
Abstract
Growing evidence shows that granulocyte macrophage colony-stimulating factor (GM-CSF) has progression-promoting potentials in certain solid tumors, which is largely attributed to the immunomodulatory function of this cytokine in tumor niches. However, little is known about the effect of GM-CSF on cancer cells. Herein, we show that chronic exposure of colon cancer cells to GM-CSF, which harbor its receptor, leads to occurrence of epithelial to mesenchymal transition (EMT), in time and dose-dependent manners. These GM-CSF-educated cancer cells exhibit enhanced ability of motility in vitro and in vivo. Furthermore, GM-CSF stimulation renders colon cancer cells more resistant to cytotoxic agents. Mechanistic investigation reveals that MAPK/ERK signaling and EMT-inducing transcription factor ZEB1 are critical to mediate these effects of GM-CSF. In specimen of CRC patients, high-level expression of GM-CSF positively correlates with local metastases in lymph nodes. Moreover, the co-expression of GM-CSF and its receptors as well as phosphorylated ERK1/2 are observed. Thus, our study for the first time identifies a progression-promoting function of GM-CSF in colon cancer by inducing EMT.
Similar content being viewed by others
Introduction
Granulocyte macrophage colony-stimulating factor (GM-CSF) is a hematopoietic growth factor produced by a variety of cell types, including macrophages, T lymphocytes, fibroblasts, endothelial cells, and keratinocytes following appropriate stimuli1. Recently, clinical data show that GM-CSF levels in the circulatory system were frequently elevated in patients suffering from colorectal cancer compared with healthy controls2,3,4,5,6. Furthermore, high GM-CSF expression in neoplastic lesions significantly correlated with local and distant metastasis, more advanced histological grade, and poor prognosis in patients with breast cancer7 and pancreatic ductal carcinoma8, 9. These disease-promoting effects of GM-CSF may be linked to modulation of immune reactions in tumor niche, involving macrophages7, 10, myeloid-derived suppressor cells9, 11, 12, and plasmacytoid predendritic cells13.
It is worthy to note that the receptors for GM-CSF are constitutively expressed on the surface of certain nonhematopoietic cancer cells14, 15, which suggests the possibility of responsiveness of cancer cells to GM-CSF challenge. Indeed, GM-CSF has been reported to support tumor growth and progression by autocrine stimulation of proliferation and migration in squamous cell carcinoma of skin or head and neck16, 17, gliomas18 and osteosarcoma19. Furthermore, it also enhances the invasive capacity of human cancer cells in vitro by increasing production and activation of several matrix metalloproteinases20,21,22. The mechanisms underlying the metastasis-promoting function of GM-CSF are still incompletely known and need to be further addressed.
Epithelial to mesenchymal transition (EMT) is a developmental process that seems to play an important role in tumor progression and metastasis in diverse solid tumors, including colorectal cancer23. The EMT phenotype is characterized by the loss of cell-to-cell adhesion with the disintegration of tight and gap junctions and a phenotypic change from an “epithelial” morphology to a motile, fibroblast-like morphology24, 25. The hallmark of EMT is the functional loss of E-cadherin, while additional cellular changes, such as reduced expression of epithelial markers cytokeratins and ZO-1, and the overexpression of mesenchymal markers N-cadherin, vimentin and fibronectin, are also often observed. Several transcription factors (TF) have been identified as master regulators of EMT, including the Snail, Zeb and Twist families, all of which interact with E-box elements located within the proximal region of the E-cadherin promoter24. Additionally, EMT-inducing TF expression is tightly regulated at different steps of transcription, translation and protein stability control by a variety of cell-intrinsic pathways as well as extracellular clues26.
In this study, we hypothesize that exogenous GM-CSF stimulation in colon cancer cells is competent to induce EMT and thereby enhance their motility in vitro and in vivo. Chronic treatment of GM-CSF or ectopic expression of GM-CSF led to the occurrence of EMT in receptor-expressing colon cancer cells, but not in receptor-negative counterparts. These GM-CSF-educated cancer cells displayed enhanced ability of migration and invasion in vitro and more metastatic nodules in a mouse model of colorectal liver metastasis. Mechanistically, MAPK/ERK signals and ZEB1 were required for GM-CSF-induced EMT phenotype. Furthermore, the exposure to GM-CSF rendered colon cancer cells more resistant to drug-mediated cell death.
Results
Chronic exposure to GM-CSF induces EMT in colon cancer cells
Firstly, we detected the constitutive expression of the receptors for GM-CSF in colon cancer cells. Indeed, in agreement with our previous study27, five of six cell lines virtually harbored two GM-CSF receptor subunits A and B, albeit different abundance (Fig. 1a and S1a). Of note, RKO cell line was devoid of subunit B of GM-CSF receptor (Fig. 1a and S1a), which was pivotal for signaling transduction upon the engagement of GM-CSF and its receptor. This cell line, thus, was utilized as GM-CSF-nonresponsive counterparts. Furthermore, we also observed the expression of GM-CSF receptors in tumor cells of specimens from CRC patients (Fig. 1b), indicating a direct responsiveness of colon cancer cells to GM-CSF. Importantly, a three-week treatment of exogenous GM-CSF resulted in EMT program in three cell lines (SW480, HCT116 and HT29), which expressed two subunits of GM-CSF receptors as described above. Specifically, chronic exposure to GM-CSF remarkably repressed E-cadherin expression and simultaneously up-regulated the expression of mesenchymal markers N-cadherin and fibronectin (Fig. 1c–e and S1b). In accord with this, the levels of EMT-regulating master regulators including SNAIL and TWIST1 were elevated dramatically following GM-CSF stimulation (Fig. 1c and e and S1b). Notably, the up-regulation of ZEB1 levels was most significant (Fig. 1c and S1b). Furthermore, the EMT-inducing effects of GM-CSF were time and dose-dependent (Fig. S1c). Intriguingly, GM-CSF stimulation did not affect the expression of epithelial and mesenchymal markers in GM-CSF-nonresponsive RKO cell line (Fig. 1c), indicating the importance of receptor-mediated signals in GM-CSF-induced EMT program. It was worthy to note that, given that RKO cell line was lack of E-cadherin28, we detected another epithelial marker Keratin 18 and showed no alteration of this marker expression following GM-CSF treatment (Fig. 1c). To further address this issue, we established the cell clones in SW480 cell lines transduced with GM-CSF-expressing construct (Fig. S2a). Compared with empty vector-transduced cell clones, GM-CSF–overexpressing clones exhibited EMT phenotypes (Fig. S2b and c), similar to those with stimulation of exogenous GM-CSF. Overall, these data suggest an EMT-inducing effect of GM-CSF exerted directly on colon cancer cells. In addition, we found that compared with its counterpart SW480 cell line, SW620 exhibited mesenchymal phenotype (Fig. S3a) and constitutively produced much more GM-CSF than SW480 (Fig. S3b). Intriguingly, blockade of GM-CSF bioactivity using neutralizing monoclonal antibodies rendered MET of SW620, including increased E-cadherin and decreased vimentin (Fig. S3c), indicating that GM-CSF may be a key factor for EMT program and maintenance.

Chronic stimulation of colon cancer cells with GM-CSF leads to EMT. (a) The subunit α and β of GM-CSF receptor in six colon cancer cell lines were examined by Western blotting. A human monocytic cell line THP-1 was used as a positive control. (b) GM-CSF receptor was detected by IHC in specimen (tumor and adjacent normal colon tissues) of CRC patients. Representative data were shown. Scale bar: 50 μm. (c) Three colon cancer cell lines (SW480, HCT116 and RKO) were stimulated with GM-CSF (25 ng/ml) for three weeks. The expression of epithelial and mesenchymal markers as well as EMT-related transcriptional factors indicated was examined by immunoblotting. Representative data of cropped blots from five independent experiments were shown. (d) The expression of E-cadherin and N-cadherin in SW480 cell line treated with GM-CSF as described above was detected by immunofluorescence. Representative images from three independent experiments were shown. Scale bar: 100 μm. (e) SW480 cell line was treated with GM-CSF as described above. The expression of EMT-related markers and transcriptional factors was examined by quantitative RT-PCR. The data were pooled from three experiments. US: unstimulated; GM: GM-CSF. *P < 0.05; **P < 0.01 vs untreated controls.
GM-CSF stimulation enhances the ability of motility in colon cancer cells
Next, we evaluated the migration and invasion of colon cancer cells with or without stimulation of GM-CSF in transwell experiments. Chronic exposure to GM-CSF, indeed, significantly augmented the capacity of migration and invasion in vitro (Fig. 2a and b). This effect was also confirmed by wound-healing assays (Fig. S4). Of note, this did not arise from the increase in the cell number following GM-CSF stimulation, as chronic exposure to GM-CSF did not render robust proliferation of cancer cells (Fig. S5). Furthermore, in a mouse model of colorectal cancer liver metastasis, these GM-CSF-educated colon cancer cells displayed more movement to target organ (Fig. 2c). Accordingly, these metastatic cancer cells exhibited mesenchymal phenotype (Fig. S6). Similar phenotype was observed in GM-CSF-overexpressing HCT116 cell line (Fig. S7a and b). Of note, upon inoculation of cancer cells into spleen, exogenous GM-CSF was removed in four-to-six week intervals. E-cadherin expression in GM-CSF-educated cancer cells isolated from splenic inoculation site, however, was much lower than that in untreated counterparts (Fig. 2d), indicating that colon cancer cells at sites of injection still harbored the mesenchymal phenotype. Thus, we can make a conclusion that GM-CSF stimulation sufficiently enhances the capacity of motility of colon cancer cells in vitro and dissemination in vivo.

GM-CSF-exposed colon cancer cells exhibit augmented motility in vitro and in vivo. (a,b) SW480 and HCT116 colon cancer cell lines were treated with GM-CSF (25 ng/ml) for three weeks. The ability of migration (a) and invasion (b) was examined by transwell experiments. Upper: representative images were shown. Bottom: the data were pooled from three experiments. (c) SW480 cell lines were treated with GM-CSF as described above and collected as well as transfused to nude mice in a model of colorectal cancer liver metastasis as described in Materials and methods. Four-to-six weeks later, livers were pooled and tumor foci per mouse were calculated. Left panel: Representative macroscopic images were shown. The arrows denoted metastatic lesions. Right panel: Representative microscopic images of primary (spleen) and metastatic (liver) tumor sites were shown. Scale bar: 50 μm. The data were pooled from three experiments. (d) Tumors in spleen of recipients were collected and E-cadherin expression was detected by immunoblotting. Cropped blots were shown. *P < 0.05; **P < 0.01; ***P < 0.001 vs untreated controls.
MAPK/ERK-ZEB1 signals are required for GM-CSF-induced EMT program
It is well-known that the binding of GM-CSF to its receptor activates at least three signaling pathways: JAK/STAT, mitogen-activated protein kinase (MAPK), and PI3K29. Thus, we detect the activation of these pathways in colon cancer cell lines upon GM-CSF stimulation. The results showed that the activation of STAT3/5 was not seen in GM-CSF-pulsed SW480 cells, and the PI3K/AKT signaling also was not disturbed (data not shown). In contrast, the phosphorylation of MAPK/ERK and NF-κB was visible in SW480 cells upon stimulation with GM-CSF (Fig. 3a), which was consistent with our previous study using a murine colon carcinoma cell line27. Furthermore, to define the signaling pathways required for GM-CSF-induced EMT program, ERK1 and 2 in SW480 cells were knockdown by RNA interference (Fig. 3b). Deletion of ERK1 or 2 had no significant impact on EMT marker expression in resting cancer cells (Fig. 3b). Conversely, ERK1 or 2 knockdown significantly abrogated EMT program following GM-CSF stimulation (Fig. 3b), including up-regulation of E-cadherin and down-regulation of N-cadherin and fibronectin expression. Moreover, the motility of GM-CSF-educated cancer cells was impaired pronouncedly when ERK1 or 2 was deleted (Fig. 3c and d). These findings indicate that ERK1 or 2 plays a nonredundant role in GM-CSF-induced EMT program.

GM-CSF induces EMT program through MAPK/ERK-ZEB1 signaling pathway. (a) SW480 cell line was stimulated with GM-CSF (25 ng/ml) for 5–120 min. At the indicated timepoints, the proteins were extracted and the phosphorylation of ERK1/2 and NF-κB p65 subunit was detected by immunoblotting. Representative data of cropped blots from three independent experiments were shown. (b) ERK1 and 2 in SW480 cell line were knockdown by RNA interference and cells were stimulated with GM-CSF (25 ng/ml) for three weeks. The expression of E-cadherin, N-cadherin, fibronectin and ZEB1 was examined by immunoblotting. Cropped blots were shown. (c,d) ERK1 and 2-knockdown SW480 cell line was treated with GM-CSF as described above. The ability of migration (c) and invasion (d) was detected by transwell experiments. (e) ZEB1 in SW480 cell line was knockdown by RNA interference and cells were stimulated with GM-CSF as described above. The expression of E-cadherin, N-cadherin, fibronectin and ERK1/2 was examined by immunoblotting. Cropped blots were shown. (f) ZEB1-knockdown SW480 cell line was treated with GM-CSF as described above. The ability of migration and invasion was detected by transwell experiments. Upper: representative images were shown. Bottom: the data were pooled from two experiments. EV: empty vector. **P < < 0.01; ***P < 0.001 vs EV controls.
Intriguingly, ZEB1 expression was dramatically suppressed in ERK1 or 2-knockdown cancer cells with stimulation of GM-CSF (Fig. 3b), suggesting that ZEB1 may represent a key factor to mediate EMT program following GM-CSF stimulation. To address the contribution of ZEB1 to GM-CSF-induced EMT program, ZEB1 was knockdown in SW480 cells (Fig. 3e). As expected, knockdown of ZEB1 did not significantly impact E-cadherin expression in resting SW480 cells, but drastically perturbed GM-CSF-induced EMT program (Fig. 3e). Consistently, the migration and invasion of GM-CSF-educated cancer cells decreased when ZEB1 was knockdown (Fig. 3f). Notably, the amounts of total ERK1/2 protein and their activation were not affected in ZEB1-silenced cancer cells with or without GM-CSF stimulation (Fig. 3e and data not shown). Therefore, MAPK/ERK-ZEB1 signaling pathways are critical for GM-CSF-induced EMT program.
Chronic stimulation of colon cancer cells with GM-CSF renders chemoresistance
EMT is well-known to intimately correlate with acquisition of drug resistance30. To determine whether chronic exposure to GM-CSF led to resistance to cytotoxic compounds, SW480 cells with or without GM-CSF stimulation were treated with three first-line chemotherapeutic agents for colorectal cancer in the clinic. The results showed that, although GM-CSF stimulation did not affect cell viability, GM-CSF-educated cancer cells were resistant obviously to fluorouracil (5-FU), oxiliplatin or irinotecan-mediated cell death, by SRB assays (Fig. 4a and S8a) and Annexin V/PI analysis (Fig. 4b and S8b). This was also reflected by decreased cleavage of caspase 3 and PARP in GM-CSF-stimulated cancer cells, two key proteins which were involved in drug-induced apoptosis (Fig. 4c). Furthermore, knockdown of ERK1/2 or ZEB1 attenuated chemoresistant potentials of GM-CSF-educated cancer cells (Fig. 4d and e).

GM-CSF-exposed colon cancer cells exhibit chemoresistance. (a,b) SW480 cell line was stimulated with GM-CSF (25 ng/ml) for three weeks and then treated with fluorouracil (5-FU) at the titrated concentrations for three days. Cell vitality was detected by SRB assays (a) and flow cytometry (b). (c) SW480 cell line was exposed to GM-CSF as described above and treated with 5-FU at different doses indicated for 24 hours. Total and cleavaged caspase 3 and PARP proteins were examined by immunoblotting. Cropped blots were shown. (d,e) SW480 cell line with knockdown of ERK1, 2 (d) or ZEB1 (e) was stimulated with GM-CSF as described above and treated with 5-FU (25 μM) for three days. Cell vitality was detected by SRB assays. The data were pooled from three independent experiments. One-way ANOVA methods were used to determine statistical significance for cell viability test. **P < 0.01; ***P < 0.001 vs untreated or EV controls.
High GM-CSF expression in tumor tissues correlates with local metastasis
To determine whether our findings are clinically relevant, colon specimens were collected from patients with colorectal cancer. Thirty-eight percentages (25/65) of specimens were positive for GM-CSF staining (Fig. 5a). The expression of GM-CSF in stroma and/or cancer cells was observed, depending on individuals (Fig. 5a). The receptors A and B for GM-CSF were positively stained in cancer cells in 12% (8/65) of specimens (Fig. 5b). Importantly, high GM-CSF expression in cancer samples significantly correlated with lymph node metastasis (Fig. 5c). Furthermore, the receptor-positive cancer samples with high GM-CSF expression frequently harbored activated ERK1 and 2 (Fig. 5d). These data are consistent with the notion that GM-CSF promotes the motility of cancer cells and metastasis by activating MAPK/ERK signaling pathways.

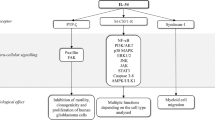
Intimate correlation of GM-CSF expression in CRC specimen with invasion/metastasis. (a) GM-CSF expression in tumor tissues of CRC patients was detected by immunohistochemistry. Representative images were shown. Scale bar: 50 μm. The percentages of GM-CSF-positive specimen in the cohorts were calculated. (b) The subunit α and β of GM-CSF receptor in tumor specimen were examined by immunohistochemistry. Representative images were shown. Scale bar: 50 μm. The percentages of CRC specimen with GM-CSF receptor-positive in cancer cells in the cohorts were calculated. (c) CRC patients were divided into two cohorts: with lymph node (LN) metastasis and without LN metastasis. The percentages of CRC specimen with positive staining of GM-CSF were calculated. LN met: lymph node metastasis. (d) The expression of phosphorylated ERK1/2 (P-ERK1/2) in CRC specimen was detected by immunohistochemistry. The percentages of P-ERK1/2-positive specimen in the indicated groups were calculated. Representative images (three cases per group) were shown. Scale bar: 50 μm. (e) The outline of our hypothesis as following: GM-CSF in tumor niches drives EMT program in receptor-expressing colon cancer cells and thereby promotes invasion/distant dissemination as well as resistance to chemotherapy. *P < 0.05.
Discussion
In a previous work we showed that GM-CSF stimulation in colon cancer cell lines could augment the migration and invasion in vitro 27. Here, we have addressed mechanistic clues underlying these effects, by analyzing EMT-inducing potentials of GM-CSF. Of note, it has been reported that GM-CSF contributes to acquisition of the invasive phenotypes in breast cancer cells by educating infiltrating macrophages in microenvironment7. In this study, we provide evidence for the first time that GM-CSF has an effect on the induction of EMT program in receptor-expressing colon cancer cells through activating MAPK/ERK-ZEB1 signaling pathways. These data may provide mechanistic explanation for recent observations that high GM-CSF contents in serum of CRC patients significantly correlated with poor prognosis3.
However, the relevance of GM-CSF to CRC progression and prognosis remains controversial. Nebiker et al. reported that GM-CSF production by tumor cells was associated with lower T stage and significantly longer survival as well as favorable prognosis in mismatch-repair proficient colorectal cancer31. This observation coincides with a previous report showing that 100% of the patients with human colon tumors that overexpressed GM-CSF and its receptors survived at least 5 years after diagnosis32. These divergences may be reconciled by the concept that the function of GM-CSF in tumor niche is context-dependent. GM-CSF stimulation alone is potent to drive M1 polarization of macrophage and maturation of dendritic cells, two populations mounting robust anti-tumor immune responses. On the other hand, cancer cell-derived GM-CSF can shape immunosuppressive microenvironment by promoting the generation and accumulation of myeloid suppressor cells and tumor-associated macrophages7, 9. The latter indicates that synergistic effects of other soluble factor (s) from cancer cells or stroma may be indispensible for GM-SCF-induced tumor-promoting environment. Besides immunomodulation of tumor-surrounding stromal components, we showed a stimulatory effect of GM-CSF on cancer cells themselves. Clinical analysis also supports the metastasis-promoting role of this cytokine. These data are not in agreement with a previous study that colorectal tumors substantially overexpressed GM-CSF and this cytokine had an immune-independent antitumor effect on receptor-positive cancer cells32. It should to be noted that the expression of proinflammatory cytokines such as IL-17A, IL-6, IL-22 was dramatically increased in lesions of colorectal cancer and their high expression significantly correlated with cancer progression and poor prognosis33,34,35. As one member of proinflammatory mediators, it seems plausible that high expression of GM-CSF in tumor is related to more invasive phenotype and poor outcome. Considering a strongly positive correlation of high GM-CSF expression in tumor with distant dissemination and unfavorable prognosis in several types of solid cancers, further investigations of GM-CSF role in CRC development and progression in larger cohorts of CRC samples are warranted.
Mechanistic investigations in this study reveal that MAPK/ERK signaling pathways are required for GM-CSF-induced EMT program and metastasis. Although several signaling pathways have been shown to be activated upon the engagement of GM-CSF with its receptor in hematopoietic cells, MAPK/ERK signals appear to dominantly mediate bioactivity of GM-CSF in epithelial cells, which is consistent with our previous results in a murine colon carcinoma cell line27. Furthermore, MAPK/ERK signaling pathways are clearly linked to tumor invasion and metastasis in response to a variety of intracellular and extracellular stimuli in several types of solid cancers including colorectal cancer22, 36,37,38,39,40. Of note, we showed that ERK1 and 2 are equivalently important for GM-CSF-induced EMT program, as knockdown of ERK1 or 2 had similar effects on the expression of EMT markers and cell motility. This is not consistent with a recent study showing that ERK2 but not ERK1 is critical for a oncogene placenta-specific 8-driven EMT phenotype in colorectal cancer41, suggesting that ERK1 and/or ERK2-mediated EMT lies on upstream clues. We also identified the activation of NF-κB in colon cancer cells upon GM-CSF challenge. The involvement of this key transcriptional factor in GM-CSF-induced EMT need to be addressed in the future through inactivation of NF-κB by ectopic overexpression of IκBα in cancer cells.
As described in previous studies that demonstrated a key role of ZEB1 in mediating EMT and tumor invasiveness in colorectal42, 43 and lung cancer44, we showed that ZEB1 was also a critical mediator for GM-CSF-driven EMT. ZEB1 expression seems to be regulated by MAPK/ERK signals, because knockdown of ERK1 or 2 decreased ZEB1 expression in GM-CSF-stimulated cancer cells. As previously reported45, 46, the activation of MAPK/ERK pathway induced Ets1 and Fra1 expression in cancer cells, and the latter activated ZEB1 promoter and induced its expression, which may provide some clues for the exact regulation of ZEB1 expression by MAPK/ERK signaling pathway in our model. In addition, one of metastasis-promoting function of ZEB1 is reported to enhance MMP secretion47, which was increased in GM-CSF-overexpressing HT-29 colon cancer cell lines as described recently22. Thus, ZEB1 could regulate colon cancer invasiveness by inducing EMT and MMP production.
It is well established that chronic exposure to a variety of growth factors or cytokines is linked to acquisition of chemoresistance in solid tumors48. The role of GM-CSF, however, in this process is unknown. In this paper, we identified GM-CSF as a chemoresistance-inducing factor, at least in colorectal cancer. Given the bioactivity of GM-CSF in other types of tumors, it is plausible that GM-CSF induces chemoresistant phenotype in receptor-expressing cancer cells. Furthermore, we showed that chemoresistance induced by chronic stimulation with GM-CSF was tightly associated with EMT, which is consistent with the notion that EMT contributes to drug resistance, which is validated in many types of tumors. Accordingly, since knockdown of ERK1/2 or ZEB1 led to the failure of GM-CSF-driven EMT, it is reasonable that inhibition of MAPK/ERK-ZEB1 signals also abrogates acquisition of chemoresistance. Notably, the contribution of MAPK/ERK-ZEB1 signaling pathway to multidrug resistance has been reported in numerous types of cancer, including breast cancer49, hepatocellular carcinoma50, and glioblastoma51. Therefore, inhibition of MAPK/ERK signals using small-molecular compounds may be favorable to reverse chemoresistance in certain settings. The details in the regulation of chemoresistant traits in cancer cells by MAPK/ERK-ZEB1 pathways need to be further addressed in the future, although GM-CSF stimulation greatly suppresses chemo-induced active cleavage of caspase and PARP.
In conclusion, in this study we demonstrate that GM-CSF is an EMT-inducing factor by binding its receptor on the surface of cancer cells through activating MAPK/ERK-ZEB1 signaling pathway (Fig. 5e). Of note, our data showed that EMT occurred on day 7 after GM-CSF stimulation, instead of day 3, indicating that products of early response genes within 3 days are not sufficient to induce EMT. Therefore, it may be true that early signaling pathways activated by GM-CSF involving MAPK/ERK are important to induce other EMT-inducing factors such IL-6, IL-1β and VEGF, whose consistent productions act in concert to induce EMT features. In support of this assumption, we showed previously that GM-CSF potently induced other proinflammatory cytokine expression in a murine colon cancer cell line27. Thus, further work is warranted to determine whether any one of them is responsible for mediating GM-CSF effect on EMT by adding neutralizing antibodies to some of these factors during the culture.
Currently, functional contribution of GM-CSF to cancer progression and metastasis is controversial, which may be due to dual effects of GM-CSF on immune cells and non-immune cells. Considering that recombinant GM-CSF are routinely used to correct neutropenia subsequent to chemotherapy and radiation and that adjuvant GM-CSF treatment has been suggested to occasionally enable tumor growth, detection of GM-CSF receptor expression in cancer cells is warranted in the future prior to GM-CSF treatment, to better define eligile patient’s cohorts.
Methods
Tissue samples
Primary CRC tissue samples were obtained from 65 patients undergoing surgical resection at the Department of Surgery, Zhengzhou Yihe Hospital from February 2010 to June 2011. Sections were reviewed by two experienced pathologists to verify the histologic assessment. All the specimens were adenocarcinoma. Prior informed consent was obtained and the study protocol was approved by the Ethics Committee of Zhengzhou Yihe Hospital. All experiments were performed in accordance with relevant guidelines and regulations.
Cell culture and drug treatment
Human colon adenocarcinoma cell lines (SW480, SW620, HT29, HCT116, DLD-1, RKO) and human acute monocytic leukemia cell line THP-1 were purchased from American Type Culture Collection and maintained in appropriate medium according to product specification, supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 100 mg/ml streptomycin. Cells were seeded in 6-well culture plates (1 × 106/ml/well) and stimulated with recombinant human GM-CSF (Peprotech) at 1–100 ng/ml for 1–3 weeks. To blocking GM-CSF activity, neutralizing anti-GM-CSF monoclonal antibody or isotype IgG (1 μg/ml, Biolegend) was added into the culture and maintained for one week. Then cells were collected and centrifuged at 1500 rpm for 5 minutes and resuspended in serum-free medium. In some settings, cells were treated with fluorouracil (5-FU), oxiliplatin and irinotecan at indicated concentration for 72 hours, which were purchased from Selleckchem. After incubation, cells were collected for analysis.
Experimental liver metastasis
The surgical procedure was performed according to splenic injection model as described previously52. Briefly, 5- to 6-week-old athymic nude mice (Jackson Laboratory) were anesthetized with ketamine/dexmedetomidine admixture and the spleen was exteriorized through a left lateral flank incision. Tumors were established by intrasplenic injection of 2 × 106 cells suspended in 50 μl of serum-free media using one 27-gauge needle. The injection site of the spleen was pressed with a cotton stick wet in iodine-polividone solution in order to remove extravasated cells and ensure homeostasis. The peritoneum and skin were closed in a single layer using surgical thread. Mice were sacrificed at four-to-six weeks post-injection and the burden of metastatic liver tumors was calculated. Primary (splenic) tumor mass was dissected and the protein was extracted conventionally for immunoblotting analysis. All animal experiments were performed in accordance with international guidelines for the care and use of laboratory animals and approved by the Animal Ethics Committee of the Institute of Basic Medical Sciences.
Transwell migration and invasion assays
In vitro cell migration and invasion assays were performed using transwell chambers with polyethylene terephthalate membrane (24-well inserts, 8.0 μm; Corning). For the migration assay, 2.5 × 104 cells were added to the top chambers. For the invasion assays, 5 × 104 cells were seeded to the top chambers coated with Matrigel (BD Biosciences). Complete medium was added to the bottom wells. After incubation for 36 hours, the noninvasive cells were removed with a cotton swab. The cells that had migrated through the membrane and adhered to the lower surface of the membrane were fixed with methanol for 10 minutes and stained with crystal violet solution (0.1%). For quantification, the cells were counted using a microscope from five randomized fields at ×200 magnification.
Wound-healing assay
Cell migration was also assessed using an in vitro scratch wound healing assay. Cells were starved overnight with serum-free RPMI1640 medium and a scratch was made using a sterile 1-mL pipette tip. The cell debris was removed with PBS and cells were cultured with GM-CSF (25 ng/ml) for 24 hours. An inverted microscope was used to observe cell migration.
Cell proliferation assays
Cell growth was assessed by Sulforhodamine B (SRB) assay. Cells (5 × 103 cells per well in 100 μl medium) were seeded into 96-well plates in triplicate and exposed to recombinant human GMCSF (Peprotech) at titrated concentrations. After incubation for indicated time, 50 μl of 30% trichloroacetic acid was added and incubated for 60 min at 4 °C. After washing and drying the plate, 100 μl of 0.4% SRB was added for 30 min. The plates were rinsed with 0.1% acetic acid and air-dried, after which 100 μl of Tris base (10 mM/L) was added, and the plates were shaken for 5 min. The SRB value was measured at a wavelength of 590 nm.
Immunoblotting
Samples were homogenized and sonicated in RIPA lysis buffer (Santa Cruz Biotech.), supplemented with protease inhibitors. After centrifugation at 20,000 g for 15 min, 30 μg of the supernatants were separated on 10% SDS-polyacrylamide gel and transferred onto an Immunobilon-P Transfer membrane (Millipore). After being blocked with 5% skim milk, the membrane was incubated with primary antibodies at1:1000 dilution. Rabbit anti-GAPDH antibody was used as an internal control (purchased from Tianjin Sungene Biotech.). ImmunoPure peroxidase-conjugated anti-rabbit IgG were used as secondary antibodies. The blotted membrane was then treated with the Super Signal West Dura Extended Duration Substrate (Pierce) and signals were detected by LAS-3000 mini CCD camera (Fuji Film). Primary antibodies used for immunoblotting were purchased from Cell Signaling.
Immunohistochemistry
The spleen and liver was removed, fixed in 10% neutral-buffered formalin solution, embedded in paraffin, and cut into tissue sections, which were stained with hematoxylin and eosin. Paraffin-embedded sections were deparaffinized and immersed in 80 °C water bath in 10 mM sodium citrate buffer with 0.1% Tween 20 overnight for antigen unmasking. Slides were incubated with primary antibody against CSF2RA (1:100, Biolegend), CSF2RB (1:80, Lifespan Bioscience), GM-CSF (1:150, Novus Biologicals), P-ERK1/2 (1:400, Cell Signaling), E-cadherin (1:100, Cell Signaling) or Fibronectin (1:100, Cell Signaling) in PBS containing 1% BSA and 10% goat serum. Biotinylated secondary antibodies (Dako) were added and incubated at room temperature for 1 hour. Streptavidin-HRP (BD Pharmingen) was added, and after 40 min the sections were stained with DAB substrate and counterstained with hematoxylin.
Immunofluorescence
Cells were seeded onto glass coverslips in 24-well plates, washed with PBS, fixed in 4% formaldehyde solution and permeabilized with 0.2% Triton X-100/PBS. Cells were blocked with 2% BSA in PBS for 30 min. Coverslips were incubated with primary antibodies for 1 hour, followed by incubation with phycoerythrin (PE)-conjugated secondary antibodies for 1 hour, and then stained with DAPI. Finally, coverslips were observed under a fluorescence microscope.
Generation of stable knockdown or knockin cell line
ShRNA plasmids targeting ERK1/2 and non-targeting shRNA control plasmid were kindly provided by Robert J. coffey (Department of Medicine, Vanderbilt University Medical Center)41. ShRNA plasmid targeting ZEB1 and human GM-CSF expression plasmid were purchased from Santa Cruz Biotech. SW480 or HCT116 colon cancer cell lines (1 × 107/ml/well) were seeded in 6-well culture plates and transfected with plasmids indicated above using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Stable cell lines were selected with 2 μg/ml puromycin.
Apoptosis asaays
Apoptosis was quantified using fluorescein isothiocyanate (FITC)-Annexin V and propidium iodide (PI), according to the manufacturer’s protocol (ApoDETECT FITC-Annexin V Kit; Zymed Laboratories Inc.). Cells were stained with 5 μl of FITC-Annexin V diluted 1:10 in buffer, and 2.5 μl of PI. After incubation for 15 minutes, cells were analyzed by flow cytometry with standard FACScan equipment (Becton Dickinson Co.).
ELISA for GM-CSF
GM-CSF levels in the supernatants were measured using ELISA kits (eBiosciences) according to the manufacturer’s protocol.
Quantitative RT-PCR
RNA was extracted and reverse-transcribed into cDNA using the SuperScript III First Strand cDNA synthesis system (Invitrogen). cDNA was synthesized from 0.5 μg RNA using random hexamer primers and SuperScript III (Invitrogen). Real-time RT-PCR was performed on a Bio-Rad iCycler to quantify mRNA levels. The primers for real-time were listed in Supplementary Table 2. All reactions were performed in triplicate. The data were analyzed using Q-Gene software and expressed as fold change mean normalized expression (MNE) from control value. MNE is directly proportional to the amount of RNA of the target gene relative to the amount of RNA of the reference gene, GAPDH.
Statistics
Data represent means of multiple determinations and were analyzed using the GraphPad Prism statistical PC program (GraphPad Software, San Diego, California, USA) by means of paired t test. One-way analysis of variance (ANOVA test) was used to define differences in cell viability in cultures treated with various concentrations of cytotoxic agents. A value of P < 0.05 was considered statistically significant.
References
Fleetwood, A. J., Cook, A. D. & Hamilton, J. A. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol 25, 405–28 (2005).
Mroczko, B., Szmitkowski, M., Wereszczynska-Siemiatkowska, U., Okulczyk, B. & Kedra, B. Pretreatment serum levels of hematopoietic cytokines in patients with colorectal adenomas and cancer. Int J Colorectal Dis 22, 33–8 (2007).
Chen, Z. Y. et al. A prognostic classifier consisting of 17 circulating cytokines is a novel predictor of overall survival for metastatic colorectal cancer patients. Int J Cancer 136, 584–92 (2015).
Taghipour Fard Ardekani, M. et al. Evaluation of Pre-Treatment Serum Levels of IL-7 and GM-CSF in Colorectal Cancer Patients. Int J Mol Cell Med 3, 27–34 (2014).
Mroczko, B., Szmitkowski, M., Okulczyk, B. & Piotrowski, Z. Granulocyte-macrophage-colony stimulating factor in patients with colorectal cancer. Folia Histochem Cytobiol 39(Suppl 2), 110–1 (2001).
Demirci, U. et al. Serum granulocyte macrophage-colony stimulating factor: a tumor marker in colorectal carcinoma? Asian Pac J Cancer Prev 10, 1021–4 (2009).
Su, S. et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 25, 605–20 (2014).
Pylayeva-Gupta, Y., Lee, K. E., Hajdu, C. H., Miller, G. & Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–47 (2012).
Bayne, L. J. et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–35 (2012).
Rigo, A. et al. Macrophages may promote cancer growth via a GM-CSF/HB-EGF paracrine loop that is enhanced by CXCL12. Mol Cancer 9, 273 (2010).
Pak, A. S. et al. Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin Cancer Res 1, 95–103 (1995).
Wu, P. et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40, 785–800 (2014).
Ghirelli, C. et al. Breast Cancer Cell-Derived GM-CSF Licenses Regulatory Th2 Induction by Plasmacytoid Predendritic Cells in Aggressive Disease Subtypes. Cancer Res 75, 2775–87 (2015).
Mueller, M. M. & Fusenig, N. E. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 4, 839–49 (2004).
Mroczko, B. & Szmitkowski, M. Hematopoietic cytokines as tumor markers. Clin Chem Lab Med 42, 1347–54 (2004).
Obermueller, E., Vosseler, S., Fusenig, N. E. & Mueller, M. M. Cooperative autocrine and paracrine functions of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in the progression of skin carcinoma cells. Cancer Res 64, 7801–12 (2004).
Gutschalk, C. M., Herold-Mende, C. C., Fusenig, N. E. & Mueller, M. M. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor promote malignant growth of cells from head and neck squamous cell carcinomas in vivo. Cancer Res 66, 8026–36 (2006).
Mueller, M. M. et al. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol 155, 1557–67 (1999).
Thacker, J. D., Dedhar, S. & Hogge, D. E. The effect of GM-CSF and G-CSF on the growth of human osteosarcoma cells in vitro and in vivo. Int J Cancer 56, 236–43 (1994).
Pei, X. H., Nakanishi, Y., Takayama, K., Bai, F. & Hara, N. Granulocyte, granulocyte-macrophage, and macrophage colony-stimulating factors can stimulate the invasive capacity of human lung cancer cells. Br J Cancer 79, 40–6 (1999).
Park, S., Kim, E. S., Noh, D. Y., Hwang, K. T. & Moon, A. H-Ras-specific upregulation of granulocyte colony-stimulating factor promotes human breast cell invasion via matrix metalloproteinase-2. Cytokine 55, 126–33 (2011).
Gutschalk, C. M. et al. GM-CSF enhances tumor invasion by elevated MMP-2, -9, and -26 expression. Cancer Med 2, 117–29 (2013).
Bates, R. C. & Mercurio, A. M. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther 4, 365–70 (2005).
Peinado, H., Olmeda, D. & Cano, A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7, 415–28 (2007).
Kang, Y. & Massague, J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118, 277–9 (2004).
Zheng, H. & Kang, Y. Multilayer control of the EMT master regulators. Oncogene 33, 1755–63 (2014).
Wang, Y. et al. Tumor-derived GM-CSF promotes inflammatory colon carcinogenesis via stimulating epithelial release of VEGF. Cancer Res 74, 716–26 (2014).
Breen, E., Steele, G. Jr. & Mercurio, A. M. Role of the E-cadherin/alpha-catenin complex in modulating cell-cell and cell-matrix adhesive properties of invasive colon carcinoma cells. Ann Surg Oncol 2, 378–85 (1995).
Martinez-Moczygemba, M. & Huston, D. P. Biology of common beta receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J Allergy Clin Immunol 112, 653–65; quiz 666 (2003).
Arumugam, T. et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 69, 5820–8 (2009).
Nebiker, C. A. et al. GM-CSF Production by Tumor Cells Is Associated with Improved Survival in Colorectal Cancer. Clin Cancer Res 20, 3094–106 (2014).
Urdinguio, R. G. et al. Immune-dependent and independent antitumor activity of GM-CSF aberrantly expressed by mouse and human colorectal tumors. Cancer Res 73, 395–405 (2013).
Tosolini, M. et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 71, 1263–71 (2011).
Knupfer, H. & Preiss, R. Serum interleukin-6 levels in colorectal cancer patients–a summary of published results. Int J Colorectal Dis 25, 135–40 (2010).
Jiang, R. et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 13, 59 (2013).
Liu, H. et al. PTP1B promotes cell proliferation and metastasis through activating src and ERK1/2 in non-small cell lung cancer. Cancer Lett 359, 218–25 (2015).
Han, H. B. et al. PBX3 promotes migration and invasion of colorectal cancer cells via activation of MAPK/ERK signaling pathway. World J Gastroenterol 20, 18260–70 (2014).
Uekita, T. et al. Oncogenic Ras/ERK signaling activates CDCP1 to promote tumor invasion and metastasis. Mol Cancer Res 12, 1449–59 (2014).
Wang, H. C., Chiang, W. F., Huang, H. H., Shen, Y. Y. & Chiang, H. C. Src-homology 2 domain-containing tyrosine phosphatase 2 promotes oral cancer invasion and metastasis. BMC Cancer 14, 442 (2014).
Yu, H. et al. Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clin Cancer Res 20, 3434–45 (2014).
Li, C. et al. Excess PLAC8 promotes an unconventional ERK2-dependent EMT in colon cancer. J Clin Invest 124, 2172–87 (2014).
Sanchez-Tillo, E. et al. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA 108, 19204–9 (2011).
Xiong, H. et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem 287, 5819–32 (2012).
Ahn, Y. H. et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest 122, 3170–83 (2012).
Sinh, N. D., Endo, K., Miyazawa, K. & Saitoh, M. Ets1 and ESE1 reciprocally regulate expression of ZEB1/ZEB2, dependently on ERK1/2 activity, in breast cancer cells. Cancer Sci (2017).
Shin, S., Dimitri, C. A., Yoon, S. O., Dowdle, W. & Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 38, 114–27 (2010).
Bae, G. Y. et al. Loss of E-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget 4, 2512–22 (2013).
Wilson, T. R. et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–9 (2012).
Chen, T. et al. Dasatinib reverses the multidrug resistance of breast cancer MCF-7 cells to doxorubicin by downregulating P-gp expression via inhibiting the activation of ERK signaling pathway. Cancer Biol Ther 16, 106–14 (2015).
Guan, J. et al. Involvement of extracellular signal-regulated kinase/mitogen-activated protein kinase pathway in multidrug resistance induced by HBx in hepatoma cell line. World J Gastroenterol 10, 3522–7 (2004).
Siebzehnrubl, F. A. et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5, 1196–212 (2013).
Buchanan, F. G. et al. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA 103, 1492–7 (2006).
Acknowledgements
We thank Dr. Robert J. coffey (Department of Medicine, Vanderbilt University Medical Center) for gifting shRNA plasmids targeting ERK1/2. This work was supported by grants from the National Key Basic Research Program of China (2015CB553704, 2013CB530506), the National Natural Science Foundation of China (81672803, 81472647, 81272320), Beijing Natural Science Foundation (7132151), Service Industry Scientific Research of National Health and Family Planning Commission of China (2015SQ00192), and Tianjin Municipal Basic Research Priorities Program (15JCZDJC34700).
Author information
Authors and Affiliations
Contributions
Y.C., Z.Z., Y.C. and Z.L. X.D. conceived and carried out experiments. R.W., H.X., C.H., B.S., J.F., R.G., and Y.L. analyzed the data. H.P., G.H., and G.C. supervised the project. Y.C. and G.C. wrote the paper and prepared the figures. All authors reviewed the manuscript and approved the submission.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Zhao, Z., Chen, Y. et al. An epithelial-to-mesenchymal transition-inducing potential of granulocyte macrophage colony-stimulating factor in colon cancer. Sci Rep 7, 8265 (2017). https://doi.org/10.1038/s41598-017-08047-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08047-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
