
Abstract
We have previously shown that in vitro transduction with bovine adeno–associated viral (BAAV) vectors restores connexin expression and rescues gap junction coupling in cochlear organotypic cultures from connexin–deficient mice that are models DFNB1 nonsyndromic hearing loss and deafness. The aims of this study were to manipulate inner ear connexin expression in vivo using BAAV vectors, and to identify the optimal route of vector delivery. Injection of a BAAV vector encoding a bacterial Cre recombinase via canalostomy in adult mice with floxed connexin 26 (Cx26) alleles promoted Cre/LoxP recombination, resulting in decreased Cx26 expression, decreased endocochlear potential, increased hearing thresholds, and extensive loss of outer hair cells. Injection of a BAAV vector encoding GFP-tagged Cx30 via canalostomy in P4 mice lacking connexin 30 (Cx30) promoted formation of Cx30 gap junctions at points of contacts between adjacent non-sensory cells of the cochlear sensory epithelium. Levels of exogenous Cx30 decayed over time, but were still detectable four weeks after canalostomy. Our results suggest that persistence of BAAV-mediated gene replacement in the cochlea is limited by the extensive remodeling of the organ of Corti throughout postnatal development and associated loss of non-sensory cells.
Similar content being viewed by others

Introduction
Up to 50% of prelingual hearing impairment is linked to the DFNB1 locus on chromosome 13q11–q121, which comprises the genes encoding two structurally and functionally related gap junction proteins, Cx26 (GJB2) and Cx30 (GJB6)2, 3. In mice, the coordinated expression of Cx26 and Cx304, 5 has been shown to depend on the spacing of their surrounding chromosomal region6. Cx26 and Cx30 colocalize in supporting and epithelial cells of the organ of Corti, in basal and intermediate cells of the stria vascularis, and in type 1 fibrocytes of the spiral ligament7,8,9, forming vast gap junction networks that can be visualized directly, using voltage imaging, even during the first postnatal week10. Gap junctions are dynamic structures and connexins turn over rapidly, with half lives of 2–5 hours11, 12.
Epithelial and supporting cells of the cochlear sensory epithelium (hereafter collectively referred to as non-sensory cells) provide trophic and mechanical support to sensory hair cells, which perform mechanotransduction13, i.e. the conversion of sound-evoked mechanical stimuli applied to their hair bundle into graded changes of their membrane potentials known as receptor potentials14. Mechanotransduction and generation of receptor potentials depend on the potential difference, known as endocochlear potential, between endolymph (a K+ rich fluid that fills scala media) and perilymph (a Na+ rich fluid akin to cerebrospinal fluid that fills scala tympany and scala vestibuli)15, 16. The indispensable endocochlear potential, which in mice exceeds 100 mV17, is generated by the activity of gap-junction coupled cells in the lateral wall (spiral ligament and stria vascularis)18, 19 and provides the driving force required for K+ influx from endolymph to hair cell cytosol via mechanically activated channels in the hair bundle20, 21. Another essential prerequisite for generating receptor potentials is electrical insulation of the hair cells from the rest of sensory epithelium22. Consistent with this requirement, neither the outer nor the inner hair cells express any connexin23,24,25. Inner hair cell depolarization drives glutamate release at their synaptic pole and consequent excitation of afferent neurotransmission along the auditory nerve26,27,28. Outer hair cell receptor potentials activate prestin in their basolateral membrane29,30,31,32 supporting mechanical amplification of basilar membrane movements33, 34 and increasing hearing sensitivity and frequency selectivity35, 36.
Virus−mediated gene delivery is largely used for human gene therapy, and clinical trials are being carried out to treat cancer, cardiovascular disorders, monogenic disorders, neurological disorders37, and eye diseases38. Gene delivery to the mammalian inner ear has been performed with a variety of viral vectors, including herpes simplex type I virus and vaccinia virus, lentiviruses, retroviruses and adenoviruses, with limited success and several shortcomings39,40,41. Furthermore, an important component of a future therapeutic intervention plan is the optimal route of vector delivery to the inner, and this has not been uniquely identified yet42,43,44,45,46,47.
Gene delivery studies with adeno–associated virus (AAV) were performed in mice in utero 48, 49, as well as in young guinea pigs50,51,52. Subsequent work provided proof-of-principle evidence that intracochlear delivery of AAV vectors can mediate restoration of hearing in mice lacking vesicular glutamate transporter 3 in inner hair cells (VGLUT3)53 and partial restoration in mice lacking transmembrane channel–like 1 (TMC1), a protein known to affect the permeation properties of sensory transduction channels in both inner and outer hair cells54. However, outer hair cell function was not restored in that study. Recently, a synthetic adeno-associated viral vector (Anc80L65) with high tropism for both types of hair cells was delivered via round window membrane injections to treat a knock-in mouse model for Usher syndrome type 1 C55.
Here, we used BAAV vectors4, 5, 49, 56,57,58 to deliver reporter and connexin genes to the inner ear of adult as well as neonatal mice. Our results in wild type and transgenic mice indicate that BAAV, administered via canalostomy42, 46, 59,60,61, is highly effective in transducing cochlear non−sensory cells in vivo, however the expression level of the transgene is insufficient to sustain adequate connexin expression from postnatal development to adulthood.
Results
Canalostomy is a viable and safe route for gene delivery to the mouse cochlea
To validate canalostomy as a potential route to target cochlear structures, we delivered bromophenol blue or fluorescent wheat germ agglutinin dissolved in PBS in the semicircular canal of P25 or P4 wild type mice (C57BL6/N), resulting in widespread distribution of the injected agent without visible damage to cochlear duct structures (Fig. 1a,b). In a subsequent set of experiments, we injected Dulbecco’s Modified Eagle Medium/Nutrient Mixture F−12 (DMEM/F12) via canalostomy in wild type mice at P25 (2.5 μl) or P4 (1.0 μl). Four weeks later, we assessed hearing performance by recording auditory brainstem responses (ABRs), which are electrical signals evoked from the brainstem following the presentation of sound stimuli62. We measured the IV wave thresholds of the ABR for click and tone burst stimuli of 8, 14, 20, 26, 32 kHz and found no differences in injected mice compared with non injected controls (Fig. 1c). These results indicate that delivery of DMEM/F12 to the inner ear via canalostomy causes no adverse effects on hearing and could function as an effective route of vector delivery.

In vivo fluid delivery via canalostomy to mouse cochlea. (a,b) Midmodiolar cochlear sections from P25 (a) and P4 (b) mice injected with fluorescent wheat germ agglutinin (which labels cell membranes) examined by confocal microscopy; scale bars: 50 μm. Insets: representative light microscopy images of cochleae from P25 (a) or P4 (b) mice injected with bromophenol blue, dissolved in PBS; images were acquired 5 min after canalostomy; scale bars: 500 μm; arrowheads point to the base (black) and apex (white) of the cochlea. BM: basilar membrane; GER, greater epithelial ridge; IHC: inner hair cell; IS: inner sulcus; LER, lesser epithelial ridge; LM: spiral limbus; OHC: outer hair cells; OS: outer sulcus; OSL, osseous spiral lamina; RM: Reissner’s membrane; SL: spiral ligament; SV: stria vascularis; TM: tectorial membrane. (c) Hearing thresholds determined by analysis of auditory brainstem responses (ABR) in wild type mice injected at P25 (red), P4 (cyan) and non-injected controls (black); ordinates are sound pressure levels (SPL) relative to 20 µPa: Error bars represent standard error of the mean (s.e.m.).
In vivo transduction with BAAV vectors via canalostomy achieves widespread expression of a reporter gene in non-sensory cells of the mouse cochlear duct
To evaluate the efficacy of in vivo transduction via canalostomy, we delivered a previously tested reporter gene vector, BAAVβ−actin−GFP56, 58 prepared in DMEM/F12, to the inner ear of P25 wild type mice. Four weeks later, we processed the cochlea for confocal immunofluorescence imaging (Fig. 2). No β−actin−GFP signal was detected in cochlear sensory hair cells, whereas diffuse expression was clearly visible in spiral limbus and spiral prominence, two structures populated by fibrocytes, as well as in non−sensory cells of the cochlear sensory epithelium. In particular, the majority of Hensen’s and Claudius cells, some pillar cells and inner sulcus cells expressed β−actin−GFP. Extensive transgene expression was also evident in the lateral wall of the cochlea, namely in the spiral ligament, stria vascularis and supra-strial zone (Fig. 2).

Confocal immunofluorescence imaging of cochlear cross–sections from mice injected at P25 with BAAVβ-actin-GFP. Color code: β-actin-GFP, green; actin filaments, red; nuclei, blue. BC: Böttcher cells; BM: basilar membrane; Bo: bone; HCR: hair cell region; IHC: inner hair cell; IS: inner sulcus; LM: spiral limbus; OHC: outer hair cells; OS: outer sulcus; OSL: osseus spiral lamina; SL: spiral ligament; SP: spiral prominence; SSZ: supra-strial zone; SV: stria vascularis. Scale bars: 50 μm.
Prior work in adult guinea pigs used cochleostomy as route of administration and reported a higher efficiency of expression of BAAVβ-actin-GFP when delivered into scala media compared to a scala tympani approach; however the scala media approach resulted in hair cells loss58. We replicated these experiments in mice injected at P25 and noted expression of the transgene not only in scala media but also in scala tympani and scala vestibuli, accompanied by an alteration of the cochlear structure and the Reissner’s membrane, highlighting the limitation of this technique for clinical applications (Figure S1).
Altogether, these experiments suggest canalostomy as a preferred route for BAAV-mediated transgene delivery to the mouse cochlea.
BAAV–driven Cre–Lox recombination abates Cx26 in the cochlea of adult Cx26loxP/loxP mice
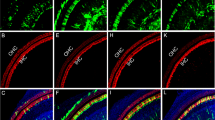
Inner ear connexins play a crucial developmental role and are essential for the maturation of sensory hair cells, despite the fact that hair cells do not express any connexin63. The conventional gene knockout approach is unsuitable for postnatal studies on Cx26 because homozygous knockout mice die in utero due to insufficient transplacental uptake of glucose64. Here, we investigated the role of Cx26 in the maintenance of sensory cells utilizing BAAV for the timed and localized knockout of Cx26 based on the Cre/loxP system65, 66 using a canalostomy route of delivery. Cre-Lox recombination is an irreversible process which does not require sustained protein expression, as it needs to take place only once and requires a limited amount of Cre recombinase65, 66. We engineered a BAAV vector encoding GFP-tagged bacterial Cre recombinase under the CMV promoter (BAAVCre-IRESGFP). We prepared the vector in DMEM/F12 and injected it via canalostomy to the inner ear of P25 Cx26loxP/loxP mice. Confocal immunofluorescence imaging of cochlear midmodiolar sections obtained four weeks after canalostomy showed BAAVCre–IRESGFP caused a dramatic reduction of Cx26 immunofluorescence signals in the lateral wall of Cx26loxP/loxP mice (n = 4) (Fig. 3a), but had no effect on tissue morphology, cell viability and connexin expression in the inner ear of wild type C57BL/6 N mice (n = 3) used as controls (Fig. 3b). Cre-mediated excision of Cx26 was comparatively less effective in the organ of Corti, as indicated by the presence of residual Cx26 expression (Fig. 3c and d for control). q-PCR analysis of whole cochlea samples confirmed a 43.5 ± 2.2% overall reduction of Cx26 transcript levels with respect to the contra lateral non–injected ear (n = 4; p < 0.001, paired t test). Based on these immunofluorescence results, we conclude that the majority of Cx26 loss occurred in fibrocytes of the spiral ligament. Consistent with this conclusion, the endocochlear potential was significantly lower in Cx26loxP/loxP mice injected with BAAVCre–IRESGFP at P25 (26 ± 10 mV, n = 3) compared to non–injected Cx26loxP/loxP controls (106 ± 3 mV, n = 11; p < 0.001, ANOVA). Inner hair cells, pillar cells and Deiters’ cells were maintained in all transversal sections, spanning the whole length of the cochlear duct. Instead, we observed a 92 ± 16% (n = 3) loss of outer hair cells (Fig. 3e and f for control). In agreement with these results, we found remarkably increased hearing thresholds in Cx26loxP/loxP mice injected with BAAVCre–IRESGFP at P25, whereas thresholds in C57BL/6 N mice injected at P25 with BAAVCre–IRESGFP were indistinguishable from non–injected Cx26loxP/loxP controls (Fig. 4). Therefore, we conclude that: (1) the Cre recombinase encoded by the BAAV vector and delivered via canalostomy acted specifically on the floxed alleles; (2) BAAV-based vectors permit efficient manipulation of floxed genes in the mammalian inner ear in vivo; (3) survival of cochlear outer hair cells depends on Cx26 expression in the lateral wall.

Confocal immunofluorescence imaging of cochlear cross–sections from mice injected at P25 with BAAVCre–IRESGFP viral vectors. Color code: Cx26, green; actin filaments, red; nuclei, blue. (a,b) Stria vascularis (SV) and spiral ligament (SL). (c,d) organ of Corti; HCR, hair cell region. (e,f) close–up view of the HCR from c,d, respectively. DCs: Deiters’ cells; IHC: inner hair cell; OHCs: outer hair cells; PCs: pillar cells. Scale bars: 50 μm in (a–d); 25 μm in (e,f).

Hearing thresholds determined by analysis of auditory brainstem responses (ABR) in mice injected with BAAVCre–IRESGFP and their respective controls. Error bars represent s.e.m.
Cx30 expression is restored in the cochlea of Cx30Δ/Δ mice following in vivo delivery of BAAVCx30GFP via canalostomy at P4
In the experiments described above we have demonstrated that BAAV vectors can promote reduction of connexin expression at a specific time point in the cochlea of adult Cx26loxP/loxP mice. Although this is useful to explore the function of inner ear connexins in hearing, restoration of connexin expression in DFNB1 mouse models is clearly of primary importance for future therapeutic applications.
Targeted ablation of Cx26 in the mouse inner ear (Cx26Sox10Cre, Cx26OtogCre) leads to irreversible cell loss in the organ of Corti either before or around the onset of hearing5, 67, which in mice occurs at P1268. A similar problem affects also Cx30−/− mice, which show hearing loss at all frequencies accompanied by complete absence of endocochlear potential69, 70. Therefore it is clear that any intervention must be scheduled before irreversible damage to the organ of Corti, due to lack of connexin expression, takes place. Previously, we succeeded in restoring connexin expression and rescued intercellular coupling in vitro, by transducing cochlear organotypic cultures from P5 Cx26Sox10Cre or Cx30−/− mice with BAAV vectors encoding respectively Cx26 or Cx304, 5. Here, we used a BAAVCx30GFP vector4, which encodes GFP-tagged mouse Cx30 under the control of the CMV promoter, prepared in DMEM/F12, and injected it to P4 Cx30–/– mice via canalostomy. Auditory thresholds of injected mice, measured four weeks after surgery, remained super imposable to those of untreated Cx30–/– mice (p < 0.001, ANOVA) (Figure S2a).
The hearing loss phenotype exhibited by Cx30−/− mice depends on the cumulative effect of deletion of Cx30 and 3’ insertion of the lacZ and neo genes6, which are associated with dramatically reduced Cx26 levels4. Previous work showed restoration of hearing and prevention of hair cell death in Cx30−/− mice in which extra copies of the Cx26 gene were transgenically expressed from a modified bacterial artificial chromosome71. Therefore we injected Cx30–/– mice with a BAAV vector espressing Cx26CFP via canalostomy at P4, but also this treatment remained ineffective (Figure S2a). q–PCR analyses using primers specific for GFP tag to monitor transgene expression at 2, 3, 6, and 30 days after canalostomy, showed that transcript levels of Cx26CFP were maximal 2 days after gene delivery and thereafter decreased continuously. Thirty days after canalostomy, transgene expression was only 15.5 ± 4.5% (p < 0.005, ANOVA) of its initial peak (Figure S2b).
These negative outcomes might depend, at least in part, on disruption of the cytoarchitecture of the sensory epithelium and the stria vascularis in the Cx30−/− mouse model69, 70. Therefore, we decided to also test the Cx30Δ/Δ Cx30 knock out strain. Despite being ubiquitously deprived of Cx30, hearing is normal in Cx30Δ/Δ mice6 and their sensory epithelium is well preserved (Figure S3). We administered the BAAVCx30GFP vector prepared in DMEM/F12 to P4 Cx30Δ/Δ pups via canalostomy, and four weeks later we measured ABR thresholds. Compared to non-injected age-matched Cx30Δ/Δ controls, threshold values were comparable or even slightly lower in Cx30Δ/Δ mice injected with BAAVCx30GFP (Fig. 5). Therefore, we conclude that delivery of exogenous Cx30 gene via BAAV and canalostomy does not cause any hearing loss.

Hearing thresholds of adult Cx30Δ/Δ mice four weeks after delivery of BAAVCx30GFP viral vector at P4 and their respective age-matched non-injected controls. Error bars represent s.e.m.; differences are significant for 32 kHz (p = 0,022) and click responses (p = 0,043) (two tailed t-test).
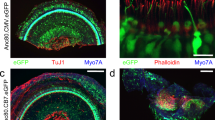
The cochleae of these injected mice were studied by immunofluorescence four weeks after vector delivery via canalostomy. In order to avoid cross-reactivity of the anti Cx30 antibody with the residual Cx26 expressed by Cx30Δ/Δ mice, we used an anti-GFP antibody that selectively targets the transgene product delivered by the BAAV vector. Confocal imaging showed extensive gap junction plaques formed by the Cx30GFP fusion protein at points of contacts between adjacent non-sensory cells of the sensory epithelium (Figs 6 and 7). Cx30GFP signals were also detected in the supra-strial zone (Fig. 7); we speculate that BAAV spread by transcytosis57 to accumulate in that zone traversing the lateral wall. Clearly, this viral fraction was lost as far as Cx30GFP expression in the critical sensory epithelium is concerned. Of note, all hair cells (both inner and outer) were preserved in this tissue after viral transduction and did not express Cx30GFP. These results indicate that recombinant Cx30GFP proteins traffic correctly to the plasma membrane of cochlear non-sensory cells not only in vitro 4 but also in vivo (Figs 6 and 7). However, Cx30GFP immunofluorescence signals in transduced cochlear epithelia were considerably weaker than the corresponding signals due to native Cx30 in age-matched non–injected wild type C57BL/6 N control mice (Figs 6 and 7). To address this issue, we quantified the viral genome copy number still present in the injected cochleae using primers specific for the CMV promoter. We found that the CMV signal was almost undetectable 6 days after transgene delivery via canalostomy (Fig. 8).

Cx30 expression in the organ of Corti of Cx30Δ/Δ mice four weeks after delivery of BAAVCx30GFP viral vector at P4. Shown are whole mount preparations from injected Cx30Δ/Δ mice (Cx30Δ/Δ + BAAVCx30GFP) and non-injected wild type age-matched controls (WT). The green signal was generated by immunostaining against GFP (Cx30Δ/Δ + BAAVCx30GFP) and native Cx30 (WT), respectively; red, actin filaments; blue, nuclei. BC: Böttcher cells CC: Claudius’ cells; DC: Deiters’ cells; OHC, outer hair cells. Scale bars: 50 μm.

Cx30 expression in cochlear outer sulcus and supra-strial zone of Cx30Δ/Δ mice four weeks after delivery of BAAVCx30GFP viral vector at P4. Shown are midmodiolar sections from injected Cx30Δ/Δ mice (Cx30Δ/Δ + BAAVCx30GFP) and non-injected wild type (WT) age-matched controls. Dashed arrows indicate gap junction plaques formed by recombinant Cx30GFP. The green signal was generated by immunostaining against GFP (Cx30Δ/Δ+ BAAVCx30GFP) and native Cx30 (WT), respectively. red, actin filaments; blue, nuclei. BC: Böttcher cells; CC: Claudius’ cells; DC: Deiters’ cells; RM: Reissners’ membrane; SP: spiral prominence; SSZ: supra-strial zone; SV: stria vascularis. Scale bars: 50 μm.

Quantification of BAAV genome following in vivo delivery at P4 via canalostomy to the mouse cochlea. CMV level was measured by q-PCR (see Methods) 2, 3 and 6 days after canalostomy and normalized to the level detected on day 2; error bars represent s.e.m.
Discussion
Viral transduction performed in adult Gjb2 conditional knock out mice failed to correct hearing function72, 73, whereas an early intervention in newborn mice produced limited Cx26 reinstatement and only partial rescue of hearing73. Here, we have assayed the potential of BAAV vectors encoding Cx30 to rescue protein expression in Cx30 KO mice in vivo by transduction at P4. To our knowledge, this is the first report of a clear and widespread expression of exogenous Cx30 (tagged with GFP) that persisted in cells of the cochlear sensory epithelium four weeks after virus delivery via canalostomy. However, Cx30GFP expression was visibly lower than native Cx30 in wild type controls. This is partly due to the spread of BAAV particles to type V fibrocytes present in the supra-strial zone74, probably via transcytosis57 from scala media through the later wall, reducing the therapeutic index of the vector dose. Furthermore, we noted a decrease in BAAV genome level following transduction, which was almost undetectable already 6 days after delivery via canalostomy at P4. In contrast, BAAV-mediated gene delivery via canalostomy in wild type mice injected at P25 achieved extensive expression of a reporter gene (β−actin−GFP) that was maintained at high levels for at least one month.
Recent work with AAV serotypes indicates that restoration of hearing is within reach of cochlear gene therapy53,54,55. However, targeting the post-natal 3500 or so inner hair cells, or even the three to four-fold more numerous outer hair cells, is facilitated by the fact that both types of postnatal sensory cells are post-mitotically differentiated75, 76 and numerically stable from P0 onward77,78,79,80. Achieving early, stable and widespread expression of exogenous connexins in the far more abundant, heterogeneous and developmentally evolving populations of non-sensory cells in the sensory epithelium and lateral wall of the post-natal cochlea is more problematic. To better appreciate the inherent difficulties, one should consider that both sensory and non-sensory cells of the cochlear sensory epithelium originate from postmitotic progenitor cells, which undergo terminal mitosis between E12 and E1475, 76. A single row of inner hair cells develop from the lateral margin of the greater epithelial ridge (GER), whereas three to four rows of outer hair cells, and lateral non–sensory cells, develop from the lesser epithelial ridge (LER)77,78,79,80. At birth (P0), differentiated hair cells are found from the basal to the apical cochlear turn and thereafter form a life-long stable population75,76,77,78,79,80, however postnatal development continues81. Programmed cell death82,83,84,85 (type-I, or apoptosis, as well as type II, or autophagic cell death) plays a crucial role in tissue remodeling, specification of cell fate and differentiation86,87,88. Recently, autophagy-related genes Becn1, Atg4g, Atg5 and Atg9 were shown to be expressed in the mouse cochlea from late embryonic developmental stages (E18.5) to adulthood, and up-regulated as the postnatal inner ear gains functional maturity89. Whereas cytological changes that occur in the LER have not been studied extensively75, the abundant non-sensory cells that populate the GER are replaced by the far fewer cuboidal cells of the inner sulcus during the first two postnatal weeks77,78,79,80. Cells positive for activated caspase-3, one of the cysteine proteases that play essential roles in programmed cell death, were detected in the GER between P7 and P13, and appeared progressively along the cochlear duct from base to apex90. The GER persisted throughout all turns of the cochlea in 2-week-old mice lacking caspase-3, resulting in hyperplasia of supporting cells, degeneration of hair cells, and severe hearing loss, suggesting that caspase-3-dependent apoptosis is necessary for the development and formation of a properly functioning auditory system in mammals91. Administration of thyroid hormone (T3) to wild type mice on P0 and P1 advanced the overall program of apoptosis and remodeling by about 4 days, suggesting initiation of apoptosis by a receptor-mediated process in conjunction with other unknown signals90, 92. Given the extensive remodeling and cell turn over, near 100% transduction of non-sensory cells will be required in therapeutic intervention for connexin related disorders.
Various laboratories reported different values for the time required to reach maximal transgene expression level after viral delivery to the developing inner ear72, 93, possibly reflecting differences in the transduction pathway of the different AAV serotypes used in those studies. Recombinant AAV vectors are well known to remain mostly episomal and be lost quickly even after one round of cell replication94.
In summary, using three different viral vectors in wild type and transgenic mice, we show here that BAAV works efficiently as a gene manipulation tool and permits to control gene expression in non-sensory cells of the inner ear, in vivo. BAAV viral transduction provides efficient in vivo gene delivery to cochlear non−sensory cells via canalostomy, however rescue of DFNB1 phenotype requires early and stable expression of connexins, which is not afforded by the current generation of these viral vectors. Our results suggest that the persistence of BAAV-mediated gene replacement in the cochlea is limited by the extensive non-sensory cell loss, which occurs in the organ of Corti throughout postnatal development88. Future work is needed to improve vector production and reformulation that will allow higher concentrations of the BAAV vectors to achieve the near 100% transduction necessary in this application in the critical postnatal period that precedes acquisition of hearing in mice.
Methods
Transgenic mice and genotyping
All experimental protocols were approved by the Ethical Committee of Padua University (Comitato Etico di Ateneo per la Sperimentazione Animale, C.E.A.S.A.) Project n.58/2013, Protocol n. 104230, date 10/12/2013) and by the Italian Ministry of Health (DGSAF 0001276-P-19/01/2016). The methods were carried out in accordance with the relevant guidelines and regulations.
Mice of either sex were used. Cx26loxP/loxP mice67 and Cx3Δ/Δ mice6 were maintained on a pure C57BL/6 N background. Cx26loxP/loxP mice were genotyped by screening for the presence of the loxP insertions on extracted mouse tail tips using the following primers:
Cx26F 5′-TTTCCAATGCTGGTGGAGTG-3′;
Cx26R 5′-ACAGAAATGTGTTGGTGATGG-3′.
A combination of three different primers was used to identify the Cx30 interruption due to LacZ insertion in Cx30−/− mice
Cx30 F: 5′-GGTACCTTCTACTAATTAGCTTGG-3′,
Cx30 R: 5′-AGGTGGTACCCATTGTAGAGGAAG-3′,
Cx30lacZ: 5′-AGCGAGTAACAACCCGTCGGATTC-3′.
Genotyping of the Cx30 fl allele was performed by PCR analysis using a primer binding in the Cx30 transcribed sequence and a primer binding upstream of the first loxP site
Gjb6R 5′-TTCCCTATGCTGGTAGAGTGCTTGT-3′;
Gjb6F 5′-GCAGTAACTTATTGAAACCCTTCACCT-3′.
Genotyping of the Cx30 Δ allele was performed using the Gjb6F and a primer binding downstream of the third loxP site
Gjb6ΔR: 5′-CCCACCATCAAGGTTGAACT-3′.
BAAV Production and Quantification
Hek293T cells, grown in DMEM/F12 supplemented with 5% FBS and 1% P/S, were transfected with three or four required plasmids (transgene vector, pAd12, and bovine adeno–associated virus (BAAV)–RepCap or AAV2–Rep plus BAAV-Cap). BAAVCx30GFP vector plasmids contained AAV-5 inverted terminal repeats (ITRs) whereas BAAVβ-actinGFP and BAAVCre-IRESGFP plasmids contained AAV–2 ITRs. Forty–eight hours after transduction, cells were harvested by scraping in TD buffer (140 mM NaCl, 5 mM KCl, 0.7 mM K2HPO4, 25 mM Tris-HCl pH 7.4) and the cell pellet was concentrated by low–speed centrifugation. Cells were lysed in TD buffer containing 0.5% deoxycholate and 100 U/ml DNase (Benzonase, Sigma) and incubated for 30 min at 37 °C. Following 10 min low speed centrifugation, viral particles were purified by CsCl gradient and dialyzed in DMEM/F12 (vehicle) with 10 K MWKO dialysis cassette (Pierce, Cat. No. 66383). Biological activity was confirmed on packaging cells95. Particle titers, determined by q-PCR, were in the range of 1012−1013 particles/ml. For viral titration, a dilution of the viral preparation was added to a q-PCR reaction mixture containing 1× SYBR Green Master Mix (Applied Biosystems/Applera, Milan, Italy) and 0.25 pmol/μl forward and reverse primers. Amplification was measured using a sequence detector (ABI 7700, Applied Biosystems). Specific primers for CMV were designed with the Primer Express program (Applied Biosystems):
CMV f 5′-CATCTACGTATTAGTCATCGCTATTACCAT-3′,
CMV r 5′–TGGAAATCCCCGTGAGTCA-3′.
Following denaturation at 96 °C for 10 min, cycling conditions were 96 °C for 15 s, 60 °C for 1 min for 40 cycles. The absolute quantification of the viral DNA in each sample was determined by generating a dilution series of a standard DNA.
Canalostomy
For canalostomy, as well as all other surgical procedures described in this article, mouse body temperature was kept at 38 °C by a feedback-controlled heating pad. P25 mice were anaesthetized with an intraperitoneal injection of zolazepam (25 mg/g) and xylazine (10 mg/g) whereas P4 pups were anaesthetized with xylazine 0.45 µg/g and zolazepam 0.15 µg/g diluted in physiological solution. Supplemental doses were administered as needed. After induction of anesthesia, mice were placed under a dissection microscope and the posterior (P25) or lateral (P4) semicircular canal was exposed by a dorsal post-auricular approach. For P25 mice, a hole was made with the tip of a 33 G needle, softly removing a part of the bony shell of the canal. Bromophenol-blue (SIGMA, product number B−5525), Alexa594® conjugated wheat germ agglutinin (10 μg/ml; ThermoFisher, catalogue # W11262) dissolved in phosphate buffered saline (PBS) or viral solution diluted in DMEM/F12 (vehicle; Gibco®, catalogue # 11320074) were injected into the endolymphatic space (2.5 μl injected volume, 3 nl/s injection speed) with a micropump–controlled micro syringe (WPI, art.no. NANOFIL-100) equipped with a 36 G needle (WPI, art.no. NF36BV-2). For every transduction experiment, we injected ~109 viral particles. To avoid fluid leakage during injection, the needle inserted in the semicircular canal of P25 mice was sealed with a drop of dental cement (Temrex Interface Light Cured Cavity Liner and Base, Product n. 7100), which was rapidly cured with blue light from a LED source (Mini Led, Satelec, F02641). Ten minutes after injection, the needle was slowly removed and the hole was closed with dental cement. To test the efficacy of delivery (Fig. 1), animals were sacrificed 10 minutes after injection and the excised cochlea was examined by light and confocal microscopy.
In vivo electrophysiology
Endochlear potential was measured 30 days after surgery both in injected and contralateral ear. Mice were anaesthetized with 0.01 ml/g body weight of 20% urethane (SIGMA, product number 94300) and the potential difference between the scala media and a reference silver/silver chloride pellet under the dorsal skin was recorded96.
To record ABRs, mice were anaesthetized with an intraperitoneal injection of zolazepam (25 mg/g) and xylazine (10 mg/g) and submitted to clicks and tone pips at 8, 14, 20, 26, 32 kHz62.
q-PCR
Total RNA was extracted from freshly dissected whole cochleae with RNeasy mini kit (Qiagen, Cat. No. 74106) and retrotranscripted with oligo–dT12–18 primers (ThermoFisher, catalogue # 18418012) using Omniscript RT kit (Qiagen, Cat. No. 205111). Total DNA was extracted from whole cochleae using GenElute Mammalian Genomic DNA miniprep kit (Sigma cat. # G1N10). q-PCR was performed on cDNA or DNA to amplify Cx26 and/or GFP and was normalized to GAPDH. Gene expression relative to GAPDH was estimated according to the method described by Pfaffl97. Amplification was carried out using Power SYBR Green (Applied Biosystems, Cat. No. 4367659) on the ABI 7700 sequence detection system equipped with ABI Prism 7700 SDS software (Applied Biosystems) through the following amplification cycles: 50 °C: 2′, 95 °C: 10′, 95 °C: 15′, 60 °C: 1′ (40 cycles). Primers used are as follows:
CMV f 5′-CATCTACGTATTAGTCATCGCTATTACCAT–3′,
CMV r 5′-TGGAAATCCCCGTGAGTCA-3′.
mCx26 f: 5′-TCACAGAGCTGTGCTATTTG-3′
mCx26r: 5′-ACTGGTCTTTTGGACTTTCC-3′
GFPf: 5′-TCCGCCATGCCCGAAGGCTA-3′
GFPf: 5′-CGGTTCACCAGGGTGTCGCC-3′
mGAPDHf: 5′-ATGTGTCCGTCGTGGATCTGAC-3′
mGAPDHr: 5′-AGACAACCTGGTCCTCAGTGTAG-3′
hGAPDHf: TGCACCACCAACTGCTTAGC
hGAPDHr: GGCATGGACTGTGGTCATGAG
Immunohistochemistry and confocal imaging
Mice injected at P25 or P4 were sacrificed four weeks after injection. Cochleae were extracted and processed for immunohistochemistry as previously described5. Briefly, samples were fixed in 4% paraformaldehyde and decalcified in ethylenediaminetetraacetic acid (EDTA, 0.3 M). Specimens were included in 3% agarose dissolved in PBS and cut in 100 µm thickness steps using a vibratome (VT 1000 S, Leica). Tissue slices were permeabilized with 0.1% Triton X–100, dissolved in bovine serum albumin 2% solution. Cx30 and Cx26 were stained respectively with a rabbit polyclonal Cx30 and Cx26 selective antibodies (10 μg/ml, ThermoFisher, catalogue # 712200 for Cx30 and catalogue # 512800 for Cx26). A rabbit polyclonal GFP selective antibody (10 μg/ml, ThermoFisher, catalogue # A11122) was used to distinguish the exogenous fusion proteins from endogenous proteins. Secondary antibodies (10 μg/ml, Alexa Fluor® 488 goat anti-rabbit IgG, ThermoFisher, catalogue # A11008) were applied overnight at room temperature whilst F–Actin was stained by incubation with AlexaFluor 568 phalloidin (1U/ml, ThermoFisher, Cat. No. A12380) and nuclei were stained with 4′,6–diamidino–2–phenylindole (DAPI, ThermoFisher, Cat. No. D1306) (1:200). Samples were mounted onto glass slides with a mounting medium (FluorSaveTM Reagent, Merk, Cat. No. 345789) and analyzed using a confocal microscope (TCS SP5, Leica) equipped with an oil–immersion objective (40× HCX PL APO 1.25 N.A., Leica).
References
Smith, R. J. H. & Jones, M. K. N. in GeneReviews® [Internet]. Available from https://www.ncbi.nlm.nih.gov/books/NBK1272/ (eds R. A. Pagon et al.) (1998).
del Castillo, F. J. & del Castillo, I. The DFNB1 subtype of autosomal recessive non-syndromic hearing impairment. Frontiers in bioscience: a journal and virtual library 16, 3252–3274 (2011).
Zonta, F., Polles, G., Zanotti, G. & Mammano, F. Permeation pathway of homomeric connexin 26 and connexin 30 channels investigated by molecular dynamics. Journal of biomolecular structure & dynamics 29, 985–998, doi:10.1080/073911012010525027 (2012).
Ortolano, S. et al. Coordinated control of connexin 26 and connexin 30 at the regulatory and functional level in the inner ear. Proceedings of the National Academy of Sciences of the United States of America 105, 18776–18781, doi:10.1073/pnas.0800831105 (2008).
Crispino, G. et al. BAAV mediated GJB2 gene transfer restores gap junction coupling in cochlear organotypic cultures from deaf Cx26Sox10Cre mice. PloS one 6, e23279, doi:10.1371/journal.pone.0023279 (2011).
Boulay, A. et al. Hearing is normal without connexin30. The Journal of neuroscience: the official journal of the Society for Neuroscience 33, 430–434, doi:10.1523/JNEUROSCI.4240-12.2013 (2013).
Lautermann, J. et al. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell and tissue research 294, 415–420 (1998).
Lautermann, J., Frank, H. G., Jahnke, K., Traub, O. & Winterhager, E. Developmental expression patterns of connexin26 and -30 in the rat cochlea. Developmental genetics 25, 306–311 (1999).
Forge, A., Marziano, N. K., Casalotti, S. O., Becker, D. L. & Jagger, D. The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell communication & adhesion 10, 341–346 (2003).
Ceriani, F. & Mammano, F. A rapid and sensitive assay of intercellular coupling by voltage imaging of gap junction networks. Cell communication and signaling: CCS 11, 78, doi:10.1186/1478-811X-11-78 (2013).
Thomas, T. et al. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. Journal of cell science 118, 4451–4462, doi:10.1242/jcs.02569 (2005).
Esseltine, J. L. & Laird, D. W. N.-G. Connexin and Pannexin Cell Biology. Trends in cell biology. doi:10.1016/j.tcb.2016.06.003 (2016).
Fettiplace, R. & Kim, K. X. The physiology of mechanoelectrical transduction channels in hearing. Physiological reviews 94, 951–986, doi:10.1152/physrev.00038.2013 (2014).
Cody, A. R. & Russell, I. J. The response of hair cells in the basal turn of the guinea-pig cochlea to tones. The Journal of physiology 383, 551–569 (1987).
Nin, F. et al. The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proceedings of the National Academy of Sciences of the United States of America 105, 1751–1756, doi:10.1073/pnas.0711463105 (2008).
Patuzzi, R. Ion flow in stria vascularis and the production and regulation of cochlear endolymph and the endolymphatic potential. Hearing research 277, 4–19, doi:10.1016/j.heares.2011.01.010 (2011).
Yamasaki, M., Komune, S., Shimozono, M., Matsuda, K. & Haruta, A. Development of monovalent ions in the endolymph in mouse cochlea. ORL; journal for oto-rhino-laryngology and its related specialties 62, 241–246, doi:27753 (2000).
Wangemann, P., Liu, J. & Marcus, D. C. Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hearing research 84, 19–29 (1995).
Adachi, N. et al. The mechanism underlying maintenance of the endocochlear potential by the K+ transport system in fibrocytes of the inner ear. The Journal of physiology 591, 4459–4472, doi:10.1113/jphysiol.2013.258046 (2013).
Beurg, M., Goldring, A. C., Ricci, A. J. & Fettiplace, R. Development and localization of reverse-polarity mechanotransducer channels in cochlear hair cells. Proceedings of the National Academy of Sciences of the United States of America 113, 6767–6772, doi:10.1073/pnas.1601067113 (2016).
Wu, Z. et al. Mechanosensory hair cells express two molecularly distinct mechanotransduction channels. Nature neuroscience 20, 24–33, doi:10.1038/nn.4449 (2017).
Mistrik, P., Mullaley, C., Mammano, F. & Ashmore, J. Three-dimensional current flow in a large-scale model of the cochlea and the mechanism of amplification of sound. Journal of the Royal Society, Interface / the Royal Society 6, 279–291, doi:10.1098/rsif.2008.0201 (2009).
Forge, A. et al. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. The Journal of comparative neurology 467, 207–231, doi:10.1002/cne.10916 (2003).
Jagger, D. J. & Forge, A. Compartmentalized and signal-selective gap junctional coupling in the hearing cochlea. The Journal of neuroscience: the official journal of the Society for Neuroscience 26, 1260–1268 (2006).
Majumder, P. et al. ATP-mediated cell-cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signal 6, 167–187, doi:10.1007/s11302-010-9192-9 (2010).
Housley, G. D., Marcotti, W., Navaratnam, D. & Yamoah, E. N. Hair cells–beyond the transducer. The Journal of membrane biology 209, 89–118, doi:10.1007/s00232-005-0835-7 (2006).
Wichmann, C. & Moser, T. Relating structure and function of inner hair cell ribbon synapses. Cell and tissue research 361, 95–114, doi:10.1007/s00441-014-2102-7 (2015).
Fuchs, P. A. Time and intensity coding at the hair cell’s ribbon synapse. The Journal of physiology 566, 7–12 (2005).
Zheng, J. et al. Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–155, doi:10.1038/35012009 (2000).
Liberman, M. C. et al. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419, 300–304 (2002).
Johnson, S. L., Beurg, M., Marcotti, W. & Fettiplace, R. Prestin-driven cochlear amplification is not limited by the outer hair cell membrane time constant. Neuron 70, 1143–1154, doi:10.1016/j.neuron.2011.04.024 (2011).
Gorbunov, D. et al. Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nature communications 5, 3622, doi:10.1038/ncomms4622 (2014).
Mammano, F. & Ashmore, J. F. Reverse transduction measured in the isolated cochlea by laser Michelson interferometry. Nature 365, 838–841 (1993).
Robles, L. & Ruggero, M. A. Mechanics of the mammalian cochlea. Physiological reviews 81, 1305–1352 (2001).
Nobili, R., Mammano, F. & Ashmore, J. How well do we understand the cochlea? Trends Neurosci 21, 159–167 (1998).
Ashmore, J. Cochlear outer hair cell motility. Physiological reviews 88, 173–210 (2008).
Giacca, M. & Zacchigna, S. Virus-mediated gene delivery for human gene therapy. J Control Release 161, 377–388, doi:10.1016/j.jconrel.2012.04.008 (2012).
Bakondi, B. et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Molecular therapy: the journal of the American Society of Gene Therapy 24, 556–563, doi:10.1038/mt.2015.220 (2016).
Luebke, A. E., Rova, C., Von Doersten, P. G. & Poulsen, D. J. Adenoviral and AAV-mediated gene transfer to the inner ear: role of serotype, promoter, and viral load on in vivo and in vitro infection efficiencies. Advances in oto-rhino-laryngology 66, 87–98, doi:10.1159/000218209 (2009).
Moser, T. Gene therapy for deafness: How close are we? Science translational medicine 7, 295fs228, doi:10.1126/scitranslmed.aac7545 (2015).
Kohrman, D. C. & Raphael, Y. Gene therapy for deafness. Gene therapy 20, 1119–1123, doi:10.1038/gt.2013.39 (2013).
Kawamoto, K., Oh, S. H., Kanzaki, S., Brown, N. & Raphael, Y. The functional and structural outcome of inner ear gene transfer via the vestibular and cochlear fluids in mice. Molecular therapy: the journal of the American Society of Gene Therapy 4, 575–585 (2001).
Praetorius, M. et al. A novel vestibular approach for gene transfer into the inner ear. Audiology & neuro-otology 7, 324–334 (2002).
Iizuka, T. et al. Noninvasive in vivo delivery of transgene via adeno-associated virus into supporting cells of the neonatal mouse cochlea. Human gene therapy 19, 384–390, doi:10.1089/hum.2007.167 (2008).
Xia, L., Yin, S. & Wang, J. Inner ear gene transfection in neonatal mice using adeno-associated viral vector: a comparison of two approaches. PloS one 7, e43218, doi:10.1371/journal.pone.0043218 (2012).
Fukui, H. & Raphael, Y. Gene therapy for the inner ear. Hearing research 297, 99–105, doi:10.1016/j.heares.2012.11.017 (2013).
Chien, W. W., McDougald, D. S., Roy, S., Fitzgerald, T. S. & Cunningham, L. L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. The Laryngoscope 125, 2557–2564, doi:10.1002/lary.25317 (2015).
Bedrosian, J. C. et al. In vivo delivery of recombinant viruses to the fetal murine cochlea: transduction characteristics and long-term effects on auditory function. Molecular therapy: the journal of the American Society of Gene Therapy 14, 328–335 (2006).
Sheffield, A. M. et al. Viral vector tropism for supporting cells in the developing murine cochlea. Hearing research (2011).
Konishi, M., Kawamoto, K., Izumikawa, M., Kuriyama, H. & Yamashita, T. Gene transfer into guinea pig cochlea using adeno-associated virus vectors. The journal of gene medicine 10, 610–618, doi:10.1002/jgm.1189 (2008).
Wang, H. et al. Efficient cochlear gene transfection in guinea-pigs with adeno-associated viral vectors by partial digestion of round window membrane. Gene therapy 19, 255–263, doi:10.1038/gt.2011.91 (2012).
Budenz, C. L. et al. Differential effects of AAV.BDNF and AAV.Ntf3 in the deafened adult guinea pig ear. Scientific reports 5, 8619, doi:10.1038/srep08619 (2015).
Akil, O. et al. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally Mediated Gene Therapy. Neuron 75, 283–293, doi:10.1016/j.neuron.2012.05.019 (2012).
Askew, C. et al. Tmc gene therapy restores auditory function in deaf mice. Science translational medicine 7, 295ra108, doi:10.1126/scitranslmed.aab1996 (2015).
Pan, B. et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nature biotechnology 35, 264–272, doi:10.1038/nbt.3801 (2017).
Di Pasquale, G. et al. A novel bovine virus efficiently transduces inner ear neuroepithelial cells. Molecular therapy: the journal of the American Society of Gene Therapy 11, 849–855 (2005).
Di Pasquale, G. & Chiorini, J. A. AAV transcytosis through barrier epithelia and endothelium. Molecular therapy: the journal of the American Society of Gene Therapy 13, 506–516 (2006).
Shibata, S. B., Di Pasquale, G., Cortez, S. R., Chiorini, J. A. & Raphael, Y. Gene transfer using bovine adeno-associated virus in the guinea pig cochlea. Gene therapy 16, 990–997 (2009).
Praetorius, M., Baker, K., Weich, C. M., Plinkert, P. K. & Staecker, H. Hearing preservation after inner ear gene therapy: the effect of vector and surgical approach. ORL; journal for oto-rhino-laryngology and its related specialties 65, 211-214, doi:73117 (2003).
Okada, H. et al. Gene transfer targeting mouse vestibule using adenovirus and adeno-associated virus vectors. Otology & neurotology: official publication of the American Otological Society, American Neurotology Society [and] European Academy of Otology and Neurotology 33, 655–659, doi:10.1097/MAO.0b013e31825368d1 (2012).
Gassner, D., Durham, D., Pfannenstiel, S. C., Brough, D. E. & Staecker, H. Canalostomy as a surgical approach for cochlear gene therapy in the rat. Anatomical record 295, 1830–1836, doi:10.1002/ar.22593 (2012).
Scimemi, P., Santarelli, R., Selmo, A. & Mammano, F. Auditory brainstem responses to clicks and tone bursts in C57 BL/6J mice. Acta otorhinolaryngologica Italica: organo ufficiale della Societa italiana di otorinolaringologia e chirurgia cervico-facciale 34, 264–271 (2014).
Johnson, S. L. et al. Connexin-Mediated Signaling in Nonsensory Cells Is Crucial for the Development of Sensory Inner Hair Cells in the Mouse Cochlea. The Journal of neuroscience: the official journal of the Society for Neuroscience 37, 258–268, doi:10.1523/JNEUROSCI.2251-16.2017 (2017).
Gabriel, H. D. et al. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. The Journal of cell biology 140, 1453–1461 (1998).
Sauer, B. & Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proceedings of the National Academy of Sciences of the United States of America 85, 5166–5170 (1988).
Orban, P. C., Chui, D. & Marth, J. D. Tissue- and site-specific DNA recombination in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 89, 6861–6865 (1992).
Cohen-Salmon, M. et al. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Current biology: CB 12, 1106–1111 (2002).
Ehret, G. Development of absolute auditory thresholds in the house mouse (Mus musculus). Journal of the American Audiology Society 1, 179–184 (1976).
Cohen-Salmon, M. et al. Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proceedings of the National Academy of Sciences of the United States of America 104, 6229–6234 (2007).
Teubner, B. et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Human molecular genetics 12, 13–21 (2003).
Ahmad, S. et al. Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proceedings of the National Academy of Sciences of the United States of America 104, 1337–1341 (2007).
Yu, Q. et al. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene therapy 21, 71–80, doi:10.1038/gt.2013.59 (2014).
Iizuka, T. et al. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Human molecular genetics 24, 3651–3661, doi:10.1093/hmg/ddv109 (2015).
Hibino, H. & Kurachi, Y. Molecular and physiological bases of the K+ circulation in the mammalian inner ear. Physiology 21, 336–345 (2006).
Kelley, M. W. Cellular commitment and differentiation in the organ of Corti. The International journal of developmental biology 51, 571–583, doi:10.1387/ijdb.072388mk (2007).
Schimmang, T. & Pirvola, U. Coupling the cell cycle to development and regeneration of the inner ear. Seminars in cell & developmental biology 24, 507–513, doi:10.1016/j.semcdb.2013.04.004 (2013).
Lim, D. J. & Anniko, M. Developmental morphology of the mouse inner ear. A scanning electron microscopic observation. Acta oto-laryngologica. Supplementum 422, 1–69 (1985).
Lim, D. & Rueda, J. in Development of auditory and vestibular systems - 2 (1st edition) (ed R. Romand) 33-58. (Elsevier Science Publishing Co., 1992).
Roth, B. & Bruns, V. Postnatal development of the rat organ of Corti. II. Hair cell receptors and their supporting elements. Anatomy and embryology 185, 571–581 (1992).
Hinojosa, R. A note on development of Corti’s organ. Acta Otolaryngol 84, 238–251 (1977).
Walters, B. J. & Zuo, J. Postnatal development, maturation and aging in the mouse cochlea and their effects on hair cell regeneration. Hearing research 297, 68–83, doi:10.1016/j.heares.2012.11.009 (2013).
Nishikori, T., Hatta, T., Kawauchi, H. & Otani, H. Apoptosis during inner ear development in human and mouse embryos: an analysis by computer-assisted three-dimensional reconstruction. Anatomy and embryology 200, 19–26 (1999).
Nikolic, P., Jarlebark, L. E., Billett, T. E. & Thorne, P. R. Apoptosis in the developing rat cochlea and its related structures. Brain research. Developmental brain research 119, 75–83 (2000).
Kamiya, K., Takahashi, K., Kitamura, K., Momoi, T. & Yoshikawa, Y. Mitosis and apoptosis in postnatal auditory system of the C3H/He strain. Brain research 901, 296–302 (2001).
Aburto, M. R., Sanchez-Calderon, H., Hurle, J. M., Varela-Nieto, I. & Magarinos, M. Early otic development depends on autophagy for apoptotic cell clearance and neural differentiation. Cell Death Dis 3, e394, doi:10.1038/cddis.2012.132 (2012).
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758, doi:10.1016/j.cell.2011.10.033 (2011).
Mizushima, N. & Levine, B. Autophagy in mammalian development and differentiation. Nature cell biology 12, 823–830, doi:10.1038/ncb0910-823 (2010).
Mammano, F. & Bortolozzi, M. Ca2+ signaling, apoptosis and autophagy in the developing cochlea: milestones to hearing acquisition. Cell calcium. doi:10.1016/j.ceca.2017.05.006 (2017).
de Iriarte Rodriguez, R., Pulido, S. Rodriguez-de la Rosa, L., Magarinos, M. & Varela-Nieto, I. Age-regulated function of autophagy in the mouse inner ear. Hearing research 330, 39–50, doi:10.1016/j.heares.2015.07.020 (2015).
Peeters, R. P., Ng, L., Ma, M. & Forrest, D. The timecourse of apoptotic cell death during postnatal remodeling of the mouse cochlea and its premature onset by triiodothyronine (T3). Molecular and cellular endocrinology 407, 1–8, doi:10.1016/j.mce.2015.02.025 (2015).
Takahashi, K. et al. Caspase-3-deficiency induces hyperplasia of supporting cells and degeneration of sensory cells resulting in the hearing loss. Brain research 894, 359–367 (2001).
Knipper, M. et al. Distinct thyroid hormone-dependent expression of TrKB and p75NGFR in nonneuronal cells during the critical TH-dependent period of the cochlea. Journal of neurobiology 38, 338–356 (1999).
Wang, Y. et al. Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. The journal of gene medicine 15, 123–133, doi:10.1002/jgm.2701 (2013).
Nakai, H., Storm, T. A. & Kay, M. A. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver in vivo. Journal of virology 74, 9451–9463 (2000).
Kaludov, N., Handelman, B. & Chiorini, J. A. Scalable purification of adeno-associated virus type 2, 4, or 5 using ion-exchange chromatography. Human gene therapy 13, 1235–1243 (2002).
Steel, K. P. & Barkway, C. Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development 107, 453–463 (1989).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic acids research 29, e45 (2001).
Acknowledgements
This work was supported by Fondazione Telethon (grant GGP13114 to F.M.) and the CNR (Project DSB.AD009.001.004 / INVECCHIAMENTO IBCN to F.M).
Author information
Authors and Affiliations
Contributions
G.C. performed B.A.A.V. production, in vivo canalostomy, immunofluorescence, q-PCR, statistical analysis, participated in the study design and drafting of the manuscript. F.G.R. performed in vivo canalostomy, A.B.R. and E.P. recordings, and statistical analysis. M.C. performed B.A.A.V. production. V.Z. superintended mouse colony maintenance, performed in vivo canalostomy, ABR recordings and statistical analysis. M.P. performed in vivo canalostomy. G.D.P. and J.A.C. provided BAAV constructs. J.A.C. and F.M. conceived of the study, participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crispino, G., Galindo Ramirez, F., Campioni, M. et al. In vivo genetic manipulation of inner ear connexin expression by bovine adeno-associated viral vectors. Sci Rep 7, 6567 (2017). https://doi.org/10.1038/s41598-017-06759-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06759-y
This article is cited by
-
Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome
Gene Therapy (2022)
-
Gene Therapy for Congenital Hearing Loss
Current Otorhinolaryngology Reports (2022)
-
Gene therapy via canalostomy approach preserves auditory and vestibular functions in a mouse model of Jervell and Lange-Nielsen syndrome type 2
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
