
Abstract
Saliva has attracted attention as a diagnostic fluid due to the association of oral microbiota with systemic diseases. However, the lack of standardised methods for saliva collection has led to the slow uptake of saliva in microbiome research. The aim of this study was to systematically evaluate the potential effects on salivary microbiome profiles using different methods of saliva collection, storage and gDNA extraction. Three types of saliva fractions were collected from healthy individuals with or without the gDNA stabilising buffer. Subsequently, three types of gDNA extraction methods were evaluated to determine the gDNA extraction efficiencies from saliva samples. The purity of total bacterial gDNA was evaluated using the ratio of human β-globin to bacterial 16S rRNA PCR while 16S rRNA gene amplicon sequencing was carried out to identify the bacterial profiles present in these samples. The quantity and quality of extracted gDNA were similar among all three gDNA extraction methods and there were no statistically significant differences in the bacterial profiles among different saliva fractions at the genus-level of taxonomic classification. In conclusion, saliva sampling, processing and gDNA preparation do not have major influence on microbiome profiles.
Similar content being viewed by others

Introduction
As a biospecimen, saliva is less utilised in a Clinical Chemistry Laboratory compared to tissue, blood, urine and faecal matter despite being the most easily accessible non-invasive body fluid1. This is in part due to the lack of standardised saliva sample collection protocols and our limited knowledge about the diurnal variability of biomolecules in saliva2. Unlike other biospecimens, saliva sample types (whole-mouth unstimulated saliva, acid and mechanically stimulated saliva, oral swab and oral rinse) may differ in their composition and may have an impact on the analytes to be detected3. In addition, the relatively low abundance of biomolecules in saliva makes it more challenging albeit using advanced technologies4,5,6,7. To date, salivary biomarkers of potential diagnostic value has been identified and validated for both oral and systemic diseases8,9,10,11,12,13,14,15. Currently, salivary DNA based methods are used in many diagnostic laboratories for mutations and polymorphisms studies relating to cancers and hereditary disorders16.
The microbial communities resident at different sites of the human body are widely recognised for their roles in protecting, initiating, and facilitating disease pathogenesis, and the oral cavity is relatively understudied in this regard. Reliable characterisation of these microbial communities in various disease states should also support the development of new saliva-based diagnostics and therapeutics17,18,19,20. Historically, oral microbiota research has heavily depended on microbial specific cultures although many researchers adapted to cultivation-independent molecular techniques to more holistically assess microbiome (the collective genomes of microorganisms) changes21, 22. The advent of high-throughput genomic (g)DNA sequencing methods has revolutionized the field of human microbiome research, with particular focus to date on the gut microbiome using faecal samples.
In 2012, the International Human Microbiome Standards (IHMS) were concerned that the sample collection, processing and gDNA preparation of faecal samples may influence the data generated in human metagenomics research studies. Supported by the European Commission, a suite of sample collection and processing procedures were tested and gDNA preparation was carried out by IHMS contributors across 12 different countries. Contributors were asked to extract gDNA from the provided faecal samples using their own laboratory procedures as well as other standard protocols from literatures. After a thorough investigation such as the gDNA yield, quality and recovery of diversity and specific bacterial taxa, a set of 14 standard operating procedures were designed to help optimise data quality and comparability in the human microbiome field (http://www.microbiome-standards.org). As the next-stage forward, this study will investigate the influence of sample collection, processing and gDNA preparation with regards to saliva samples.
Aims of this study were three-fold: (i) to investigate the influence of saliva collection method on microbial analysis; (ii) to evaluate the most efficient bacterial gDNA extraction protocol from human saliva and; (iii) to determine the best saliva fraction for oral microbial studies. While different saliva collection methods are known to influence the composition of saliva biomolecules, we demonstrate that it does not contribute significantly to the oral microbiome profile. In addition, the overall bacterial gDNA yield was not affected by different extraction protocols when repeated bead-beating with lysis-buffer was implemented. However, the Maxwell® 16 LEV blood DNA kit was able to significantly increased the purity of the bacterial gDNA.
Methods and Materials
Study cohort and sample collection
This study was approved by the Queensland University of Technology (HREC no.: 1400000617) Medical Ethical Institutional Board and informed consent was obtained from all participants. All methods in this study were performed in accordance with the relevant guidelines and regulations. We have recruited normal healthy controls (n = 40) from the general population based on stringent recruitment criteria (Supplementary Data 1) to minimise baseline variations that may potentially affect the experimental endpoints. All volunteers (between 20 to 30 years of age) were self-reported to be in good general health with no underlying diseases, not receiving local and/or systemic antibiotics and no history of smoking and drinking habits.
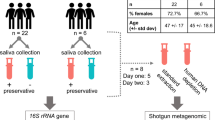
Volunteers were asked to refrain from eating and drinking for an hour prior to donating saliva samples as per our previous work9, 10, 12. The volunteers were asked to sit in a comfortable position and were asked to rinse their mouths with bottled water to remove food debris. A set of two spit saliva samples were collected from ten volunteers (total samples, n = 20) in a 50 mL sterile Falcon tube (Becton, Dickinson and Company, New Jersey, USA) and an OMNIgene tube (DNA Genotek Incorporation, Ontario, Canada), respectively. Spit samples were collected by asking volunteers to forcefully spit saliva (not sputum) directly into the saliva collection devices (OMNIgene includes a separate mouth-piece that can be connected to the devise for saliva spitting and drooling proposes) (Fig. 1a). OMNIgene contained 1 mL of stabilising buffer, hence the maximum saliva sample that can be collected is 1 mL (OMNIgene tube capacity is 2 mL). Since 200 μL of saliva sample is required per extraction, a 1:1 mixture of spit sample collected from 50 mL sterile Falcon tube and 1 x phosphate-buffered saline (PBS) was used to have similar saliva sample volumes for microbial analysis.

Study design overview. From Fig. 1a, Maxwell® 16 LEV Blood Kit was found to be the most efficient bacterial gDNA extraction method when spit samples were collected in 50 mL sterile Falcon tube. Hence, OMNIgene and other salivary bacterial gDNA extraction methods were excluded from the second part of the study (Fig. 1b).
Similarly three sets of saliva fractions were collected from a different cohort of 30 normal healthy volunteers (total samples, n = 90) in the order of spit, drool and oral rinse with five minute intervals. Drool samples were collected by asking volunteers to pool saliva in the mouth and expectorate naturally into a 50 mL sterile Falcon tube while spit samples were collected as described above. Oral rinse samples were collected by asking volunteers to swish and gargle with 10 mL of 0.9% (w/v) saline solution (Baxter International Incorporate, Illinois, USA) for a minute and expectorate into a 50 mL sterile Falcon tube as previously published (Fig. 1b)8. We have also tested the reverse order of sample collection to ensure that the order of collection does not influence the microbial diversity.
After collection, all samples were transported back to the laboratory on dry ice. Spit and drool samples were aliquoted evenly into 1.5 mL Eppendorf tubes (Eppendorf, Hamburg, Germany) and stored at −80 °C. Oral rinse samples were further processed by centrifugation at 1000 × g for 15 minutes at 4 °C to separate the cellular pellet from cell-free salivary supernatant. Cellular pellets were resuspended in sterile 1 x PBS and aliquoted evenly into 1.5 mL Eppendorf tubes and stored at −80 °C.
Maxwell® 16 LEV blood DNA kit
Saliva samples (200 μL irrespective of the fraction) were subjected to centrifugation at 16000 × g for 10 minutes at 4 °C (to reduce bacterial activities in saliva samples) to separate the cellular pellet from cell-free supernatant. A volume of 500 μL of in-house lysis buffer (200 mL of 0.5 M sodium chloride, 0.05 M trisaminomethane at pH8, 0.05 M ethylenediaminetetraacetic acid at pH 8 and 4% sodium dodecyl sulfate) was added to the cellular pellet and mixed well by vortexing. The samples were then transferred to a 2 mL conical screw cap microtube with zirconimum beads (combination of 0.3 g of 0.1 mm and 0.1 g of 0.5 mm, Daintree Scientific, St. Helens, Tasmania, Australia) and the cells were disrupted using the Precellys® 24 (Bertin Corporation, Rockville, USA) at 5000 × g for 3 × 60 seconds. The homogenates were then incubated within a 70 °C water-bath for 15 minutes with the contents mixed at five minute intervals by gentle inversion of the tubes. After incubation, samples were centrifuged at 15000 × g for five minutes and the supernatant fractions were transferred to sterile 1.5 mL Eppendorf tubes containing 30 μL of proteinase-K (Maxwell® 16 LEV blood DNA kit), vortexed for 30 seconds, then incubated at 56 °C for 20 minutes. Total nucleic acids were then recovered from these samples using the blood DNA columns using the Maxwell® 16 MDx Research Instrument (Promega Corporation, Wisconsin, USA). Finally, 2 μL of 10 μg RNase A (Qiagen, Hilden, Germany) was added to the samples and incubated in a 37 °C water-bath for 15 minutes for RNA digestion.
Phenol-chloroform method
The sample preparation and bacterial cell lysis for the phenol-chloroform extraction method was similar to the Maxwell® 16 LEV blood DNA method described above, with the following modifications. After the second incubation in a 56 °C water-bath for 20 minutes, equal volumes of buffer saturated phenol (Sigma Co., Victoria, Australia) was added and mixed by inverting several times and centrifuged at 18000 × g for 5 minutes at 4 °C. The aqueous layer was carefully removed into a new 1.5 mL Eppendorf tube without disturbing the interphase layer. An equal volume of chloroform:isoamyl (24:1) alcohol (Sigma Co., Victoria, Australia) mixture was added to the sample and vortexed for 5 minutes for further purification. Mixed samples were centrifuged at 18000 × g for 10 minutes at 4 °C and the aqueous layer was transferred into a new 1.5 mL Eppendorf tube. Depending on the volume of the aqueous layer, 1:10 of 3 M sodium acetate (pH 5.2) and an equal volume of 100% isopropanol were added to the sample and mixed well before incubating at −20 °C overnight. The samples were then centrifuged at 18000 × g for 10 minutes at 4 °C, the supernatant discarded and 500 μL of 70% ethanol was added to the pellet before centrifugation again at 18000 × g for 5 minutes at 4 °C. The supernatant was discarded and the gDNA samples were dried using a SpeedVac Concentrator SAVANT ISS110 (ThermoFisher Scientific, Massachusetts, USA) for 5 minutes at 60 °C. The gDNA was resuspended in 20 μL of 1 x trisaminomethane and ethylenediaminetetraacetic acid buffer and vortexed for a minute to mix well. Finally, 2 μL of 10 μg RNase A was added to the sample and incubated in a 37 °C water-bath for 15 minutes for RNA digestion.
QIAamp DNA Microbiome Kit
The sample preparation and bacterial cell lysis for the QIAamp DNA Microbiome Kit (Qiagen, Hilden, Germany) extraction method was similar to the Maxwell® 16 LEV blood DNA method. However, the proteinase K from QIAamp DNA microbiome kit was used for the second incubation. The QIAamp DNA microbiome kit extraction was carried out according to the manufacturers’ instruction and gDNA was eluted in a final volume of 50 μL. A volume of 2 μL of 10 μg RNase A was added to the gDNA sample and incubated in a 37 °C water-bath for 15 minutes for RNA digestion.
gDNA quantification
The quantity and quality of the extracted gDNA were evaluated using the Nano Drop ND-1000 spectrophotometer (ThermoFisher Scientific, Massachusetts, USA).
Quantitative PCR
Bacterial 16S rRNA primer set (1114F-5′ CGGCAACGAGCGCAACCC 3′ and 1221R-5′ CCATTGTAGCACGTGTGTAGCC 3′) and human β-globin primer set (F-5′ CAACTTCATCCACGTTCACC 3′ and R-5′ GAAGAGCCAAGGACAGGTAC 3′) were used in quantitative (q)PCR reactions to determine the ratio of bacteria to human gDNA from the extracted samples. The reaction consisted of 5 μL of 2 × iTaq™ Universal SYBR® Green Supermix (Bio-Rad Laboratories, California, USA), 200 nM of forward and reverse primers for each of the respective genes and 20 ng of gDNA template. The total reaction volume (10 μL) was subjected to qPCR amplification using the conditions of an initial denaturing stage at 95 °C for 10 minutes and followed by 30 cycles of a minute at 60 °C. Escherichia coli gDNA was used as a positive control while DNase/RNase water was used as a negative control for the qPCR assay.
16S rRNA gene amplicon library preparation and sequencing
16S rRNA gene amplicons for sequencing by Illumina MiSeq system (Illumina Incorporate, California, USA) was prepared according to the manufacturers’ method with gene-specific sequences targeting the V6 and V8 hypervariable regions of the 16S rRNA gene23. Q5® Hot Start High-Fidelity (New England Biolads, Masachusetts, USA) polymerase enzyme was used for the index PCR instead of the recommended KAPA HiFi HotStart ReadyMix (Kapa Biosystem, Massachusetts, USA) polymerase enzyme, as the former enzyme yielded superior amplification efficiency with a lower error rate. Sequencing was performed at the Australian Centre for Ecogenomics (ACE, Brisbane, Australia).
Statistical analysis
The statistical analysis for the investigation of the influence of saliva collection devices and the efficiency of saliva gDNA extraction methods were carried out by using GraphPad Prism (GraphPad Software Incorporate, California, USA). Since the quantity and quality of the extracted gDNA were not normally distributed, the non-parametric Wilcoxon matched-pairs signed rank test was used when comparing two different variables while the Friedman test was used when comparing more than two different variables. In addition, the Friedman test was also used to analyse the Ct mean variation of the bacterial 16S rRNA gene and the human β-globin gene of the gDNA extracted.
Illumina sequenced datasets were analysed using Quantitative Insights Into Microbial Ecology (QIIME) version 1.9.1 [PMID: 20383131] on the Ubuntu Linux virtual machine (Canonical Limited, London, United Kingdom). USEARCH 6.1 [PMID: 20709691] was used to perform a reference based chimera detection and removal to avoid perceived diversity24. Briefly, sequences were clustered into operational taxonomic units (OTUs) by PyNAST [PMID: 19914921] with a 97% sequence identity threshold against Greengenes core set database version 13.8 [PMID:16820507]25. An OTU table was generated with a list of prokaryotes and their respective observed OTU counts (abundance) for each sample based on the sequenced data in a biological observation matrix format. The OTU table was further edited to remove low abundance OTUs (≤0.1% of total sequences) and any sequences that were not of bacterial or archaeal origin. A subsampled OTU table was created by random sampling (without replacement) of the original OTU table based on the sample with the lowest OTU counts to account for different sequencing depths that may occur for each individual sample. The subsampled OTU table was used to calculate the α- (within-sample) and β- (between-sample) diversity metrics and to generate taxa summaries for each saliva fraction, from the phylum to genus levels of classification. Shannon index was used as an estimator for richness and evenness of the microbial community. Distance between samples was represented by the weighted (quantitative) and unweighted (qualitative) UniFrac distance metrics [PMID: 16332807] which are presented via principal component analysis (PCoA) plotting. Calypso version 5.4 (http://cgenome.net:8080/html/wiki/index.php/Calypso) was used to further analyse these data by providing statistical information on the significance of distribution for each genus as well as the combined genera in the three saliva fractions. Since the distribution of the genera between each saliva fraction is non-parametric, the Kruskal-Wallis rank test was used on Calypso to identify any significant differences in genera distribution. According to the p-values generated, there are no significant differences in the distribution of genera in spit, drool and oral rinse (Supplementary Data 6).
Results
The quantity and quality of total gDNA extracted using different saliva collection and extraction methods
There were no significant differences in the quantity and quality of extracted gDNA in saliva samples collected from the first normal healthy control cohort (n = 10) (Fig. 1a), demonstrating the resilient of salivary gDNA without the help of additional preservation system (Fig. 2). The salivary gDNA extraction was performed in triplicate and the results were consistent and reproducible (Supplementary Data 2). In contrast, the salivary gDNA quantity and quality significantly differed among the three extraction methods (Fig. 3). Irrespective of the collection method, the average gDNA quantity was high in spit samples using the Maxwell® 16 LEV blood DNA kit followed by the phenol-chloroform extraction method compared with a commercial QIAamp DNA Microbiome Kit method (Supplementary Data 3). However, the data were less variable when gDNA was extracted using the QIAamp DNA Microbiome Kit. In terms of gDNA quality, the 260/280 ratio of samples extracted with Maxwell® 16 LEV blood DNA kit were closer to the 1.8 ‘pure’ gDNA value. To ensure that the extracted gDNA is not contaminated with RNA, randomly selected gDNA samples were analysed on a 1% agarose gel electrophoresis. No RNA traces were detected by this analysis.

Extracted salivary gDNA from different saliva collection methods. Scatter plots for the average quantity and quality of the extracted gDNA triplicates from each collection and extraction method (a., b. Maxwell® 16 LEV blood DNA kit; c., d. in-house phenol-chloroform extraction and e., f. QIAamp DNA Microbiome Kit).

Extracted salivary gDNA from different bacterial gDNA extraction methods. Scatter plots for the average quantity and quality of the extracted gDNA triplicates from each collection (a., b. spit from 50 mL Falcon tube and c., d. OMNIgene) and extraction (MW represents Maxwell® 16 LEV blood DNA kit; PC represents in-house phenol-chloroform extraction and QM represents QIAamp DNA Microbiome Kit) method. Significant differences are denoted with *=p < 0.05, **p = < 0.01, ***=p < 0.001, ****=p < 0.0001 respectively.
Purity of isolated bacterial gDNA from various saliva fractions
Since there were no significant differences in the quantity and quality of gDNA extracted from the triplicate extraction as described above for each sample, gDNA was pooled and used as template for qPCR assays to determine the ratio of bacterial to human gDNA content. The qPCR assays’ efficiency were optimised using six-point serial dilution from 1.56 to 50 ng for the bacterial 16S rRNA gene and the human β-globin gene. Based on the results, the qPCR showed an average PCR efficiency of 0.99 for the bacterial 16S rRNA gene and an average efficiency of 0.98 for the human β-globin gene (Supplementary Data 4).
All spit samples were screened under the optimised condition. According to the results, Maxwell® 16 LEV blood DNA kit appeared to be a better extraction method with spit sample collected in 50 mL sterile Falcon tubes. The average ratio of threshold cycle (Ct) mean between extracted bacterial to human gDNA from spit samples collected in 50 mL Falcon tube using Maxwell® 16 LEV blood DNA kit was highest (Ct, 16.48:28.35 respectively) with multiple undetectable Ct means for the human β-globin gene (Table 1). It was noted that although the human β-globin gene was mostly undetected in extracted gDNA from spit samples collected in OMNIgene using the Maxwell® 16 LEV blood DNA kit, the average Ct mean for the bacterial 16S rRNA gene was significantly higher at 22.51, indicating a low abundance of bacterial gDNA (Table 1).
Based on these collective results, the Maxwell® 16 LEV blood DNA kit appears to provide a more enriched bacterial gDNA extraction from our samples. The average Ct mean for the bacterial 16S rRNA gene from each extraction method did not significantly differ from one another while Maxwell® 16 LEV blood DNA kit was able to significantly reduce the content of human gDNA (Table 1). We also chose to collect samples in 50 mL sterile Falcon tubes and store at −80 °C for the second part of this study, in our hands, this collection method was more compatible with our subsequent use of the Maxwell® 16 LEV blood DNA kit.
The influence of saliva fraction on microbial analysis
From the second normal healthy control cohort (n = 30) (Fig. 1b), the quantity and quality of extracted gDNA were significantly different between saliva fractions (Fig. 4a). The purity of gDNA from the saliva fractions was determined using the 16S rRNA gene and the human β-globin gene qPCR assays as previously described. There were no significant differences between the purity of bacterial gDNA extracted from spit, drool and oral rinse (Fig. 4b).

Extracted salivary gDNA from different saliva fractions. (a) Scatter plots for the quantity and quality of the extracted bacterial gDNA from each saliva fraction. Significant differences are denoted with *=p < 0.05, **=p < 0.01, ***=p < 0.001, ****=p < 0.0001 respectively. (b) Bacterial 16S rRNA and human β-globin qPCR Ct means distribution trend for the extracted gDNA from spit, drool and oral rinse.
Extracted gDNA from spit, drool and oral rinse samples from 10 randomly selected healthy individuals were subjected to 16S rRNA gene amplicon sequencing to compare the microbial diversity among different saliva fractions. Based on our data, the average length of reads is 500 bp and the OTU were subsampled to 5833 counts. After processing, 651760 high quality sequences were obtained in this study, with an average of 20651 sequences per sample. From these sequences, 6 known phyla and 30 genera were identified, and a total of 90 OTUs were detected at the 97% sequence identity threshold.
A rarefaction curve of the observed OTUs against sequences per sample was plotted for spit, drool and oral rinse to determine the efficiency of the sequencing process (Fig. 5). The Shannon index was determined for each sample, with the oral rinse samples having a consistently greater mean value (4.83) compared to spit and drool samples (4.79 and 4.77, respectively), although these differences were not statistically significant (Fig. 5). The β-diversity analyses showed no observable patterns among the spit, drool and oral rinse samples, the differences thereby primarily driven by the inter-subject variation rather than saliva fractions (Fig. 6).

Rarefaction curve for observed operational taxonomic units against sequences per sample for spit, drool and oral rinse.

Beta-diversity of salivary microbiome. Weighted (a) and unweighted (b) PCoA plot for spit, drool and oral rinse samples with respective adjacent plots emphasizing on the subjects in different representing colours.
The taxonomic profiles of spit, drool and oral rinse samples were examined based on the proportion of bacterial sequences determined at genus level (Supplementary Data 5). The five major genera found in all three saliva fractions were Streptococcus (17.5%), Prevotella (15.5%), Veillonella (15.3%), Neisseria (12.7%) and Haemophilus (10%). Calypso’s redundancy analysis was used to determine if combining the distribution pattern of all the genera from each saliva fraction into a panel could distinguish different saliva fractions (Fig. 7a). The p-value generated was 0.999, indicating that there are no significant differences. A second redundancy analysis under the same set of parameters was also carried out to investigate if the genera distribution of the three saliva fractions is distinguishable from one another (Fig. 7b). The results showed that the genera distributions from the three saliva fractions were significantly different for each individual with a p-value of 0.001, indicating the microbiome were more driven by host/environmental variations between subjects.

Salivary microbiome genera redundancy analysis on different (a) saliva fractions and (b) subjects.
Discussion
Saliva harbours bacteria shed from adhering microbial communities on various intraoral surfaces26. It is also well established that the oral cavity is colonized by numerous and diverse microorganisms27. While bacterial gDNA extraction methods have been rigidly investigated in the past, there is a paucity of data and knowledge regarding the ideal saliva fraction to be used for microbial analysis of the oral cavity28,29,30. According to Lazarevic et al.28, mechanical lysis (repeated bead-beating) is critical for greater bacterial OTU richness in saliva samples due to the rigid bacterial cell walls. This was also confirmed by Sohrabi et al.29 in addition to the incorporation of enzymatic cell lysis (repeated bead-beating in lysis buffer) to increase the purity of extracted bacterial gDNA. In this study, we incorporated the enzymatic-mechanical lysis method with current bacterial gDNA extraction kits to investigate the yield and quality of bacterial gDNA from different saliva fractions.
Our results demonstrate comparable quality and quantity of extracted gDNA from saliva samples when samples are either preserved using a stabilising buffer solution (OMNIgene) or when stored neat at −80 °C. In our hands, the Maxwell® 16 LEV Blood DNA kit with enzymatic-mechanical lysis provides the preferred method for isolating bacterial gDNA from saliva, in terms of gDNA yield, quality and ratio of bacterial:human gDNA. We have found also relatively higher α-diversity of OTUs in the oral rinse fraction as opposed to spit and drool, although these differences were not statistically significant.
Bacterial colonisation is apparent at many sites within the oral cavity, including the gingival, cervical line, tooth surface, tonsil, buccal mucosa, and tongue. As such it is important to develop a method that can access all of these sites and the diversity inherent to them27, 31, 32. Since microbes in the oral cavity travel via tongue and saliva movement, three saliva fractions were tested for microbial diversity via 16S rRNA gene amplicon sequencing31, 33. The main difference among these saliva fractions is that the muscle movements from spitting action may provide a larger microbial coverage compared with passive drooling; while oral rinse sampling provides access into the microbial populations residing within the oropharyngeal/throat area through the gargling action of the saline solution. The taxonomic profiles of the three saliva fractions were similar at genus-level and therefore can arguably be used inter-changeably for down-stream applications. However, we recommend the oral rinse method as the saliva fraction of choice when possible for future studies due to a greater α-diversity in OTUs compared to drool and split considering the inclusion of oropharyngeal regions. Furthermore, it should also be pointed out that healthy volunteers were used in this study, but in our experience an oral rinse sample is ideal in people restricted in their ability to produce saliva, such as patients with xerostomia or cancer patients who have undergone chemoradiation treatment.
The bacterial profiles identified in our study corroborated with previous findings. Bik et al.34 investigated the oral microbiome of ten individuals with healthy oral tissues and gingiva (full-mouth clinical examinations were performed by certified dentists) in both men and women between the ages of 27 to 61 years34. Whole-mouth saliva samples and dental plaque specimens were collected and pooled to create a single ‘subgingival pool’ in this study34. Similarly, Ozga et al.35 examined the oral microbiome of 57 individuals (no selection criteria) from Oklahoma, United States35. Whole-mouth saliva samples were collected from both men and women between the ages of 23 to 70 years35. The oral microbiome profiles from both of these studies were determined by 16S rRNA gene amplicon sequencing34, 35. The result from our study bears a closer resemblance with findings from Bik et al.34 probably due to the similarly stringent recruitment criteria used in subject selection34. In contrast, Ozga et al.35 selected more lenient recruitment criteria that included subjects with smoking history, bad oral hygiene and active antibiotic use35. The microbial profiles produced from all three saliva fractions were comparable with the numerically predominant taxa included in both studies despite minor differences in abundance, indicating the reliability of our dataset.
Conclusion
Saliva sample collection, processing and gDNA preparation does not significantly influence the salivary microbiome profiles provided that enzymatic-mechanical lysis has been incorporated into the bacterial gDNA extraction protocol. Based on our findings, saliva sampling for microbial research is fairly flexible compared to faecal samples. This allows for number of modifications based on the experimental set-up and down-stream application. However, to improve the purity of bacterial gDNA extracted from saliva samples, we found the Maxwell® 16 LEV Blood DNA kit provided the optimal results. The lack of a coherent and comprehensive guideline in salivary microbiome study design might be a major driver for conflicting findings, a limit inter-study comparison derived from the published literature. We hope that our findings will contribute to those efforts to standardise the saliva collection and bacterial gDNA extraction protocol for future microbiome research.
Declarations
Ethics
This study is approved by the University of Queensland’s (HREC no. 2014000679 and 2017000662) and Queensland University of Technology’s (HREC no. 1400000617) Medical Ethical Institutional Board and the Princess Alexandra Hospital’s (PAH) Ethics Review Board (HREC no. HREC/12/QPAH/381). All participants were informed consent for this study.
Availability of data and materials
All data and materials supporting the findings of this study can be found in the body of the manuscript as well as in the supplementary section.
References
na Head and Neck Cancer Biomarkers Detected in Saliva. Cancer Biol Ther 4, 6–12 (2005).
Pepe, M. S. et al. Phases of Biomarker Development for Early Detection of Cancer. J Natl Cancer Inst 93, 1054–1061 (2001).
Lucs, A. V., Saltman, B., Chung, C. H., Steinberg, B. M. & Schwartz, D. L. Opportunities and challenges facing biomarker development for personalized head and neck cancer treatment. Head Neck 35, 294–306 (2013).
Bandhakavi, S., Stone, M. D., Onsongo, G., Van Riper, S. K. & Griffin, T. J. A dynamic range compression and three-dimensional peptide fractionation analysis platform expands proteome coverage and the diagnostic potential of whole saliva. J. Proteome Res. 8, 5590–5600 (2009).
Iorgulescu, G. Saliva between normal and pathological. Important factors in determining systemic and oral health. J Med Life 2, 303–307 (2009).
Pfaffe, T., Cooper-White, J., Beyerlein, P., Kostner, K. & Punyadeera, C. Diagnostic potential of saliva: current state and future applications. Clin Chem 57, 675–687 (2011).
Malamud, D. Saliva as a diagnostic fluid. Dent Clin North Am 55, 159–178 (2011).
Chai, R. C. et al. A pilot study to compare the detection of HPV-16 biomarkers in salivary oral rinses with tumour p16INK4a expression in head and neck squamous cell carcinoma patients. BMC Cancer 16, 1–8 (2016).
Lim, Y. et al. Salivary DNA methylation panel to diagnose HPV-positive and HPV-negative head and neck cancers. BMC Cancer 16, 1–12 (2016).
Ovchinnikov, D. A. et al. DNA Methylation at the Novel CpG Sites in the Promoter of MED15/PCQAP Gene as a Biomarker for Head and Neck Cancers. Biomark Insights 9, 53–60 (2014).
Ovchinnikov, D. A. et al. Tumor-suppressor Gene Promoter Hypermethylation in Saliva of Head and Neck Cancer Patients. Transl Oncol 5, 321–326 (2012).
Salazar, C. et al. A novel saliva-based microRNA biomarker panel to detect head and neck cancers. Cell Oncol (Dordr) 37, 331–338 (2014).
Zhang, X. et al. Quantification of D-dimer levels in human saliva. Bioanalysis 5 (2013).
Zhang, X., Dimeski, G. & Punyadeera, C. Validation of an immunoassay to measure plasminogen-activator inhibitor-1 concentrations in human saliva. Biochem Med 24, 258–265 (2014).
Wong, D. T. Salivary extracellular noncoding RNA: emerging biomarkers for molecular diagnostics. Clin Ther 37, 540–551 (2015).
Nunes, L. A., Mussavira, S. & Bindhu, O. S. Clinical and diagnostic utility of saliva as a non-invasive diagnostic fluid: a systematic review. Biochem Med (Zagreb) 25, 177–192 (2015).
Guerrero-Preston, R. et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, Human Papilloma Virus infection and surgical treatment. Oncotarget (2016).
Weidlich, P., Cimões, R., Pannuti, C. M. & Oppermann, R. V. Association between periodontal diseases and systemic diseases. Braz Oral Res 22, 32–43 (2008).
Shacter, E. & Weitzman, S. A. Chronic inflammation and cancer. Oncology (Williston Park) 16, 217–226, 229; discussion 230–212 (2002).
Vogelmann, R. & Amieva, M. R. The role of bacterial pathogens in cancer. Curr Opin Microbiol 10, 76–81 (2007).
Chen, M. et al. The impact of different DNA extraction methods on the analysis of microbial diversity of oral saliva from healthy youths by polymerase chain reaction-denaturing gradient gel electrophoresis. JDS 11, 54–58 (2016).
Jenkinson, H. F. Beyond the oral microbiome. Environ Microbiol 13, 3077–3087 (2011).
Shanahan, E. R., Zhong, L., Talley, N. J., Morrison, M. & Holtmann, G. Characterisation of the gastrointestinal mucosa-associated microbiota: a novel technique to prevent cross-contamination during endoscopic procedures. Aliment Pharmacol Ther 43, 1186–1196 (2016).
Haas, B. J. et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21, 494–504 (2011).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618 (2012).
Takeshita, T. et al. Bacterial diversity in saliva and oral health-related conditions: the Hisayama Study. Sci Rep 6, 22164 (2016).
Dewhirst, F. E. et al. The human oral microbiome. J Bacteriol 192, 5002–5017 (2010).
Lazarevic, V., Gaia, N., Girard, M., Francois, P. & Schrenzel, J. Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLoS One 8, e67699 (2013).
Sohrabi, M. et al. The yield and quality of cellular and bacterial DNA extracts from human oral rinse samples are variably affected by the cell lysis methodology. J Microbiol Methods 122, 64–72 (2016).
Vesty, A., Biswas, K., Taylor, M. W., Gear, K. & Douglas, R. G. Evaluating the Impact of DNA Extraction Method on the Representation of Human Oral Bacterial and Fungal Communities. PLoS ONE 12, e0169877 (2017).
Hull, M. W. & Chow, A. W. Indigenous microflora and innate immunity of the head and neck. Infect Dis Clin North Am 21, 265–282 (2007).
Taylan, I. et al. Comparison of the Surface and Core Bacteria in Tonsillar and Adenoid Tissue With Beta-Lactamase Production. Indian J Otolaryngol Head Neck Surg 63, 223–228 (2011).
Danser, M. M., Gómez, S. M. & Van der Weijden, G. A. Tongue coating and tongue brushing: a literature review. Int J Dent Hyg 1, 151–158 (2003).
Bik, E. M. et al. Bacterial diversity in the oral cavity of ten healthy individuals. The ISME J 4, 962–974 (2010).
Ozga, A.T. et al. Oral microbiome diversity among Cheyenne and Arapaho individuals from Oklahoma. Am J Phys Anthropol (2016).
Acknowledgements
We thank Ms. Nida Murtaza for technical support with 16S rRNA gene amplicon library preparation and Dr. Naoki Fukuma for his support in the QIIME script coding process and troubleshooting. This study was supported by the Queensland Centre for Head and Neck Cancer funded by Atlantic Philanthropies, the Queensland Government, the Princess Alexandra Hospital, the Queensland University of Technology Vice Chancellor Fellowship (C.P.) and the QUT postgraduate research scholarship (Y.K.L.).
Author information
Authors and Affiliations
Contributions
Y.K.L. collected samples, compiled all epidemiological data on all subjects, conducted all experiments, data interpretation and wrote the manuscript, M.M. and M.T. contributed to the experimental design, data interpretation and revised the manuscript critically for content and C.P. participated in study concept and design, study coordination and revised the manuscript critically for content. All authors have read and approved the content of this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, Y., Totsika, M., Morrison, M. et al. The saliva microbiome profiles are minimally affected by collection method or DNA extraction protocols. Sci Rep 7, 8523 (2017). https://doi.org/10.1038/s41598-017-07885-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07885-3
This article is cited by
-
Extensive comparison of salivary collection, transportation, preparation, and storage methods: a systematic review
BMC Oral Health (2024)
-
Salivary microbiomes: a potent evidence in forensic investigations
Forensic Science, Medicine and Pathology (2024)
-
Profiling salivary miRNA expression levels in Fanconi anemia patients – a pilot study
Odontology (2024)
-
Factors influencing oral microbiome analysis: from saliva sampling methods to next-generation sequencing platforms
Scientific Reports (2023)
-
Promising applications of human-derived saliva biomarker testing in clinical diagnostics
International Journal of Oral Science (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
