
Abstract
Livestock-associated methicillin-resistant Staphylococcus aureus (LA-MRSA) is an emerging problem in many parts of the world. LA-MRSA has been isolated previously from animals and humans in the United Kingdom (UK), but the prevalence is unknown. The aim of this study was to determine the prevalence and to describe the molecular epidemiology of LA-MRSA isolated in the East of England (broadly Cambridge and the surrounding area). We accessed whole genome sequence data for 2,283 MRSA isolates from 1,465 people identified during a 12-month prospective study between 2012 and 2013 conducted in the East of England, United Kingdom. This laboratory serves four hospitals and 75 general practices. We screened the collection for multilocus sequence types (STs) and for host specific resistance and virulence factors previously associated with LA-MRSA. We identified 13 putative LA-MRSA isolates from 12 individuals, giving an estimated prevalence of 0.82% (95% CI 0.47% to 1.43%). Twelve isolates were mecC-MRSA (ten CC130, one ST425 and one ST1943) and single isolate was ST398. Our data demonstrate a low burden of LA-MRSA in the East of England, but the detection of mecC-MRSA and ST398 indicates the need for vigilance. Genomic surveillance provides a mechanism to detect and track the emergence and spread of MRSA clones of human importance.
Similar content being viewed by others

Introduction
Staphylococcus aureus is a major cause of infection in hospitals and the community. The widespread dissemination of methicillin-resistant S. aureus (MRSA) adds complexity to the treatment of infection. S. aureus also colonises and infects wild and domestic animals and livestock, and in the latter causes economically important diseases such as mastitis in ruminants, and bumble foot in poultry. Several S. aureus multilocus sequence type (MLST) lineages are associated with animals, including clonal complex (CC)1 (livestock) CC5 (avian), CC130 (multi-host), CC133 (ruminants), CC151 (ruminants), CC425 (ruminants and wild mammals), and CC398 (livestock)1,2,3,4,5,6. Adaption of S. aureus for specific animal species can be mediated by as little as a single nucleotide polymorphism (SNP)7, while genetic decay1 and acquisition of mobile genetic elements encoding host specific virulence factors such as von Willebrand factor binding protein also play an important role8, 9. Resistance to tetracycline is common in livestock-associated (LA)-MRSA, and is associated with high usage of this drug in veterinary medicine10.
While animal-adapted S. aureus has been recognised for many years, more recently it has become clear that zoonotic transmission of S. aureus is relatively common11. The emergence and spread of LA-MRSA has been exemplified by CC398 MRSA, which is associated with pigs, poultry, cattle and horses10. Since its emergence in the early 2000′s, CC398 has gone on to cause a significant and increasing burden of human disease in a number of European countries12, 13. In the UK, there have been numerous reports of the presence of LA-MRSA in livestock and animal products14,15,16,17,18 and rare cases of human infections3, 19 caused by CC398 and several MRSA lineages (CC130, CC425 and CC1943) positive for a type XI SCCmec element which encodes the mecC 20. Two recent prevalence surveys targeting the livestock-associated mecC-MRSA in human and dairy cattle estimated that mecC-MRSA was present in 2.15% (95% confidence interval (CI) 1.17–3.91%) of English and Welsh dairy farms, and caused 0.45% (95% CI 0.24–0.85%) of human MRSA infections in England16, 21. However, no estimation of the total burden of LA-MRSA associated with human infection has been conducted in the UK. Here, we used an unbiased collection of MRSA genome sequences from a single year in Cambridgeshire to estimate the burden of LA-MRSA in a defined geographical region.
Results
Screening for LA-MRSA
We screened 2,282 MRSA isolates collected from 1,465 people during a prospective study conducted at the Public Health England Clinical Microbiology and Public Health Laboratory, Cambridge University Hospitals NHS Foundation Trust in Cambridge, UK between 18 April 2012 and 17 April 2013. We initially screened genome sequence data for putative LA-MRSA based on known genetic features of LA-MRSA, namely (i) the presence of mecC or (ii) specific MLST sequence types (ST) or CCs. This identified 15 isolates from 13 people (ST97 (n = 2), ST398 (n = 1), mecC positive (n = 12, which belonged to CC130 (n = 10), ST425 (n = 1), and 1 ST1943 (n = 1) (Table 1). We then expanded the screen to include virulence factors associated with LA-MRSA (LukM/F leukotoxin22, SaPI-carried von Willebrand binding protein (vWbp)9, the allele variant of tst (toxic shock syndrome toxin (TSST) found on the bovine staphylococcal pathogenicity island (SaPIbov)23 and the avian-associated prophages: ϕAv1, φAvβ, and SaPIAv)1. This identified two isolates, both of which had been captured in the initial screen: an ST398 isolate (CBLA13 positive for the SaPI-carried vwb), and an ST1943 isolate (CBLA15 positive for the allelic variant of tst (Table 1). The minority of isolates (n = 3) were associated with clinical disease (skin and soft tissue infections in three people), the remainder being isolated from multisite screens (Table 1).
Phylogenetic analyses of LA-MRSA
S. aureus CC97 underwent a host jump from cattle into humans around 40 years ago24.To determine whether the two Cambridgeshire CC97 isolates (from a single individual) belonged to the cattle or human clade, we compared these with genomes of a global collection of 43 CC97 isolates24. A phylogenetic tree demonstrated that the two Cambridge ST97 isolates resided in the human clade A, and were most closely related to an isolate associated with a bloodstream infection in Turkey in 2007 (Fig. 1A)24. The host jump of CC97 into humans has been linked to the gain of a β-toxin-converting phage (φSa3) containing the human immune evasion cluster (IEC) of genes (staphylokinase (sak), staphylococcal complement inhibitor (scn), and chemotaxis inhibitory protein of S. aureus (chp)). The two Cambridgeshire CC97 isolates were positive for sak and scn, (IEC type E), providing further evidence of a human source (Table 1). We conclude that the Cambridgeshire ST97 isolates were not of a livestock origin, but rather a rare human adapted lineage of ST97 MRSA.

(A) Phylogenetic relationships between Cambridgeshire CC97 isolates and most closely related isolates from a global collection10. Figure shows an unrooted maximum likelihood tree generated from core genome single nucleotide polymorphisms. (B) Phylogenetic relationships between Cambridgeshire CC130 isolates. Figure shows an unrooted maximum likelihood tree generated from core genome single nucleotide polymorphisms. The two clinical isolates are highlighted in red. The two closely related isolates from the same patient are highlighted in blue.
The genetic relatedness between the Cambridgeshire mecC ST425 isolate (CBLA14) and the ST425 reference genome (an English bovine isolate, LGA251) was defined based on SNPs in the core genome3. The two isolates differed by more than 400 core genome SNPs, indicating that they were distantly related. A phylogenetic tree of the ten CC130 mecC-MRSA isolates was reconstructed based on SNPs in the core genome compared with a reference. This showed clustering that was consistent with ST designation, although isolates belonging to the same ST were not closely related (>50 SNPs different) (Fig. 1B). The exception was two ST1245 isolates (CBLA8 and CBLA10) isolated from the same patient 19 days apart that differed by only four SNPs. In contrast to the rest of the mecC-MRSA isolates, the single ST1945 (CBLA3) was positive for sak and scn, (IEC type E) suggestive of a human origin. However, ST1945 isolates from wild mice and deer in Spain have also been found to be positive for IEC type E, suggesting this may be conserved trait of ST1945 isolates, irrespective of host25, 26.
To determine the genetic relatedness of the Cambridgeshire ST398 isolate (CBLA13) to those found elsewhere, we compared this with a global collection of 89 CC398 isolates10. A phylogenetic tree demonstrated that CBLA13 resided in the livestock-associated clade and was most closely related to three isolates with a porcine origin, in subclade IIa1ii from Austria and Slovenia (data not shown). This is consistent with a livestock source for CBLA13.
Antimicrobial resistance
With one exception (CBLA12), mecC-MRSA isolates were resistant to cefoxitin, but susceptible to oxacillin based on VITEK testing, as reported previously (Table 1)27. Most mecC-MRSA isolates (11/12) were relatively susceptible, with resistance limited to β-lactam antibiotics out of the panel of agents tested. The exception (CBLA6) was resistant to erythromycin, associated with the presence of msrA, and also carried smr (formly qacC) that mediates resistance to quaternary ammonium compounds in antiseptics and disinfectants28. The single ST398 isolate (CBLA13) was resistant to erythromycin and tetracycline, which was associated with the presence of ermC, and tetK and tetM respectively. Tetracycline resistance is not unique to livestock-associated MRSA but is notable, as this is associated with livestock-associated ST39810.
Discussion
Here, we report an unbiased assessment of the prevalence of LA-MRSA isolated by a single diagnostic microbiology laboratory in the East of England. By searching for lineages and virulence genes associated with animal MRSA we identified 13 people (15 isolates) positive for MRSA, with features associated previously with LA-MRSA. Detailed genome-level analysis supported a plausible link to livestock for 12 cases (13 isolates). The two exceptions (two isolates from the same case) were ST97, which were assigned to the human clade of a lineage that underwent a host-jump from cattle to humans around 40 years ago, and were therefore unlikely to be livestock-associated24. For the remaining 13 isolates (12 mecC-MRSA (CC130, ST425 and ST1943) and 1 ST398), the evidence suggests a likely livestock source, giving an overall prevalence rate of LA-MRSA in MRSA-positive people of 0.82% (95% CI 0.47% to 1.43%).
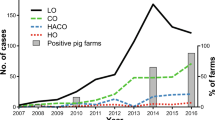
In the UK, ST398 has been detected in cattle, horses, pigs and pork meat, poultry, and in humans.12, 14, 17, 19, 29,30,31 The single ST398 isolate in this study clustered with the livestock-associated clade identified by Price and co-workers, and was most closely related to isolates from pigs in continental Europe10. Consistent with a livestock origin was the observation that the isolate carried a gene encoding a von Willebrand factor binding protein that mediates clotting of ruminant plasma, suggesting that this isolate might have ruminant source. This concurs with previous reports of ST398 in bovine milk in England17. However, vwb has also been found in three ST398 MRSA isolates from UK retail pork samples, suggesting this might be widespread in UK ST398 isolates14. The isolate was also resistant to tetracycline, which is also associated with LA ST398. We noted that the isolate harboured two different tetracycline resistance genes (tetM and tetK), which has also been reported in Scottish Human CC398 isolates19. The acquisition of tetK as part of the staphylococcal cassette chromosome mec (SCCmec) type Vc element by tetM positive LA ST398 has been demonstrated to significantly increase fitness at sub-lethal concentrations of tetracycline32. The use of tetracycline in livestock is likely to be a driver for this and might have contributed to the success of SCCmec Vc-bearing LA-MRSA CC39832. In addition, the presence of the czrC gene encoding resistance to copper and zinc, which are added to animal feed, may have also contributed to the success of LA-MRSA CC39834. Although there are currently very low levels of ST398 in humans in the UK, its presence in a range of livestock species combined with the results of mathematical modelling suggests that once established in livestock populations, ST398 would be hard to eradicate from humans33. A 66% increase in human CC398 cases in Denmark between 2004–2011 was associated with a four-fold increase in the CC398 prevalence in Danish pigs13. Similar dramatic increases in ST398 prevalence have been reported in Germany34. Recent data has also highlighted the role of humans as the vector for transmission between livestock populations including between countries35. Further systematic sampling of livestock and livestock workers for ST398 in the UK is required to better understand its prevalence and epidemiology.
The majority of LA-MRSA isolates were mecC-MRSA, demonstrating that at least in this part of England, this is the dominant form of LA-MRSA. The prevalence in this study for mecC-MRSA of 0.75% (95% CI: 0.30% to 0.92%) is close to the 0.45% (95% CI 0.24%–0.85%) identified in a 2011–2012 multicentre English prevalence study, suggesting that prevalence is currently stable21. This is similar to a prevalence estimate from Denmark (0.5%)36 but higher than large studies from Belgium (0.18%)37, Germany (0, 0.09, and 0.06%)38,39,40, and Spain (0.04%)41, though some variation exists between studies from the same countries42. Both ST1245 isolates and its single locus variant ST2574 have been identified previously in Cambridge, in a multicentre English prevalence study, suggesting that these two clones are endemic in Cambridgeshire21. Importantly, ST1245 has also been isolated from bovine milk at two different locations in England3. Cambridgeshire is an area with low livestock density, but borders East Anglia where there is high density of pig and poultry production43. The predominance of CC130 in the Cambridgeshire LA-MRSA, a clone which is commonly isolated from sheep and cattle3, 11, 44, 45, suggests that the prevalence of CC130 might be higher in parts of the UK with higher sheep and cattle densities, or alternatively the reservoirs of mecC-MRSA are still not fully understood. Indeed, mecC-MRSA has recently been reported in pigs and pig workers in Denmark46. Five of the eight ST1245 isolates were detected in May and June (Table 1) when people are more likely to be outdoors and may come in contact with livestock, but this is underpowered and future studies are required to investigate this association in a robust manner.
In summary, our data demonstrate a low burden of LA-MRSA in the East of England, but the detection of mecC-MRSA and ST398 indicates the need for vigilance. As demonstrated here and elsewhere47, genomic surveillance provides a mechanism to detect and track the emergence of MRSA clones of human importance.
Materials and Methods
Study design
To understand the molecular epidemiology and transmission pathways in a healthcare network we conducted a prospective observational cohort study between April 2012 and April 2013. We identified all individuals with MRSA-positive samples processed by the Public Health England Clinical Microbiology and Public Health Laboratory, Cambridge University Hospitals NHS Foundation Trust in Cambridge, UK. The laboratory processes samples from four Cambridgeshire hospitals (Addenbrooke’s hospital (a large university teaching hospital), the Rosie hospital (a maternity hospital), Papworth hospital (a specialist cardiothoracic hospital) and Hinchingbrooke hospital (a district general hospital) and 75 general practices in the same geographic region (broadly the area around Cambridge). Samples were from multisite screening swabs from hospital patients on admission or during a hospital stay, or from clinical specimens from hospital patients or taken from patients in general practice. All cases with MRSA isolated at least once, from either screening swabs and/or clinical specimens, were included in the study. Clinical metadata and demographic information were collected from electronic and paper medical records. In accordance with national policy at this time, universal MRSA screening (a multi-site MRSA screen of all patients on hospital admission, and weekly MRSA screening of patients in critical care units) was conducted at all four hospitals throughout the study period.
Ethics
The study protocol was approved by the National Research Ethics Service (ref: 11/EE/0499), the National Information Governance Board Ethics and Confidentiality Committee (ref: ECC 8–05(h)/2011), and the Cambridge University Hospitals NHS Foundation Trust Research and Development Department (ref: A092428).
Microbiology methods
MRSA was isolated from screening samples by directly plating swabs onto Brilliance MRSA chromogenic medium (Oxoid, Basingstoke, UK), and from all other samples by plating onto Columbia Blood Agar (Oxoid, Basingstoke, UK). S. aureus was identified using a commercial latex agglutination kit (Pastorex Staph Plus, Bio Rad Laboratories, Hemel Hempstead, UK). Antimicrobial susceptibility was determined to a panel of antibiotics (benzylpenicillin, cefoxitin, oxacillin, ciprofloxacin, erythromycin, chloramphenicol, daptomycin, fusidic acid, gentamicin, linezolid, mupirocin, nitrofurantoin, rifampicin, teicoplanin, tetracycline, tigecycline, trimethoprim, vancomycin, clindamycin, and inducible resistance to clindamycin) using the VITEK 2 instrument (bioMerieux, Marcy l’Etoile, France). Antimicrobial susceptibility results were interpreted using the European Committee on Antimicrobial Susceptibility Testing (EUCAST) criteria48.
Whole genome sequencing and bioinformatics analysis
Genomic DNA was extracted from MRSA isolates, libraries prepared and 150-bp paired end sequences determined on an Illumina HiSeq. 2000 as previously described49. Sequence data have been submitted to the European Nucleotide Archive (ENA) (www.ebi.ac.uk/ena) under the accession numbers listed in Table 1. Sequence data were assembled using a previously described pipeline50. Briefly, for each isolate the sequence reads were used to create multiple assemblies using VelvetOptimiser v2.2.551 and Velvet v1.252. The assemblies were improved by scaffolding the best N50 and contigs using SSPACE53 and sequence gaps filled using GapFiller54. Multilocus sequence types (MLST) were determined from the assemblies using MLST check (https://github.com/sanger-pathogens/mlst_check), which was used to compare the assembled genomes against the MLST database for S. aureus (http://pubmlst.org/saureus/). The presence of S. aureus virulence factors and antibiotic resistance genes were identified using BLAST against the assemblies. For phylogenetic analyses, sequence reads were mapped to a relevant reference genome (CC130 and ST425 isolates (strain LGA251, accession number FR821779), ST97 (strain MW2, accession number BA000033), ST398 (strain S0385, accession number AM990992)) using SMALT (http://www.sanger.ac.uk/science/tools/smalt-0) using the default settings to identify single nucleotide polymorphisms (SNPs). SNPs located in mobile genetic elements were removed and a maximum likelihood tree created using RAxML using the default settings and 100 bootstrap replicates55. For the ST398 phylogeny the large block of ST8 recombination present in ST398 (S0385 genomic locations: 12252 to 135180) was also removed from the ST398 alignment.
References
Lowder, B. V. et al. Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proceedings of the National Academy of Sciences of the United States of America 106, 19545–19550, doi:10.1073/pnas.0909285106 (2009).
Paterson, G. K. et al. The newly described mecA homologue, mecALGA251, is present in methicillin-resistant Staphylococcus aureus isolates from a diverse range of host species. The Journal of antimicrobial chemotherapy 67, 2809–2813, doi:10.1093/jac/dks329 (2012).
Garcia-Alvarez, L. et al. Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. The Lancet infectious diseases 11, 595–603, doi:10.1016/S1473-3099(11)70126-8 (2011).
Guinane, C. M. et al. Pathogenomic analysis of the common bovine Staphylococcus aureus clone (ET3): emergence of a virulent subtype with potential risk to public health. The Journal of infectious diseases 197, 205–213, doi:10.1086/524689 (2008).
Porrero, M. C. et al. Detection of mecC-MRSA isolates in river water: a potential role for water in the environmental dissemination. Environmental Microbiology Reports, n/a-n/a, doi:10.1111/1758-2229.12191 (2014).
Alba, P. et al. Livestock-Associated Methicillin Resistant and Methicillin Susceptible Staphylococcus aureus Sequence Type (CC)1 in European Farmed Animals: High Genetic Relatedness of Isolates from Italian Cattle Herds and Humans. PloS one 10, e0137143, doi:10.1371/journal.pone.0137143 (2015).
Viana, D. et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat Genet 47, 361–366, doi:10.1038/ng.3219 (2015).
Guinane, C. M. et al. Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome biology and evolution 2, 454–466, doi:10.1093/gbe/evq031 (2010).
Viana, D. et al. Adaptation of Staphylococcus aureus to ruminant and equine hosts involves SaPI-carried variants of von Willebrand factor-binding protein. Mol Microbiol 77, 1583–1594 (2010).
Price, L. B. et al. Staphylococcus aureus CC398: Host Adaptation and Emergence of Methicillin Resistance in Livestock. mBio 4, doi:10.1128/mBio.00520-12 (2013).
Harrison, E. M. et al. Whole genome sequencing identifies zoonotic transmission of MRSA isolates with the novel mecA homologue mecC. EMBO Mol Med. (2013).
Kock, R. et al. Livestock-Associated Methicillin-Resistant Staphylococcus aureus (MRSA) as Causes of Human Infection and Colonization in Germany. PloS one 8, e55040, doi:10.1371/journal.pone.0055040 (2013).
Larsen, J. et al. Meticillin-resistant Staphylococcus aureus CC398 is an increasing cause of disease in people with no livestock contact in Denmark, 1999 to 2011. Euro surveillance: bulletin europeen sur les maladies transmissibles=European communicable disease bulletin 20, doi:10.2807/1560-7917.es.2015.20.37.30021 (2015).
Hadjirin, N. F. et al. Detection of livestock-associated meticillin-resistant Staphylococcus aureus CC398 in retail pork, United Kingdom, February 2015. Euro surveillance: bulletin europeen sur les maladies transmissibles=European communicable disease bulletin 20 (2015).
Dhup, V., Kearns, A. M., Pichon, B. & Foster, H. A. First report of identification of livestock-associated MRSA ST9 in retail meat in England. Epidemiology and infection 143, 2989–2992, doi:10.1017/s0950268815000126 (2015).
Paterson, G. K. et al. Prevalence and properties of mecC methicillin-resistant Staphylococcus aureus (MRSA) in bovine bulk tank milk in Great Britain. The Journal of antimicrobial chemotherapy 69, 598–602, doi:10.1093/jac/dkt417 (2014).
Paterson, G. K. et al. First detection of livestock-associated meticillin-resistant Staphylococcus aureus CC398 in bulk tank milk in the United Kingdom, January to July 2012. Euro surveillance: bulletin europeen sur les maladies transmissibles = European communicable disease bulletin 17 (2012).
Hartley, H. et al. Confirmation of LA-MRSA in pigs in the UK. The Veterinary record 175, 74–75, doi:10.1136/vr.g4620 (2014).
Ward, M. J. et al. Time-scaled evolutionary analysis of the transmission and antibiotic resistance dynamics of Staphylococcus aureus CC398. Applied and environmental microbiology, doi:10.1128/aem.01777-14 (2014).
Paterson, G. K., Harrison, E. M. & Holmes, M. A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends in microbiology 22, 42–47, doi:10.1016/j.tim.2013.11.003 (2014).
Paterson, G. K. et al. Prevalence and characterization of human mecC methicillin-resistant Staphylococcus aureus isolates in England. The Journal of antimicrobial chemotherapy 69, 907–910, doi:10.1093/jac/dkt462 (2014).
Vrieling, M. et al. Bovine Staphylococcus aureus Secretes the Leukocidin LukMF′ To Kill Migrating Neutrophils through CCR1. mBio 6, doi:10.1128/mBio.00335-15 (2015).
Fitzgerald, J. R. et al. Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. Journal of bacteriology 183, 63–70, doi:10.1128/jb.183.1.63-70.2001 (2001).
Spoor, L. E. et al. Livestock Origin for a Human Pandemic Clone of Community-Associated Methicillin-Resistant Staphylococcus aureus. mBio 4, doi:10.1128/mBio.00356-13 (2013).
Gomez, P. et al. High prevalence of methicillin-resistant Staphylococcus aureus (MRSA) carrying the mecC gene in a semi-extensive red deer (Cervus elaphus hispanicus) farm in Southern Spain. Veterinary microbiology 177, 326–331, doi:10.1016/j.vetmic.2015.03.029 (2015).
Gomez, P. et al. Detection of methicillin-resistant Staphylococcus aureus (MRSA) carrying the mecC gene in wild small mammals in Spain. The Journal of antimicrobial chemotherapy 69, 2061–2064, doi:10.1093/jac/dku100 (2014).
Cartwright, E. J. et al. Use of Vitek 2 antimicrobial susceptibility profile to identify mecC in methicillin-resistant Staphylococcus aureus. Journal of clinical microbiology 51, 2732–2734, doi:10.1128/jcm.00847-13 (2013).
Littlejohn, T. G., DiBerardino, D., Messerotti, L. J., Spiers, S. J. & Skurray, R. A. Structure and evolution of a family of genes encoding antiseptic and disinfectant resistance in Staphylococcus aureus. Gene 101, 59–66 (1991).
Loeffler, A. et al. First isolation of MRSA ST398 from UK animals: a new challenge for infection control teams? The Journal of hospital infection 72, 269–271, doi:10.1016/j.jhin.2009.04.002 (2009).
GOV. UK. Livestock-associated MRSA found at a farm in East Anglia, 2013.
Paterson, G. K. et al. Incidence and characterisation of methicillin-resistant Staphylococcus aureus (MRSA) from nasal colonisation in participants attending a cattle veterinary conference in the UK. PloS one 8, e68463, doi:10.1371/journal.pone.0068463 (2013).
Larsen, J. et al. Copresence of tet(K) and tet(M) in Livestock-Associated Methicillin-Resistant Staphylococcus aureus Clonal Complex 398 Is Associated with Increased Fitness during Exposure to Sublethal Concentrations of Tetracycline. Antimicrobial agents and chemotherapy 60, 4401–4403, doi:10.1128/aac.00426-16 (2016).
Porphyre, T., Giotis, E. S., Lloyd, D. H. & Stark, K. D. A metapopulation model to assess the capacity of spread of meticillin-resistant Staphylococcus aureus ST398 in humans. PloS one 7, e47504, doi:10.1371/journal.pone.0047504 (2012).
van Alen, S. et al. In the centre of an epidemic: Fifteen years of LA-MRSA CC398 at the University Hospital Munster. Veterinary microbiology, doi:10.1016/j.vetmic.2016.01.021 (2016).
Grontvedt, C. A. et al. Methicillin-Resistant Staphylococcus aureus CC398 in Humans and Pigs in Norway: A “One Health” Perspective on Introduction and Transmission. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 63, 1431–1438, doi:10.1093/cid/ciw552 (2016).
Petersen, A. et al. Epidemiology of methicillin-resistant Staphylococcus aureus carrying the novel mecC gene in Denmark corroborates a zoonotic reservoir with transmission to humans. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 19, E16–22, doi:10.1111/1469-0691.12036 (2013).
Deplano, A., Vandendriessche, S., Nonhoff, C. & Denis, O. Genetic diversity among methicillin-resistant Staphylococcus aureus isolates carrying the mecC gene in Belgium. The Journal of antimicrobial chemotherapy 69, 1457–1460, doi:10.1093/jac/dku020 (2014).
Cuny, C., Layer, F., Strommenger, B. & Witte, W. Rare occurrence of methicillin-resistant Staphylococcus aureus CC130 with a novel mecA homologue in humans in Germany. PloS one 6, e24360, doi:10.1371/journal.pone.0024360 (2011).
Schaumburg, F. et al. Population dynamics among methicillin-resistant Staphylococcus aureus isolates in Germany during a 6-year period. Journal of clinical microbiology 50, 3186–3192, doi:10.1128/jcm.01174-12 (2012).
Becker, K., Schaumburg, F., Fegeler, C., Friedrich, A. W. & Kock, R. Staphylococcus aureus from the German general population is highly diverse. International journal of medical microbiology: IJMM 307, 21–27, doi:10.1016/j.ijmm.2016.11.007 (2017).
Garcia-Garrote, F. et al. Methicillin-resistant Staphylococcus aureus carrying the mecC gene: emergence in Spain and report of a fatal case of bacteraemia. The Journal of antimicrobial chemotherapy 69, 45–50, doi:10.1093/jac/dkt327 (2014).
Diaz, R., Ramalheira, E., Afreixo, V. & Gago, B. Methicillin-resistant Staphylococcus aureus carrying the new mecC gene–a meta-analysis. Diagnostic microbiology and infectious disease 84, 135–140, doi:10.1016/j.diagmicrobio.2015.10.014 (2016).
Maps of livestock populations in 2000 and 2010 across England. (Department for Environment, Food and Rural Affairs (DEFRA)).
Schlotter, K. et al. Multiple cases of methicillin-resistant CC130 Staphylococcus aureus harboring mecC in milk and swab samples from a Bavarian dairy herd. Journal of dairy science 97, 2782–2788, doi:10.3168/jds.2013-7378 (2014).
Vandendriessche, S. et al. Prevalence, risk factors and genetic diversity of methicillin-resistant Staphylococcus aureus carried by humans and animals across livestock production sectors. The Journal of antimicrobial chemotherapy 68, 1510–1516, doi:10.1093/jac/dkt047 (2013).
Angen, O. et al. Report of mecC-carrying MRSA in domestic swine. The Journal of antimicrobial chemotherapy 72, 60–63, doi:10.1093/jac/dkw389 (2017).
Toleman, M. S. et al. Systematic surveillance detects multiple silent introductions and household transmission of methicillin-resistant Staphylococcus aureus USA300 in the east of England. Journal of Infectious Diseases (2016).
(The European Committee on Antimicrobial Susceptibility Testing (http://www.eucast.org/), 2015).
Reuter, S. et al. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA internal medicine 173, 1397–1404, doi:10.1001/jamainternmed.2013.7734 (2013).
Page, A. J. et al. Robust high throughput prokaryote de novo assembly and improvement pipeline for Illumina data. bioRxiv, 10.1101/052688 (2016).
Velvet Optimiser: For automatically optimising the primary parameter options for the Velvet de novo sequence assembler. (2008).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829, doi:10.1101/gr.074492.107 (2008).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579, doi:10.1093/bioinformatics/btq683 (2011).
Boetzer, M. & Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol 13, R56, doi:10.1186/gb-2012-13-6-r56 (2012).
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463, doi:10.1093/bioinformatics/bti191 (2005).
Acknowledgements
We thank Lois Mlemba for assistance with data collection. We are grateful for assistance from the library construction, sequencing and core informatics teams at the Wellcome Trust Sanger Institute. Supported by grants from the UKCRC Translational Infection Research (TIR) Initiative, and the Medical Research Council (Grant Number G1000803) with contributions to the Grant from the Biotechnology and Biological Sciences Research Council, the National Institute for Health Research on behalf of the Department of Health, and the Chief Scientist Office of the Scottish Government Health Directorate (to Prof. Peacock); a Hospital Infection Society Major Research Grant, and by Wellcome Trust grant number 098051 awarded to the Wellcome Trust Sanger Institute. This work was supported by the Wellcome Trust 201344/Z/16/Z. M.E.T. is a Clinician Scientist Fellow, supported by the Academy of Medical Sciences and the Health Foundation, and by the NIHR Cambridge Biomedical Research Centre.
Author information
Authors and Affiliations
Contributions
E.M.H. and F.C. conducted the bioinformatics analysis. M.S.T. collected clinical data and contributed to the analysis. B.B. conducted the laboratory work. M.E.T. and S.J.P. designed the original study and obtained ethical and research and development approvals for the study. N.M.B. provided clinical laboratory support. M.E.T., J.P., and S.J.P. supervised the study. E.M.H. and S.J.P. wrote the manuscript. All authors contributed, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Harrison, E.M., Coll, F., Toleman, M.S. et al. Genomic surveillance reveals low prevalence of livestock-associated methicillin-resistant Staphylococcus aureus in the East of England. Sci Rep 7, 7406 (2017). https://doi.org/10.1038/s41598-017-07662-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07662-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
