
Abstract
A nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) (BHTH) ceramic with a single-phase tetragonal structure was prepared at 1400 °C using the solid-state reaction method. The analysis on the defect chemistry revealed that the real formula of BHTH is (Ba1−x Ho3x/4)(Ti1−x/4Ho x/4)O3 with Ba vacancies via electron paramagnetic resonance (EPR). Photoluminescence (PL) was investigated on the basis of excitation with different wavelength lasers. The results indicated that under 488-nm excitation, PL and Raman scattering can occur simultaneously as two distinct optical processes for BHTH ceramic powders, and the strongest PL band at 564 nm was discovered and verified to originate from the 5G6/5F1 → 5I7 transition of Ho3+ ions on the Ti sites in the BaTiO3 lattice. Upon 532- and 638-nm excitations, three PL bands of 5F4/5S2 → 5I8, 5F5 → 5I8, and 5F4/5S2 → 5I7 transitions are attributed to the contributions from Ho3+ ions on the Ba sites. The common Raman spectrum of BaTiO3 can be observed without PL disturbance using 785-nm excitation wavelength. The PL effect may provide a probe for the site occupations of Ho3+ ions in widely-used BaTiO3 dielectric ceramics co-doped with Ho3+ and other dopants.
Similar content being viewed by others

Introduction
Ho3+, as a co-dopant with other dopants (Er3+, Tm3+, Yb3+, Dy3+) in compounds, is widely used in upconversion luminescence in the filed of luminescence1,2,3,4,5,6 and in multilayer ceramic capacitors (MLCC) in the filed of dielectrics7, 8. Barium titanate (BaTiO3) is a typical dielectric material. The dielectric properties and structural information of Ho-doped BaTiO3 were systematically investigated in terms of structural modification9,10,11,12,13,14,15. Ho3+ is known to be an amphoteric dopant in BaTiO3 polycrystalline ceramic and its solid solubility is dependent on the Ba/Ti ratio and sintering temperature (T s). In the Ti-rich case, Ho3+ was considered to to occupy the Ba site to induce Ti vacancies and the solid solubility limit was relatively small, only x = ~1.4% at T s = 1400 °C; the formula was described as (Ba1−x Ho x )Ti1−x/4O3 10. Higher T s is generally required for the incorporation of Ho3+ into the Ti site because of a relatively larger ionic size of Ho3+ compared to Ti4+ (Table 1)16 and TiO6 octahedrons characteristic of the skeleton of the perovskite structure. In the Ba-rich case, the solubility limit was reported to be x = ~15% at T s = 1550 °C and the formula was described as Ba(Ti1−x Ho x )O3−x/2 with O vacancies10, 14, 15. However, Ti vacancies and O ones had not been confirmed in Ho-doped BaTiO3 and one is hard to avoid questioning a single site-occupation mode due to the amphoteric nature of Ho3+.
On the other hand, the structural information of BaTiO3 doped with most rare earths such as La17,18,19,20, Ce21, 22, Pr23, Nd24, Eu25, Tb26, Dy27, Er28, Yb29, 30, has been reported via laser Raman spectroscopy to date. However, no Raman information is known about Ho-doped BaTiO3. Because Ho usually acts as a well luminescent center in different host lattices31,32,33,34, the lack of Raman information about Ho-doped BaTiO3 is probably because the preference of strong photoluminescence (PL) caused by Ho3+ affects observation of Raman spectra, similar to Er3+ 28. As far as we know, only Battisha35 and Secu et al.36 reported the up-conversion luminescence (under 808-nm excitation) and PL (under 455-nm excitation) of Ho-doped BaTiO3. Some luminescence bands at 435 (5F1 → 5I8), 545 (5F4,5S2 → 5I8), 660 (5F5 → 5I8), and 760 nm (5F4,5S2 → 5I7) were observed. Unfortunately, XRD data show that their Ho-doped BaTiO3 samples prepared at T s = 750 and 1350 °C are not single-phase, accompanied by a secondary phase such as Ho2Ti2O7 35, 36. For this reason, the PL origin associated with the site occupations of Ho3+ ions in BaTiO3 is still an unresolved scientific problem. Moreover, a distinct exhibition between PL and Raman scattering needs to be also clarified.
In this work, a nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) ceramic was prepared at 1400 °C using the solid-state reaction method. The defect chemistry is discussed. On the basis of excitation with different wavelength lasers (488‒785 nm), photoluminescence and Raman scattering can occur simultaneously as two distinct optical processes for BHTH ceramic powders. The reasonable laser wavelength is indicated for observation for the common Raman phonon modes of Ho-doped BaTiO3. A rare non-ground-state transition corresponding to 5G6/5F1 → 5I7 of Ho3+ was discovered and associated with the site occupations of Ho3+ ions in BaTiO3, which may provide a probe for the site occupations of Ho3+ ions in BaTiO3-based dielectric ceramics.
Experimental
The initial materials were analytical-reagent chemicals of BaCO3, TiO2, and Ho2O3 (99.9%). Ho-doped BaTiO3 ceramics were prepared using the solid-state reaction method according to the nominal formula (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) named as BHTH. The weighed powders were dry-mixed and ground. Then, the powders were calcined at 1100 °C for 5 h to decarbonate. The calcined powders were mixed with a small amount of PVA aqueous solution and pressed at a pressure of 200 MPa into pellets (Φ12 mm). These pellets were sintered at T s = 1400 °C for 12 h, and cooled at a cooling rate of −200 °C/h from 1400 °C to 700 °C and then furnace-cooled to room temperature. The pellets were densified into crack-free ceramics. In addition, a nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) (BH1T) ceramic was prepared at T s = 1300 °C for 12 h for analysis on site-occupation-related photoluminescence of Ho3+ ions in the BaTiO3 lattice.
Powder X-ray diffraction (XRD) data were recorded from 20° to 85° and in steps of 0.02° using a DX-2700 X-ray diffractometer (Dandong Haoyuan) at room temperature. Lattice parameters were calculated with MS Modeling (Accelry Inc.) using Cu Kα1 radiation (λ = 1.540562 Å). Microstructure was observed using an EVOMA 10 scanning electron microscope (Zeiss) operated at 15 keV. Photoluminescence (PL) and Raman spectra of the ceramic powders were recorded using a LabRAM XploRA Raman spectrometer (Horiba Jobin Yvon) with 532- (green) and 638-nm (red) lasers and an inVia Raman spectrometer (Renishaw) equipped with 488- (blue-purple) and 785-nm (red) lasers. The laser power level was adjusted to 0.1–10% (Filter) of the normal output of 25 mW because of a huge difference in spectral intensity. The accumulation time and resolution are 2 s and 2.7 cm−1, respectively. Electron paramagnetic resonance (EPR) measurements were performed at room temperature using an EMX Plus X-band spectrometer (Bruker) operating at 9.84 GHz. The gyromagnetic factor (g) was calculated by the relationship hν 0 = gβH, where h is the Planck constant (h = 6.626 × 10−34 J·s), ν 0 is the microwave frequency, β is the Bohr magnetron (β = 9.262 × 10−24 J/T), H is the magnetic field strength.
Results and Discussion
Crystal structure, site occupation and microstructure
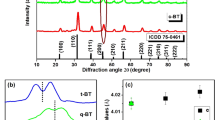
Figure 1a shows the XRD pattern of (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) ceramic (BHTH) prepared at T s = 1400 °C. BHTH has a single-phase tetragonal perovskite structure, indicating that Ho3+ ions are incorporated sufficiently into the perovskite lattice. This result is in accord with the solubility limit of x = 0.014 for (Ba1−x Ho x )Ti1−x/4O3 at T s = 1400 °C reported by Makovec et al.10.

(a) XRD pattern, (b) EPR spectrum, SEM images on (c) the surface and (d) the interior for the nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) ceramic (BHTH) sintered at a sintering temperature of T s = 1400 °C. The real formula of BHTH is expressed by (Ba1−x Ho3x/4)(Ti1−x/4Ho x/4)O3 with Ba vacancies (VBa).
The lattice parameters (a, c) and unit cell volume (V 0) is shown in Fig. 1a. It is inferred that the expansion in V 0 caused by the occupations of the Ti4+ sites by Ho3+ ions should be greater than the contraction in V 0 caused by the occupations of the Ba2+ sites by Ho3+ ions on the basis of the BO6 octahedrons skeleton characteristic of the perovskite lattice and ionic size comparisons between 12-CN Ba2+ and Ho3+ and between 6-CN Ti4+ and Ho3+ (Table 1). The V 0 of BHTH (64.44 Å3) is slightly greater than that of the tetragonal BaTiO3 (V 0-BT = 64.41 Å3) (JCPDS Cards No. 6–526), implying that Ho3+ ions enter the Ti sites in part except for some Ho3+ ions on the Ba sites.
Figure 1b shows the EPR spectrum of BHTH. Ho3+ (4f 10) is a non-Kramers ion, which is EPR silent in theory. A very strong EPR signal at g = 1.974 appears in BHTH and was observed in Dy- and Er-doped BaTiO3 27, 28. This signal is assigned to ionized Ba-vacancy defects (VBa)27, 28, 37, 38. A g = 2.004 signal associated with Ti vacancies is absent for BHTH, revealing that Ti-vacancy defects cannot be formed in the nominal (Ba1−x Ho x )Ti1−x/4O3 (BHTH) with deliberately designed Ti vacancies; Ho3+ ions transfer from the Ba sites to the Ti ones and results in appearance of Ba vacancies in BHTH. This result is in good agreement with the previous XRD result.
Figure 1c and d shows the SEM images on the surface and the interior of BHTH. The surface of the ceramic sintered exhibits an inhomogeneous microstructure (Fig. 1c) with grains (2‒20 μm), but the interior in the ceramic shows a fine-grained feature (2‒6 μm) (Fig. 1d). This illustrates that the grain growth on the surface is faster than the interior.
Defect chemistry
For the nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) ceramic (BHTH) sintered at T s = 1400 °C, the above XRD result gives evidence of a single-phase perovskite ceramic and the partial occupations of the Ti sites by Ho3+ ions (Fig. 1a); the EPR result reveals that Ti vacancies in BHTH are completely filled by Ho3+ and the transfer of Ho3+ ions from the Ba sites to the Ti ones gives rise to the presence of Ba vacancies (Fig. 1b). It is obvious that the formula cannot be described as (Ba1−x Ho x )Ti1−x/4O3 with Ti vacancies suggested by Makovec et al.10. Ho3+ is known to show an amphoteric behavior, substituting for both Ba and Ti sites in BaTiO3 10,11,12. Thus, the defects in BHTH are Ba-site Ho3+ (\({{\rm{Ho}}}_{{\rm{Ba}}}^{\bullet }\)), Ba vacancies (\({{\rm{V}}}_{{\rm{Ba}}}^{^{\prime\prime} }\)), and Ti-site Ho3+ (\({{\rm{Ho}}}_{{\rm{Ti}}}^{^{\prime} }\)). The defect notation adopts that suggested by Kröger and Vink39. The lattice electroneutrality is collectively compensated by these three types of point defects. The detailed process of point defects formation is as follows:
Ho3+ ions enter both Ba and Ti sites simultaneously, forming the self-compensated \({{\rm{Ho}}}_{{\rm{Ba}}}^{\bullet }-{{\rm{Ho}}}_{{\rm{Ti}}}^{^{\prime} }\) defect complexes, similar to \({{\rm{Dy}}}_{{\rm{Ba}}}^{\bullet }-{{\rm{Dy}}}_{{\rm{Ti}}}^{^{\prime} }\) 27 and \({{\rm{Er}}}_{{\rm{Ba}}}^{\bullet }-{{\rm{Er}}}_{{\rm{Ti}}}^{^{\prime} }\) 28.
The partial occupations of the Ti sites by Ho3+ ions result in a Ti-rich case. Accordingly, Ba vacancies are induced by the extra Ba-site Ho3+ ions excluding \({{\rm{Ho}}}_{{\rm{Ba}}}^{\bullet }-{{\rm{Ho}}}_{{\rm{Ti}}}^{^{\prime} }\) to meet the requirement of the lattice electroneutrality.
Thus, a very strong EPR signal at g = 1.974 is present in BHTH (Fig. 1b). The real formula of BHTH is determined to be (Ba1−x Ho3x/4)(Ti1−x/4Ho x/4)O3 with double-site substitutions and Ba-vacancy compensation, i.e., that at the doping level of x = 0.01, 0.75 at.% Ho3+ ions are substituted on the Ba sites and 0.25 at.% Ho3+ ions on the Ti sites. The quantitative information on the site occupations of Ho3+ ions in BHTH can be drawn from the EPR and XRD results. The defect chemistry of BHTH is the same as that of BaTiO3 doped with Dy27 and Er28. An interesting phenomenon is that Ho (AN = 67) is between Dy (66) and Er (68) in atomic number (AN).
Photoluminescence and Raman spectra
Figure 2a‒c show the photoluminescence (PL) spectra of BHTH upon excitations with 638-, 532-, and 488-nm laser lines. Energy level diagram for Ho3+ ions in BHTH is shown in Fig. 2d. In the detectable range, these PL bands originate from the 4f-4f inner-shell emission of Ho3+ in BaTiO3. Upon 638-nm excitation, Ho3+ ions are excited through one-photon absorption from 5I8 to 5F5; the strongest emission band at 653 nm (Fig. 2a) is caused by the 5F5 → 5I8 transition. The near infrared (NIR) emission band (755 nm) associated with the 5F4/5S2 → 5I7 transition might originate from the following process: Ho3+ ions are excited through one-photon absorption from the ground state 5I8 to the excited state 5F5 that relaxes nonradiatively to the lying excited state 5I7 via continuous multi-phonon relaxation processes (5F5-5I4-5I5-5I6-5I7). Subsequently, Ho3+ ions were excited again through one-photon absorption (excited state absorption (ESA) process or energy transfer (ET) process) from 5I7 to the excited state 5F3 that relaxes nonradiatively to the emitting level 5F4/5S2. Finally, the 5F4/5S2 → 5I7 transition occurs (Fig. 2d). Each emission band shows a multiplet feature because of the Stark components of the ground state 5I8 and the excited states 5F4/5S2 and 5F5 5, 31, 32, 35.

PL spectra of BHTH at room temperature, excitation with (a) 638-, (b) 532-, and (c) 488-nm laser lines. (d) Energy level diagram for Ho3+ ions in BHTH. MPA-ET represents multi-phonon assisted energy transfer. The values in bracket represent the percentage of the line intensity to the strongest emission line intensity. (e) Pure Raman spectrum is observed using 785-nm excitation wavelength. (f) Comparison of Raman spectra (stokes components) under 785- and 488-nm excitation wavelengths, shown in Raman shift and absolute wavenumber. The right figure shows the weak bands at around 500 nm in Fig. 2c inset.
Upon 532-nm excitation, three PL bands with different intensities can be observed (Fig. 2b). The emission mechanism of Ho3+ in BHTH does not go through two-step ESA or ET process, but one-step one-photon absorption from 5I8 to 5F4/5S2 that relax nonradiatively to the emitting level 5F5, which is the reason that the intensity of the NIR (755 nm) emission band under 532-nm excitation is higher than that under 638-nm excitation. BHTH exhibits strong green (545 nm) with weak red (653 nm) and NIR (755 nm) emission bands associated with 5F4/5S2 → 5I8, 5F5 → 5I8 and 5F4/5S2 → 5I7 transitions, respectively, of Ho3+ ions31, 32. The multiplet feature of bands is also caused by Stark splitting of the energy levels. It can be seen from the insets in Fig. 2a and b that the corresponding emission bands upon excitation with different wavelengths exhibit the same spectral structure.
Pure Raman spectrum was observed in BHTH using 785-nm excitation wavelength, as shown in Fig. 2f. This spectrum of BHTH, which does not show any emission transition line and is similar to that reported for the tetragonal BaTiO3 28, 40,41,42, shows four common bands, with peaks at 267 [A1 (TO2)], 304 [B1 + E(TO3 + LO2)], 521 [A1 (TO3)], and 718 cm−1 [A1 (LO3) + E (LO4)], corresponding to 802, 804, 817, and 832 nm in wavelength (Fig. 2e), respectively. No signal was present in the range of 850–1000 nm.
Upon 488-nm excitation, two bands were observed and the intensity of the main PL band at around 564 nm is 40 times that of the weak band at around 500 nm (Fig. 2c). The band at 564 nm, which has not been reported for Ho-doped BaTiO3 31, 32, was observed for BHTH (Fig. 2c). This band is attributed to the 5G6/5F1 → 5I7 transition (Fig. 2d). The following analyses give its emission mechanism, and indicate that the band at 564 nm is associated with Ho3+ ions on the Ti sites and the weak band at 500 nm originates from Raman scattering of BHTH.
It is well known that when the excitation is changed to a different wavelength, the Raman bands shift the same amount correspondingly, while the PL lines stay on the same absolute wavenumber28, 43. Upon 638- and 532-nm excitations, it can be seen that both 5F5 → 5I8 and 5F4/5S2 → 5I7 transition bands at around 653 and 755 nm (Fig. 2a and b) stay on the same position, respectively, illustrating the PL nature of these two bands. Upon 488-nm (20491.8 cm−1) and 785-nm (12738.9 cm−1) excitations, Raman band is easy to be identified by comparison of band positions. Figure 2f shows their spectra, in which the right figure shows the weak band at 500 nm in Fig. 2c inset. When the excitation is changed from 488 nm to 785 nm, the shift of 7752.9 cm−1 between two Raman signals was observed for BHTH. Moreover, both spectra exhibit the nearly same spectral structure as the tetragonal BaTiO3 (Fig. 2f). Thus, the band at around 500 nm is confirmed to originate from Raman scattering in the case of excitation at 488 nm.
The 5G6/5F1 → 5I7 transition most likely originates from ground state absorption plus phonon-assisted transition (Fig. 2d). The 5F3 energy level can be populated from 5I8 via 488-nm laser energy absorption (one-photon). As the energy differences between 5F3 and 3K8/5F2 and between 3K8/5F2 and 5G6/5F1 is about 250 and 1050 cm−1, respectively, the multi-phonon assisted energy transfer (MPA-ET) corresponding to one A1 (TO2) phonon absorption (5F3 → 3K8/5F2) and then two A1 (TO3) phonons absorption (3K8/5F2 → 5G6/5F1) can occur. The absorbed phonons can be detected in light of the Raman spectrum in Fig. 2c inset or Fig. 2f. Normally, the three-phonon absorption is enough for the energy gap between 5F3 and 5G6/5F1. Finally, the emission band at 564 nm is due to the transition from 5G6/5F1 to the second ground state level 5I7.
On the basis of the above analyses, an important finding is that PL and Raman scattering under 488-nm excitation can occur simultaneously as two distinct optical processes for BHTH ceramic powders. One can also see that the PL signals from the 5F4/5S2 → 5I8 transition under 532-nm excitation are so intense that they overwhelm the traditional Raman spectra of BaTiO3 (Fig. 2b and Fig. 2c inset). The option for laser wavelength is an important factor in observation of Raman signals.
Photoluminescent origin associated with site occupations of Ho3+ in BaTiO3
To investigate the PL origin associated with the site occupations of Ho3+ ions in the host BaTiO3 lattice, a BaTiO3 ceramic doped with Ho3+ on the Ba site is required to be prepared. In the Ti-rich case of Ba/Ti < 1, Ho3+ was considered to enter the Ba site for (Ba1−x Ho x )TiO3 sintered at T s = 1320–1360°C in air12,13,14. For this reason, we prepared a nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) ceramic (BH1T) sintered at T s = 1300 °C. BHTH and BH1T have the same stoichiometric proportions in components.
The XRD pattern and EPR spectrum of BH1T as well as a comparison in PL spectra between BHTH and BH1T under 488-nm excitation are shown in Fig. 3. BH1T also exhibits a single-phase tetragonal perovskite structure (Fig. 3a) like BHTH, implying a complete incorporation of Ho3+ ions into the host BaTiO3 lattice. The V 0 of BH1T (64.25 Å3) is less than those of BHTH (64.44 Å3) and the tetragonal BaTiO3 (64.41 Å3), suggesting that Ho3+ ions are dominantly substituted for the Ba sites.

(a) XRD pattern, (b) EPR spectrum for the nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) ceramic (BH1T) sintered at T s = 1300 °C. The real formula of BH1T is approximately expressed by (Ba1−x Ho x−δ )(Ti1−x/4Ho δ )O3 (δ is a small quantity) with a small number of Ti-site Ho3+ ions. (c) The comparison in photoluminescence spectra between BHTH and BH1T clearly reveals that Ti-site Ho3+ ions are responsible for 5G6/5F1 → 5I7 transition of Ho3+.
The EPR monitoring for BH1T shows coexistence of both Ba vacancies and Ti ones, marked by two signals at g = 1.974 and 2.004, respectively (Fig. 3b), implying that Ho3+ may transfer mutually between the Ba site and the Ti one during ceramic sintering and cooling. This behavior is similar to that of Lu3+-doped BaTiO3 44. In this case more Ho3+ ions in BH1T can be present on the Ba2+ sites relative to BHTH, resulting in a stronger donor effect. The Mn2+ sextet signal is therefore caused by the reduction of Mn4+/Mn3+ to Mn2+ impurities in BH1T45, 46.
For Ho-doped BaTiO3 ceramics prepared by Battisha and Secu et al., Ho3+ is mainly substituted for the Ba sites because of a lower sintering temperature. They observed some luminescence bands at 435 (5F1 → 5I8), 545 (5F4/5S2 → 5I8), 660 (5F5 → 5I8), and 760 nm (5F4/5S2 → 5I7)35, 36. The solid solubility limit of Ho3+ ions on the Ba sites is less than x = ~1.4% at T s = 1400 °C10. For our BHTH, 0.75 at.% Ho3+ ions are substituted on the Ba sites. Thus, the PL bands at 545, 653, and 755 nm (Fig. 2a and b) are attributed to Ho3+ ions on the Ba sites.
In a detectable wavelength range, one can see that both PL bands at around 564 nm are nearly the same in spectral structure and different in intensity for BHTH and BH1T, while there is no difference between both Raman bands in a lower-wavelength range at around 500 nm (Fig. 3c). It is obvious that the PL band at around 564 nm does not relate to Ho3+ ions on the Ba sites. The above XRD and EPR results reveal that Ho3+ ions in BH1T are dominantly substituted for the Ba sites, which is in good agreement with the site occupations of Ho3+ at a lower sintering temperature of T s = 1320–1360°C in air12,13,14. Similar to the doping behavior of Dy3+ or Er3+ in BaTiO3 27, 28, a small number of Ho3+ ions in BH1T inevitably enter the Ti sites. For our BHTH, 0.25 at.% Ho3+ ions are substituted on the Ti sites. The PL intensity is proportional to the concentration of Ho3+ ions. The PL intensity of the 5G6/5F1 → 5I7 transition of BH1T is one-twentieth of BHTH (Fig. 3c), which matches the concentration of Ho3+ ions on the Ti sites in BH1T and BHTH. Thus, Ho3+ ions on the Ti sites are responsible for the 5G6/5F1 → 5I7 transition at around 564 nm.
It is inferred from the PL intensity that 0.0125 at.% Ho3+ ions are substituted on the Ti sites in BH1T, though no quantitative information on the site occupations of Ho3+ ions on the Ti sites or Ba ones can be drawn directly from the EPR and XRD results. The real formula of BH1T may be approximately described as (Ba1−x Ho x−δ )(Ti1-x/4Ho δ )O3 (δ is a small quantity, δ = 0.0125%) with Ba vacancies and Ti ones. Hence, the change in local environment and crystal field at which Ho3+ lies plays a decisive role in PL.
PL band at 564 nm as a probe for dielectrics in MLCC
Temperature-stable dielectric materials in MLCC often adopt the core-shell structured BaTiO3 ceramics, in which a fine grain consists of a tetragonal BaTiO3 core and a cubic surface layer consisting of Ho2O3 and other oxide, such as Ho/Mg7, 47, Ho/Mn48, Ho/Zr,49, 50 Ho/Dy8 etc. The incorporation of additives ions into BaTiO3 particles will reduce dielectric permittivity and dielectric-temperature stability. The sintering process plays an important role in controlling incorporation of Ho3+ into BaTiO3 particles. Although at a high sintering temperature (e.g. T s = 1550 °C10, 15) Ho3+ ions are considered to substitute for the Ti sites, even at a lower T s Ho3+ ions inevitably enter the Ti sites in part, as observed in Fig. 3c. It is more expensive and complicated to detect the amount of Ho3+ ions dissolved in the BaTiO3 lattice using an ion implantation technique with an accelerating voltage of 500 keV and secondary-ion mass spectrometry (SIMS)11. The PL of the 5G6/5F1 → 5I7 transition is sensitive to Ho3+ on the Ti sites in BaTiO3. Thus, the application of PL under 488-nm excitation may provide a probe for the occupations of Ho3+ ions on the Ti sites in BaTiO3 ceramics co-doped with Ho3+ and other dopants. Accordingly, the application of PL under 532- or 638-nm excitations may provide a probe for the occupations of Ho3+ on the Ba site.
Conclusions
The nominal (Ba1−x Ho x )Ti1−x/4O3 (x = 0.01) (BHTH) ceramic was prepared at 1400 °C using the solid-state reaction method. BHTH exhibits a single-phase tetragonal perovskite structure. The study on the defect chemistry indicates that the defects in BHTH are Ba-site Ho3+ (\({{\rm{Ho}}}_{{\rm{Ba}}}^{\bullet }\)), Ba vacancies (\({{\rm{V}}}_{{\rm{Ba}}}^{^{\prime\prime} }\)), and Ti-site Ho3+ (\({{\rm{Ho}}}_{{\rm{Ti}}}^{\text{'}}\)). The real formula of BHTH is expressed by (Ba1−x Ho3x/4)(Ti1−x/4Ho x/4)O3. The change in local environment and crystal field at which Ho3+ lies plays a decisive role in photoluminescence (PL) of Ho3+ ions. Upon 532- and 638-nm excitations, three PL bands corresponding to 5F4/5S2 → 5I8, 5F5 → 5I8, and 5F4/5S2 → 5I7 transitions are attributed to the contributions from Ho3+ ions on the Ba sites in the BaTiO3 lattice. On the contrary, Ho3+ ions on the Ti sites are responsible for the 5G6/5F1 → 5I7 transition under 488-nm excitation, and moreover, PL and Raman scattering can occur simultaneously as two distinct optical processes. The common Raman spectrum of BaTiO3 can be observed without PL disturbance using 785-nm excitation wavelength. The PL signals under 532-nm excitation are intense enough to conceal the traditional Raman phonon modes of BaTiO3. The application of PL may provide a probe for the site occupations of Ho3+ in BaTiO3 dielectric ceramics co-doped with Ho3+ and other dopants.
References
Cao, R. et al. 2 μm emission properties and nonresonant energy transfer of Er3+ and Ho3+ codoped silicate glasses. Sci. Rep. 6(37873), 1–11 (2016).
Li, G. et al. Diode-pumped efficient laser operation spectroscopy of Tm:Ho:YVO4. Opt. Mater. 33, 937–941 (2011).
Zmojda, J. et al. Investigation of upconversion luminescence in antimony–germanate double-clad two cores optical fiber co-doped with,Yb3+/Tm3+ and Yb3+/Ho3+ ions. J. Lumin. 170, 795–800 (2016).
Mahata, M. K., Koppe, T., Kumar, K., Hofsäss, H. & Vetter, U. Demonstration of temperature dependent energy migration in dual-mode YVO4: Ho3+/Yb3+ nanocrystals for low temperature thermometry. Sci. Rep. 6(36342), 1–11 (2016).
Ryba-Romanowski, W., Komar, J., Niedzwiedzki, T., Głowacki, M. & Berkowski, M. Excited state relaxation dynamics and up-conversion phenomena in Gd3(Al,Ga)5O12 single crystals co-doped with holmium and ytterbium. J. Alloys Compd. 656, 573–580 (2016).
Zhu, Y. et al. Broad white light and infrared emission bands in YVO4:Yb3+,Ln3+ (Ln3+ = Er3+, Tm3+, or Ho3+). Appl. Phys. Express 5(092701), 1–3 (2012).
Kishi, H., Mizuno, Y. & Chazono, H. Base-metal electrode-multilayer ceramic capacitors: past, present and future perspectives. Jpn. J. Appl. Phys. 41, 1–15 (2003).
Gong, H., Wang, X., Zhang, S. & Li, L. Synergistic effect of rare-earth elements on the dielectric properties and reliability of BaTiO3-based ceramics for multilayer ceramic capacitors. Mater. Res. Bull. 73, 233–239 (2016).
Hanafi, Z. M., Ismail, F. M., Hammad, F. F. & Nasser, S. A. Structural investigation of the phase transition in perovskite titanate containing additives. J. Mater. Sci. 27, 3988–3992 (1992).
Makovec, D., Samardzija, Z. & Drofenik, M. Solid solubility of holmium, yttrium, dysprosium in BaTiO3. J. Am. ceram. soc. 87, 1324–1329 (2004).
Itoh, J. et al. Diffusion and solubility of holmium ions in barium titanate ceramics. J. Mater. Res. 19, 3512–3520 (2004).
Jeong, J., Lee, E. J. & Han, Y. H. Electrical properties of holmium-doped BaTiO3. Jpn. J. Appl. Phys. 44, 4047–4051 (2005).
Cernea, M. et al. Electrical investigations of holmium-doped BaTiO3 derived from sol-gel combustion. J. Mater. Res. 25, 1057–1063 (2010).
Jeong, J., Lee, E. J. & Han, Y. H. Effects of Ho2O3 addition on defects of BaTiO3. Mater. Chem. Phys. 100, 434–437 (2006).
Liu, Y. & West, A. R. Ho-doped BaTiO3: Polymorphism, phase equilibria and dielectric properties of BaTi1−x Ho x O3−x/2: 0 ≤ x ≤ 0.17. J. Eur. Ceram. Soc. 29, 3249–3257 (2009).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 32, 751–767 (1976).
Kchikech, M. & Maglione, M. Electron and lattice excitations in BaTiO3-La. J. Phys. Conden. Matt. 6, 10159–10170 (1994).
Morrison, F. D., Sinclair, D. C. & West, A. R. Electrical and structural characteristics of lanthanum-doped barium titanate ceramics. J. App. Phys. 86, 6355–6366 (1999).
Dobal, P. S., Dixit, A. & Katiyar, R. S. Effect of lanthanum substitution on the Raman spectra of barium titanate thin films. J. Raman Spectrosc. 38, 142–146 (2007).
Liu, Y., Feng, Y., Wu, X. & Han, X. Microwave absorption properties of La doped barium titanate in X-band. J. Alloys Compd. 472, 441–445 (2009).
Chen, M. S. et al. Stress effect on Raman spectra of Ce-doped BaTiO3 film. J. Phys. Condens. Mat. 12, 7013–7023 (2000).
Lu, D. Y., Han, D. D., Sun, X. Y., Zhuang, X. L. & Zhang, Y. F. Raman evidence for Ba-site Ce3+ in BaTiO3. Jpn. J. Appl. Phys. 52(111501), 1–4 (2013).
Lu, D. Y., Sun, X. Y., Liu, B., Zhang, J. L. & Ogata, T. Structural and dielectric properties, electron paramagnetic resonance, and defect chemistry of Pr-doped BaTiO3 ceramics. J. Alloys Compd. 615, 25–34 (2014).
Yao, Z. et al. Structure and dielectric behavior of Nd-doped BaTiO3 perovskites. Mater. Chem. Phys. 109, 475–481 (2008).
Lu, D. Y. et al. Self-compensation characteristics of Eu ions in BaTiO3. Solid State Ionics 201, 6–10 (2011).
Lu, D. Y. Self-adjustable site occupations between Ba-site Tb3+ and Ti-site Tb4+ ions in terbium-doped barium titanate ceramics. Solid State Ionics 276, 98–106 (2015).
Lu, D. Y. & Cui, S. Z. Defects characterisation of Dy-doped BaTiO3 ceramics via electron paramagnetic resonance. J. Eur. Ceram. Soc. 34, 2217–2227 (2014).
Lu, D. Y. et al. Abnormal Raman spectra in Er-doped BaTiO3 ceramics. J. Raman Spectrosc. 45, 963–970 (2014).
Amami, J. et al. Second harmonic generation and Yb3+ cooperative emission used as structural probes in size-driven cubic-tetragonal phase transition in BaTiO3 sol–gel nanocrystals. J. Lumin. 119–120, 383–387 (2006).
Ganguly, M., Rout, S. K., Ahn, C. W., Kim, I. W. & Kar, M. Structural, electrical and optical properties of Ba(Ti1−x Yb4x/3)O3 ceramics. Ceram. Int. 39, 9511–9524 (2013).
Kumar, G. A., Martinez, A., Mejia, E. & Eden, J. G. Fluorescence and upconversion spectral studies of Ho3+ in alkali bismuth gallate glasses. J. Alloys Compd. 365, 117–120 (2004).
Babu, S., Seshadri, M., Balakrishna, A., Reddy Prasad, V. & Ratnakaram, Y. C. Study of multicomponent fluoro-phosphate based glasses: Ho3+ as a luminescence center. Physica B 479, 26–34 (2015).
Pandey, A. & Swart, H. C. Luminescence investigation of visible light emitting Ho3+ doped tellurite glass. J. Lumin. 169, 93–98 (2016).
Valiev, U. V. et al. Study of the line intensity in the optical and magnetooptical spectra in holmium-containing paramagnetic garnets. Opt. Mater. 51, 42–49 (2016).
Battisha, I. K. Visible up-conversion luminescence in Ho3+: BaTiO3 nano-crystals prepared by sol gel technique. J. Sol-Gel Sci. Techn. 30, 163–172 (2004).
Secu, M., Cernea, M., Secu, C. E. & Vasile, B. S. Structural characterization and photoluminescence of nanocrystalline Ho-doped BaTiO3 derived from sol–gel method. J. Nanopart. Res. 13, 3123–3128 (2011).
Kolodiazhnyi, T. & Petric, A. Analysis of point defects in polycrystalline BaTiO3 by electron paramagnetic resonance. J Phys Chem Solids 64, 953–960 (2003).
Dunber, T. D., Warren, W. L., Tuttle, B. A., Randall, C. A. & Tsur, T. Electron paramagnetic resonance investigation of lanthanide-doped barium titanate: dopant site occupancy. J. Phys. Chem. B 108, 908–917 (2004).
Kröger, F. A. & Vink, H. J. Solid State Physics III (Academic Press, New York, 1956).
DrDomenico, M., Wemple, J. S. H., Porto, S. P. S. & Rauman, R. P. Raman spectrum of single-domain BaTiO3. Phys. Rev. 174, 522–530 (1968).
Gupta, S. Investigations of micro-stress and phase transition in so-gel-derived multideposited coatings of barium titanate using Raman spectroscopy. J. Raman Spectrosc. 33, 42–49 (2002).
Buscaglia, V. et al. Raman and AFM piezoresponse study of dense BaTiO3 nanocrystalline ceramics. J. Eur. Ceram. Soc. 25, 3059–3062 (2005).
Gajović, A., Tomašić, N., Djerdj, I., Su, D. S. & Furić, K. Influence of mechanochemical processing to luminescence properties in Y2O3 powder. J. Alloys Compd. 456, 313–319 (2008).
Lu, D. Y. & Liu, T. T. Dielectric properties and defect chemistry of (Ba1−x La x )(Ti1−x Lu x )O3 ceramics. J. Alloys Compd. 698, 967–976 (2017).
Ikushima, H. & Mayakawa, S. Electron spin resonance Mn2+ in BaTiO3. J. Phys. Soc. Jpn. 19, 1986 (1964).
Müller, K. A., Berlinger, W., Blazey, K. W. & Albers, J. Electron paramagnetic resonance of Mn4+ in BaTiO3. Solid State Commun. 61, 21–25 (1987).
Kishi, H. et al. The effect of rare-earth (La, Sm, Dy, Ho and Er) and Mg on the microstructure in BaTiO3. J. Eur. Ceram. Soc. 19, 1043–1046 (1999).
Jeong, J., Lee, E. J. & Han, Y. H. Defect chemistry and electrical degradation of BaTiO3 co-doped with Ho and Mn. J. Eur. Ceram. Soc. 27, 1159–1163 (2007).
Sagar, R., Hudge, P., Madolappa, S., Kumbharkhane, A. C. & Raibagkar, R. L. Electrical properties and microwave dielectric behavior of holmium substituted barium zirconium titanate ceramics. J. Alloys Compd. 537, 197–202 (2012).
Jha, P. A. & Jha, A. K. Effect of holmium substitution on structural and electrical properties of barium zirconate titanate ferroelectric ceramics. Ceram. Int. 40, 5209–5216 (2014).
Acknowledgements
This work was supported by the projects of the National Natural Science Foundations of China (Grant No. 21271084) and of Jilin Province (20160101290JC), and Changbai Mountain Scholar Distinguished Professor (2015047). The authors would like to thank Dr. Zhenyu Liu in The Hong Kong Polytechnic University for the help of photoluminescence analyses.
Author information
Authors and Affiliations
Contributions
D.X.G. conducted the synthesis, XRD, SEM, EPR, Raman, and PL measurements of Ho-doped BaTiO3 ceramics. D.Y.L. supervised the work, carried out the analysis of all the data, and wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, DY., Guan, DX. Photoluminescence associated with the site occupations of Ho3+ ions in BaTiO3 . Sci Rep 7, 6125 (2017). https://doi.org/10.1038/s41598-017-06521-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06521-4
This article is cited by
-
Investigation on energy storage properties and thermally stable dielectric constant for high temperature electronic device applications in the holmium substituted Na0.5Bi0.5TiO3
Journal of Materials Science: Materials in Electronics (2021)
-
Studies on structural, electronic and magnetic properties of La3+ ion-substituted Ho2FeMnO6 double perovskite compounds
Journal of Materials Science: Materials in Electronics (2021)
-
Environmentally sustainable facile synthesis of nanocrystalline holmium hafnate (Ho2Hf2O7): Promising new oxide-ion conducting solid electrolyte
SN Applied Sciences (2020)
-
Photoluminescence activity of BaTiO3 nanocubes via facile hydrothermal synthesis
Journal of Materials Science: Materials in Electronics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
