUL56-LZ (h = α helix).
A deletion or targeted mutations of 671WMVVKYMGFF680 pUL56 domain affects viral replication in MRC-5 cells
To evaluate the importance of the pUL56 predicted domain for viral replication, we produced by “en passant” mutagenesis recombinant EGFP-virus with complete deletion of UL56(671-680) or point mutations in this sequence. Analysis of HCMV genome confirmed that UL56 sequence does not encode a gene on the other strand19. Thus, mutations in the virus are silent on the other strand and thus cannot impact the function of another gene expressed from the other strand. To ensure that no other mutations that could have a negative impact on viral replication was introduced in the BAC backbone during the manipulations, we performed NGS sequencing on both the original BAC and the mutants. The deletion was found in 100% of the mutants BAC sequences whereas other SNPs were located in genes non essential for viral replication and represent less than 30% of the sequences both in the original BAC and in the mutants.
Unlike the wild-type HCMV-BAC, eleven days after the transfection of human fibroblasts (MRC-5 cells), we observed no foci of cytopathic effect for the mutant which has a deletion of 671WMVVKYMGFF680 sequence (Fig. 2). This deletion dramatically impaired viral replication and propagation in cell-culture. In the same way, recombinant EGFP-viruses with a single or a combination of mutations among W671A, Y676A, F679A and F680A do not produce progeny virion as well. These residues were selected for mutagenesis because they are perfectly or for the less highly conserved (i.e. replaced by another aromatic amino acid) among all the 20 herpesviruses homologues of pUL56 (Fig. 1). To check if these deletion or mutations may disrupt another step of the viral replication, immunostaining assays were performed to detect proteins produced at immediate early and late stages of viral cycle (IE and late proteins). Expression of immediate early (IEA) and late (gB) viral genes were detected indicating that mutations have no impact on viral gene expression (Fig. 3). Therefore W671, Y676, F679 and F680 within pUL56(671-580) are critical amino acids for viral replication.

Plaque formation assay in MRC-5 cells after transfection of HCMV-BAC GFP UL56 WT (AD169) or recombinant virus strains with a deletion, a combination of mutations or a single mutation. Green fluorescent foci (white arrow) were observed with the wild-type HCMV-BAC GFP UL56 and single infected cells (grey arrow) were observed with the other recombinants viruses. Eleven days after transfection of human fibroblasts, we observed no cytopathic effect for either the mutant with a deletion of 671WMVVKYMGFF680 sequence, or a combination of mutations or single mutations. These mutations dramatically impaired viral replication and propagation in cell-culture.

Fragment 671WMVVKYMGFF680 of pUL56 is not required for viral gene expression. MRC-5 were transfected with HCMV-BAC WT or the mutant with a deletion of 671WMVVKYMGFF680 sequence. Five days after transfection, immunostaining was performed for immediate early (I.E.A) (white arrow) and late (gB) (grey arrow) viral proteins.
pUL56(671-680) is necessary for pUL89 association
HEK293 were transfected with SC784 and pCI-neo His-89 expression plasmids and protein-protein interactions were carried out by the Alpha assay. This technology represents a powerful method to highlight protein-protein interactions20, 21. Since we have no virion production for mutant viruses, we chose to study in vitro biochemical interactions after protein overexpression in HEK cells which allow introduction of tags (HA and His) for the Alpha assay.
Alpha assay needs both acceptor and donor beads. For this study, HA-coated Donor beads and His-coated Acceptor were used. A singlet of oxygen diffuses from Donor bead to the Acceptor bead, resulting in light production at 615 nm. In the absence of a specific biological interaction between proteins, singlet molecules produced by the Donor bead cannot be detected beyond 200 nm from the Acceptor bead (Fig. 4). First step consisted in verifying the interaction between pUL56-WT and pUL89-WT as a valuable positive control. Alpha assays with 3xHA-pUL56 and 6xHis-pUL89 results in the production of over 9,000 relative light units (RLU), over two-fold more than negative controls (3xHA-pUL56 or 6x His-pUL89) (Fig. 5). pUL56 depleted of its W671-F680 fragment was in turn soaked with pUL89-WT and their affinity assessed by Alpha analysis. The lack of pUL56(671-680) decreased the interaction signal by 50% which is significant in this assay. These data strongly suggest that 671WMVVKYMGFF680 is necessary for interaction with pUL89.

Scheme of an Alpha protein-protein interaction assay, using HA-coated Donor beads, His-coated Acceptor beads, 3xHA-pUL56, 6x His-pUL89. In an Alpha interaction assay, one protein is captured on the Donor beads, and the other protein is captured on the Acceptor beads. In case of interaction, the Donor bead is brought into proximity of the Acceptor bead, and excitation of the Donor bead will result in signal generation dependent on the presence of an interaction.

Determination of pUL56 binding domains for the interaction with pUL89. (A) Analysis of protein production for alpha assay. Immunoblot was performed using the anti-His antibody for pUL89-His or the anti-HA antibody for pUL56 and pUL56-HA Del W671-F680 and secondary rabbit anti-mouse HRP conjugated antibody. (B) Alpha assay results. The Alpha assay for the binding of full-length pUL89 (His-pUL89, 1.5E + 03 nM) was performed with 5E + 02 nM wild-type pUL56 (HA-pUL56) or a deletion mutant of pUL56 (HA-pUL56 Del W671-F680). As a negative control, proteins were used alone and a reaction was performed without proteins (mock). Two measures for each reaction were performed in duplicate.
Discussion
Protein-protein interactions are essential for several biological pathways such as herpesviruses DNA-packaging. Terminase subunits are proteins forming a hetero-oligomeric complex involved in this process. The HCMV terminase complex is composed of the large subunit pUL56 and the small subunit pUL89. Four additional HCMV proteins, namely, pUL51, pUL52, pUL77, pUL93 contribute also to this process6,7,8,9,10. To date, the structural knowledge of herpesviruses terminases is poorly understood including interactions inside its molecular assembly. Previous studies suggest that the large subunit pUL56 has an essential role in this process and contains several functional patterns as a zinc finger domain and a C-terminal nuclear localization signal (NLS)22, 23. Although pUL56-Cter is sufficient to interact with pUL89(580-600) subunit13, precise moieties of pUL56 constituting the interface against pUL89 are still unknown12, 13.
In the present study, we show that deletion of pUL56(671-680) abolishes HCMV replication. We then furtherly checked the impact of this sequence on interaction with pUL89 using Alpha technology. Consistent with our hypothesis, deletion of residues W671 to F680 drastically affects the interaction between pUL56 and pUL89. We propose that the peptide WMVVKYMGFF is crucial for the interaction with pUL89 and thereby for DNA-packaging. Interestingly, this motif is close to the pUL56 region carrying the ATP-binding site (amino acids 709 to 723)11. It has been previously demonstrated that the ATPase activity responsible for HCMV DNA translocation into capsids is only associated with pUL56 and is enhanced by up to 30% when pUL56 is associated with pUL8912. In this study, we show a close proximity in the pUL56 sequence between interaction site with pUL89 and the ATP-binding site. Moreover, it is important to highlight that interaction locus W671 to F680 is near to the point mutation A662V selected under tomeglovir (Bay38-4766), a non-nucleoside inhibitor of HCMV24.
The terminase complex is highly CMV-specific, as no counterpart in mammalian cells exists, and thus represents a promising therapeutic strategy for new antivirals development. This has been confirmed by the recent development of the terminase inhibitor letermovir in the transplant setting3. However, its precise site of action in the terminase complex is not yet understood. Clues are offered by a large number of letermovir resistance mutations in UL56 that have been identified, clustered at UL56 codons 231–36925, 26, and the uncommon selection of UL89 D344E under letermovir, which combines with UL56 mutations to increase the overall level of drug resistance. This hints at the possibility of regions of pUL56 and pUL89 that are close to a small molecule drug binding site. Other terminase inhibitors preferentially select for UL89 mutations, such as D344E for benzimidazole compounds, along with UL56 mutations at loci such as codons 204 and 662, suggesting yet other possibilities for subunit interactions. A better understanding of all these potential interactions between terminase subunits of HCMV could be valuable when studying the mechanism of action of drugs and the design of new antivirals such as peptidomimetics27, antibodies or small molecules that target the interaction domain between these essential viral proteins. Indeed, alteration of protein-protein interaction could be used as a way of inhibition of HCMV replication. A modified peptide based on the WMVVKYMGFF scaffold could serve as molecular target-decoy by interacting with pUL89 and so disrupt the interaction between pUL56 and pUL89. Moreover, we could consider combinations tests of peptides or antibodies with currently available anti-HCMV drugs.
In conclusion, the data from the present study demonstrated that the pUL56 sequence 671WMVVKYMGFF680 is necessary for its interaction with pUL89 and could constitute a good target to suppress this interaction and thus block HCMV replication.
Materials and Methods
Identification of conserved patterns and secondary structure prediction
The pUL56 amino acid sequence of reference strain AD16928 was aligned with the sequences of 21 homologous proteins from other herpesviruses, as described in Supplementary Table 1. Alignments were performed with Clustal Omega (Ω) multiple sequence alignment (MSA) tool provided by the EMBL-EBI bioinformatics web and programmatic tools framework29,30,31. Secondary structure prediction was carried by Phyre2 web portal32.
Cells and bacterial strains
Human fibroblasts MRC-5 (Biomerieux, France) were cultivated at 37 °C in 5% CO2 and grown in minimal essential medium (MEM) containing 10% fetal bovine serum with antibiotics. HEK293 (ATCC® CRL-1573™) were cultivated at 37 °C in 5% CO2 and grown in minimal essential medium (MEM) containing 10% fetal bovine serum with antibiotics. E. coli strain DH5α and StellarTM (Clontech, USA) were used for cloning procedures. E. coli strain GS1783 was used for BAC mutagenesis33.
BAC mutagenesis and reconstitution of mutant viruses
Conserved domains were deleted by “en passant” mutagenesis, a two-step markerless Red recombination system for BAC mutagenesis in E. coli strain GS1783. UL56 point mutations were introduced into an EGFP-expressing HCMV-BAC33 to generate several mutants (primers used for mutagenesis are described in Supplementary Table 2). Presence of mutations in UL56 gene of each virus was confirmed by sequencing prior to transfection. The HCMV-BAC contains an enhanced green fluorescent protein (EGFP) gene in the unique short region and was derived from parental strain pHB5, a BAC-cloned genome of the CMV laboratory strain AD16933. The impact of different mutations on viral growth was assessed using transfection of mutated HCMV-BAC into human fibroblasts MRC-5 using liposomal reagent TransfastTM (Promega, USA) following manufacturer’s instructions34.
Library construction and whole-genome DNA sequencing
After HCMV-BAC preparation, amplicons were purified using magnetic beads (Agencourt AMPure XP) and fragmented using the Ion Xpress Plus DNA Fragment Library Preparation kit (Life Technologies). Barcodes adapters were ligated to fragment ends and 250 bp fragments were collected. The library was PCR amplified, then sequenced on the Ion Proton with the Ion Sequencing kit (Life Technologies). Bases callings were performed with Torrent Suite Software version 5.0.2. Mutations were obtained using Torrent Variant Caller using Somatic variant frequency and AD169_ATCC as reference. Mutations were then filtered against reference (Wild-type HCMV-BAC) using vcftools version 0.1.13.
Viral immediate early and late protein expression
A transfection of mutated HCMV-BAC into human fibroblasts MRC-5 using liposomal reagent TransfastTM (Promega, USA) was performed. Cells were fixed at 5 days post transfection, and immunostaining was performed for viral immediate early (anti-IE1 antibody; Argene, France) and late (anti-gB antibody; Abcam, United Kingdom) proteins in transfected cells.
Plasmids construction for Alpha analysis
For protein production, the SC784 expression plasmid encoding full-length amino-terminal 3xHA-tagged pUL56 and driven by an upstream HCMV major immediate early promoter was cloned in vector pGEM3z. In-Fusion® (Clontech, USA) kit was used following manufacturer’s instructions to clone several UL56 mutants from source HCMV-BAC in SC784 plasmid. ORF encoding pUL89 is composed of two exons separated by an intron. Both exons were generated by assembling PCR from AD169 strain and cloned into pCI-neo (Promega, USA) with His tag to obtain pCI-neo His-pUL89. Transformations were performed in DH5∝ cells. The nucleotide sequence of all constructs generated was verified by Sanger sequencing prior to use.
Transfection and proteins purification
HEK293 were transfected with the appropriate expression vectors using liposomal reagent TransfastTM following manufacturer’s instructions, washed and lysed 48 h later with CelLytic M (Sigma-Aldrich, USA). Lysates were cleared by centrifugation.
For purification of HA-tagged pUL56, the cell-free reaction was performed with Anti-HA Immunoprecipitation Kit according to the manufacturer’s protocol (Sigma-Aldrich, USA).
For purification of His-tagged pUL89, the cell-free reaction was performed with Ni resin (Clontech, USA).
All proteins were concentrated approximately 5-fold using Pall centrifugal filters (Pall, USA), and protein concentration was determined by the Bradford method using bovine serum albumin (Sigma-Aldrich, USA) as standard protein.
Western blotting
SDS-PAGE was performed under reducing conditions on Mini-PROTEAN TGX Stain-Free gels (BioRad, USA). Proteins were then transferred onto a Trans-Blot Turbo PVDF Western blotting membrane (BioRad, USA). Antibody dilutions were 1:1,000 for the mouse anti-HA antibody (catalogue number: 2367, Cell Signaling, USA), mouse anti-His antibody (catalogue number: 2366, Cell Signaling, USA) and secondary anti-mouse horseradish peroxidase (HRP)-linked antibody (catalogue number: 7076, Cell Signaling, USA). Signals were visualized using the Substrat HRP Immobilon Western (Merck Millipore, USA) and a ChemiDoc imager (BioRad, USA).
Protein/Protein interaction analysis by Alpha
Alpha (Amplified Luminescent Proximity Homogeneous Assay) experiments were conducted according to the manufacturer’s protocol (PerkinElmer, USA). Five μL of transfected MRC-5 lysate with HCMV-BAC is first disposed in wells of a 96-well AlphaPlate. The final concentration of each proteins was optimized to obtain the best value of interaction. Ten μL of each purified protein were combined (to give a final assay concentration of 500 nM of 3xHA-pUL56 and 1,5 µM of 6xHis-pUL89). Ten μL and 15 μL of 10 mg/mL of donor beads and acceptor beads, respectively, were added and incubated for 1 hour. Plates were read on a PerkinElmer EnVisionTM plate reader using an excitation wavelength of 680 nm and emission detection was set at 615 nm.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Alain, S. et al. Detection of ganciclovir resistance after valacyclovir-prophylaxis in renal transplant recipients with active cytomegalovirus infection. J. Med. Virol. 73, 566–573 (2004).
Hantz, S. et al. Drug-resistant cytomegalovirus in transplant recipients: a French cohort study. J. Antimicrob. Chemother. 65, 2628–2640 (2010).
Lischka, P. et al. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 54, 1290–1297 (2010).
Melendez, D. P. & Razonable, R. R. Letermovir and inhibitors of the terminase complex: a promising new class of investigational antiviral drugs against human cytomegalovirus. Infect. Drug Resist 8, 269–277 (2015).
Borst, E. M., Wagner, K., Binz, A., Sodeik, B. & Messerle, M. The essential human cytomegalovirus gene UL52 is required for cleavage-packaging of the viral genome. J. Virol. 82, 2065–2078 (2008).
Borst, E. M. et al. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J. Virol. 87, 1720–1732 (2013).
Borst, E. M. et al. The Essential Human Cytomegalovirus Proteins pUL77 and pUL93 are Structural Components Necessary for Viral Genome Encapsidation. J. Virol, doi:10.1128/JVI.00384-16 (2016).
Köppen-Rung, P., Dittmer, A. & Bogner, E. Intracellular distributions of capsid-associated pUL77 of HCMV and interactions with packaging proteins and pUL93. J. Virol, doi:10.1128/JVI.00351-16 (2016).
DeRussy, B. M. & Tandon, R. Human cytomegalovirus pUL93 is required for viral genome cleavage and packaging. J. Virol. doi:10.1128/JVI.02382-15 (2015).
DeRussy, B. M., Boland, M. T. & Tandon, R. Human Cytomegalovirus pUL93 Links Nucleocapsid Maturation and Nuclear Egress. J. Virol, doi:10.1128/JVI.00728-16 (2016).
Scholz, B., Rechter, S., Drach, J. C., Townsend, L. B. & Bogner, E. Identification of the ATP-binding site in the terminase subunit pUL56 of human cytomegalovirus. Nucleic Acids Res 31, 1426–1433 (2003).
Hwang, J.-S. & Bogner, E. ATPase activity of the terminase subunit pUL56 of human cytomegalovirus. J. Biol. Chem. 277, 6943–6948 (2002).
Thoma, C. et al. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry (Mosc.) 45, 8855–8863 (2006).
Couvreux, A. et al. Insight into the structure of the pUL89 C-terminal domain of the human cytomegalovirus terminase complex. Proteins 78, 1520–1530 (2010).
Nadal, M. et al. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. USA 107, 16078–16083 (2010).
Eilers, M., Patel, A. B., Liu, W. & Smith, S. O. Comparison of helix interactions in membrane and soluble alpha-bundle proteins. Biophys. J. 82, 2720–2736 (2002).
Ansari, S. & Helms, V. Statistical analysis of predominantly transient protein-protein interfaces. Proteins 61, 344–355 (2005).
Scheffczik, H., Savva, C. G. W., Holzenburg, A., Kolesnikova, L. & Bogner, E. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res 30, 1695–1703 (2002).
Bradley, A. J. et al. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J. Gen. Virol. 90, 2375–2380 (2009).
Ullman, E. F. et al. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. USA 91, 5426–5430 (1994).
Waller, H., Chatterji, U., Gallay, P., Parkinson, T. & Targett-Adams, P. The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J. Virol. Methods 165, 202–210 (2010).
Champier, G. et al. Putative functional domains of human cytomegalovirus pUL56 involved in dimerization and benzimidazole D-ribonucleoside activity. Antivir. Ther. 13, 643–654 (2008).
Giesen, K., Radsak, K. & Bogner, E. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J. Gen. Virol 81, 2231–2244 (2000).
Buerger, I. et al. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J. Virol. 75, 9077–9086 (2001).
Goldner, T. et al. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 85, 10884–10893 (2011).
Chou, S. Rapid In Vitro Evolution of Human Cytomegalovirus UL56 Mutations That Confer Letermovir Resistance. Antimicrob. Agents Chemother. 59, 6588–6593 (2015).
Walker, M. P., Yao, N. & Hong, Z. Promising candidates for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs 12, 1269–1280 (2003).
Chee, M. S. et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol 154, 125–169 (1990).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
McWilliam, H. et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res 41, W597–600 (2013).
Li, W. et al. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43, W580–584 (2015).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Borst, E. M., Hahn, G., Koszinowski, U. H. & Messerle, M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J. Virol. 73, 8320–8329 (1999).
Hantz, S. et al. Novel DNA polymerase mutations conferring cytomegalovirus resistance: input of BAC-recombinant phenotyping and 3D model. Antiviral Res. 98, 130–134 (2013).
Acknowledgements
The authors gratefully acknowledge M. Gaschet for her helpful advice for library construction, E. Guérin and V. Tilloy from the technical facilities BISCEm of the Limoges University for their help in NGS analysis. This work was granted by the University of Limoges and the Inserm. G. Ligat received financial support from the National Reference Center for Cytomegaloviruses, the CHU Limoges, and Inserm. S. Chou was supported by U.S. NIH grant AI116635 and Department of Veterans Affairs research funds.
Author information
Authors and Affiliations
Contributions
G. Ligat designed and performed research experiments, statistical analysis, wrote the manuscript and prepared figures. C. Jacquet participated in research experiments. S. Chou designed the plasmid for pUL56 wild-type production. A. Couvreux performed sequences alignment. S. Alain and S. Hantz coordinated the research and manuscript writing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ligat, G., Jacquet, C., Chou, S. et al. Identification of a short sequence in the HCMV terminase pUL56 essential for interaction with pUL89 subunit. Sci Rep 7, 8796 (2017). https://doi.org/10.1038/s41598-017-09469-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09469-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
