
Abstract
Interspecific fungal antagonism occurred commonly in the interaction zone of different white rot fungi. This competitive interaction could markedly influence the metabolic pathway of intracellular metabolites, which was associated with the fungal morphology change and growth restriction. So far, it remains unknown on intracellular metabolite regulation during fungal competitive interaction. Herein, we performed the metabolomic analysis of the in vivo metabolite changes during competitive interaction between each two of the three white rot fungi Trametes versicolor, Pleurotus ostreatus and Dichomitus squalens and identified differential metabolites in the interaction zone compared to each two isolates. Many metabolites in the carnitine, lipid, ethylene and trehalose metabolic pathways were significantly up-regulated. These metabolic pathways are all involved in defensive response to abiotic and/or biotic stressful condition.
Similar content being viewed by others

Introduction
Competition is the most common type of interaction occurring between white rot fungi1,2,3. When different fungal species confront with each other, direct combative interactions between mycelia to defend or compete for resources are always accompanied by changes of mycelial morphology and secretion of extracellular metabolites1, 3, 4. These metabolites have been identified including alcohols, aldehydes, ketones, terpenes and aromatic compounds, most of which are important in defensive response to interspecific antagonism in the interaction zones5. As it has been reported that oxidative stress can be induced during the interspecific interaction6, some oxidative enzymes were enhanced in the interaction zone to mediate the oxidative stress by removing reactive oxygen species (ROS), such as laccase and peroxidases7, 8, which also implied another extracellular signaling and metabolic pathway.
Metabolomics-based approach has been used extensively for understanding metabolites respond to environmental stimuli in fungi, bacteria and plants9,10,11,12,13,14. Metabolomics is increasingly being applied in identifying the target metabolites produced by fungi during stressful condition. Nowadays, some studies showed that the secreted metabolites in the interaction zone of two fungi species caused related mycelial morphological phenotypes, suggesting the changed metabolites could be also involved in the defense response against abiotic stress15,16,17,18,19,20. Some potentially novel metabolites belonging to natural products were induced and detected during co-culturing of fungi on solid media, and these metabolites were released in response to antagonistic interactions17, 19, 21, 22.
There was no report focusing on the differences of in vivo cellular metabolites, especially those serve as a composition of cell membrane and cell wall structure, which can affect the morphology of the cells during fungal antagonistic interaction. Until now, the intracellular metabolic regulation mechanism against stressful condition are still less characterized. It has been known that various pathways can be motivated to enhance the competitive potential of each species1, 3, which might involve signal molecules, growth inhibitors or toxins, and their by-products in this barrage zones3, 16, 18. Therefore, even though the two fungi confronting on the plate were different species, there may be common strategies in response to the interspecific interaction.
In order to study the differential metabolic response between competitive mycelial interaction zones and corresponding isolates, as well as to unravel the effects on the in vivo metabolic network, we performed the metabolomics analysis of interaction zone between each two of the three white rot fungi by ultra-performance liquid and/or gas chromatography coupled to mass spectrometry (UPLC–MS and GC–MS) based on metabolomics approach. The three white rot fungi Trametes versicolor (Tv), Dichomitus squalens (Ds) and Pleurotus ostreatus (Po) were chosen based on their significant interspecific laccase induction. The study is important to reveal differential levels of intracellular metabolites when fungal mycelia interact with each other, which can be applied into the further analysis of metabolite signaling research.
Results
Synergistically changed metabolites in all three interactions
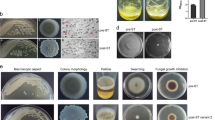
The metabolomic dataset was comprised of total 279 compounds of known metabolites from the three white rot fungi isolates Tv, Ds and Po and their interaction zones (Abbreviated as TvDs, TvPo, PoDs; Fig. 1). The false discovery rate and statistical significance were calculated to represent the multiple comparisons between interaction zones and the relevant isolates (Supplementary Table S3). Moreover, the significances of the differential metabolites were confirmed by a pairwise comparison to the t-test. The total identified differential metabolites with intensity values were listed in Supplementary Table S1.

Paired-culture on Sc agar plate among Tv, Ds and Po.
The detected 279 compounds covered various metabolic pathways such as metabolic pathways of purine, pyrimidine, amino acid, central carbon, TCA, sugar, glyoxylate, alkaloid, photosynthetic, phenylpropanoid, and flavonoid metabolism. Based on the metabolomics data, we compared compounds which met the statistical criterion for significance (p-valve ≤ 0.05) in the three interaction zones of TvDs, TvPo and PoDs to their respective isolate zones of Tv, Ds and Po. The result showed that in most cases, there were more compounds in the interaction zone relatively increased, not decreased, comparing to the isolates. Only the Ds isolate relative to TvDs interaction zone had more increased compounds (Fig. 2). Moreover, over half of the increased compounds in the interaction zone were synergistically increased in all three interaction zones when compared to both isolates. For instance, 65 synergistically increased compounds and only 3 synergistically decreased compounds in TvPo comparing with isolates of Tv or Po(Fig. 2).

Significant changes of compounds in each interaction zone compare to the two isolates. Ratios as: up-regulated numbers/down-regulated numbers, p-value ≤ 0.05 was taken as significant.
Interestingly, these synergistically changed metabolic pathways showed some similarities (Fig. 3). In the interaction zone of TvDs, the changed (increased) pathways were all shared with TvPo or PoDs, and the number of decreased pathways in TvDs was more than that in TvPo and PoDs. Four unique decreased pathways can be seen in TvDs, including ascorbate metabolism, fatty acid, dicarboxylate, glutamate family (alpha-ketoglutarate derived) and photorespiration pathways. In the interaction zone of TvPo, the number of increased pathways was more than that in TvDs and PoDs. Six increased pathways were unique in TvPo, including C5 branched dibasic acid metabolism, calvin cycle and pentose phosphate, CoA metabolism, free fatty acid, glutathione metabolism and photorespiration pathways. Only three increased pathways were unique in the interaction zone of PoDs, including benzenoids, Branched Chain Amino Acids (OAA derived) and oxylipins pathways. Otherwise, there were also three unique decreased pathways in PoDs, including free fatty acid, glycolysis and sucrose, glucose and fructose metabolism pathways (Fig. 3).

Summary of the pathway-level increase (red box) and decrease (green box) of metabolism in three interaction zones with significant data combined from synergistically changed metabolites compared to both isolates. Pathways in yellow and white fonts represent unique and shared pathways, respectively.
To analyze these significantly changed metabolites more globally, we also investigated them in all interaction zones among the three different white rot fungi (Fig. 4). We found 16 metabolites synergistically increased and only one synergistically decreased compounds in all interaction zones, the pathway classification and fold-change calculations for all 17 synergistic metabolites were shown in Table 1. According to the result, the aromatic amino acid tryptophan was one of the most strongly induced compounds in all three interactions. Notably, the fold change of TvPo and PoDs compared to Po isolate reached up to 119.94 and 71.73, respectively. The major commonality of other compounds was that most of the compounds were associated with catabolic processes, including branched chain amino acids metabolism, carnitine metabolism, glycerolipids metabolism and phospholipids metabolism. Two groups of compounds stood out as being overrepresented, they were group of various carnitine derivatives and another group of lysolipids and glycerolipid catabolites.

Synergistic changes in all three interaction zones relative to both isolates. Red arrow means increase, green arrow means decrease.
Carnitine metabolism
Carnitine conjugates are derived both from the catabolism of branched chain amino acids and fatty acid oxidation23, 24 (Figure S2A,B). Carnitine is used to transport fatty acids across membranes, as well as serves as an alternative acyl-group receiver to CoA in order to buffer the CoA pool, typically under stressful conditions when CoA becomes limited25. As is shown in Fig. 5, not only the carnitine increased in all interaction zones, the catabolic products acetylcarnitine (C2), propionylcarnitine (C3), isobutyrylcarnitine (C4), 2-methylbutyrylcarnitine (C5) and hydroxybutyrylcarnitine also significantly increased in all three interactions, which reflected the cellular catabolism of amino acids and fatty acid activation. The result implied that the interaction of the three white rot fungi may trigger various catabolic processes to intensify tricarboxylic acid (TCA) cycle for energy provision.

Carnitine esters significantly increase in all interaction zones. (A) Carnitine; (B) Hydroxybutyrylcarnitine; (C) Acetylcarnitine; (D) Propionylcarnitine; (E) isobutyrylcarnitine; (F) 2-methylbutyrylcarnitine.
Lipid metabolism
Membrane catabolism and remodeling occurs through the action of various lipases that release fatty acids from phospholipids, triacylglycerols (TAGs), diacylglycerols (DAGs) and other complex lipid26, 27. Although the larger complex lipids (TAGs, DAGs, cardiolipins) were not detected, many lipolysis products belonging to the group of lysolipids and monoacylglycerols significantly increased in all three interaction zones relative to each isolates. The lipolysis products included glycerophosphocholine (GPC; Fig. 6D) and its derivatives 1-linoleoylglycerol, 1-linoleoyl-GPE and 1-linoleoyl-GPI (Fig. 6A–C). GPC is generated by the degradation of phosphatidyl choline and is regarded as a very powerful osmolyte, as it is often induced under condition of osmotic stress and is served to stabilize proteins subjected to strong gradients of water potential28. This result suggested the existence of active lipolysis in the interaction zones of the three fungi.

Lipolysis products significantly increase in all interaction zones. (A) 1-linoleoylglycerol; (B) 1-linoleoyl-GPE; (C) 1-linoleoyl-GPI; (D) glycerophosphocholine (GPC).
Ethylene metabolism
There were only few compounds which exhibited opposite induction effects in the different combinations of interaction zones and isolates. One of the strongest inductions in the dataset, but only in two (TvPo and TvDs) of the three interactions, was the induction of the ethylene precursor 1-aminocyclopropanecarbolylic acid (ACC) (Fig. 7A). Ethylene is known as a stress hormone, which can be induced by a variety of stress signals, such as chemicals, metals and pathogen infection29. It has been reported that stress stimulation for ethylene synthesis was accompanied by the generation of reactive oxygen species (ROS) that could cause damage to cellular organelles by triggering lipid peroxidation30, 31. Although ethylene was not measured in this study, the high levels of its precursor ACC was detected in the TvPo and TvDs interactions, and it was significantly induced in these two interactions. Even in the TvPo interaction, the ACC was increased more than 130-fold relative to each isolates (Fig. 7A). By contrast, the accumulation of ACC was not observed in the PoDs interaction. However, the level of cyanoalanine, a by-product of ethylene synthesis that scavenges the hydrogen cyanide generated in the oxidation of ACC to form ethylene32, was higher in the PoDs interaction zone than in either TvPo or TvDs (Supplementary Table S3). This result suggested that ethylene may be an induced product in all interactions. However, the fact that the precursor ACC was not accumulated in PoDs revealed that there may be differential regulation mechanism of ACC oxidase in the interaction of PoDs compared with TvPo and TvDs (Figure S2D).

The metabolites only significantly increase in two interactions. (A) 1-aminocyclopropanecarbolylic acid (ACC); (B) Trehalose-6-phosphate, (C) UDP-glucose, (D) UDP-galactose.
Trehalose metabolism
Another pathway that was significant for only two interactions (TvDs and TvPo) was the UDP-sugars and trehalose pathway (Figure S2C). UDP-sugars like UDP-glucose and UDP-galactose, acting as the primary metabolic intermediates for the enzymatic biosynthesis of carbohydrates, were involved in cell wall biosynthesis and nucleotide sugars metabolism33. In addition, UDP-glucose is the direct precursor of trehalose-6-phosphate (T-6-P). T-6-P can serve to be key regulator of cell wall biosynthesis in fungi, and it was an active component that regulated the trehalose metabolic pathway34. Based on the antioxidant activity of trehalose, T-6-P played a key role in protecting cells from oxidative injuries, especially in cell membranes35. We noticed that UDP-glucose, UDP-galactose and T-6-P were synergistically increased in both TvDs and TvPo interactions (Fig. 7B–D). However, as trehalose showed decreased change in all three interactions (Supplementary Table S3), there possibly were other metabolic mechanism to degrade trehalose when cell viability was threatened under interspecific antagonism.
Discussion
Change of metabolites in vivo during interactions among three white rot fungi Trametes versicolor, Pleurotus ostreatus and Dichomitus squalens were analysis in our study, and the metabolomic approach adopted in this study provided a broader understanding of fungal territory and resource competition during mycelial interaction by relating to the up-regulation of intracellular metabolites. There were some interesting changes on specific metabolite pathways in response to antagonistic interaction. Among the detected 279 metabolites, most of them were up-regulated in all three interactions. They were involved in the pathways of branched chain amino acids, carnitine metabolism, nicotinate and nicotinamide metabolism, glutamate family, glycerolipids, phospholipids, serine family, sucrose, glucose, fructose metabolism and TCA cycle (Figure S1). The result implied that the common metabolic processes and reactions were more active in these pathways. Importantly, except nicotinate and nicotinamide metabolism, all the other induced pathways belonged to three super pathways: carbohydrate, amino acids and lipid pathways (Supplement Table S3), indicating that a broad array of metabolic reactions with energy production could be triggered to whelm competitor in the interaction zone where fungal mycelia were overlapped.
According to the study, almost half of the compounds exhibited “synergistic” effects, which indicated they were higher or lower in the interaction zones than in either of the individual isolates. Interestingly, 17 compounds showed the same synergistic effects for all three fungal interactions, especially the most strongly induced compound tryptophan. As an aromatic amino compound, tryptophan was reported to be able to significantly induce laccase production of different fungi species36. In addition, many evidences revealed that laccase played an important role in a defensive response against stressful condition37,38,39,40 and could be significantly increased during the interaction among many white-rot fungi8, 41,42,43,44,45. Therefore, the accompanying induction of laccase was probably correlated with significant increase of tryptophan during interspecific mycelial interaction.
It has been known that a series of antagonistic responses could be triggered to compete for nutrient during combative interaction between two fungal species1,2,3. These responses were involved in intensifying fat catabolism, provision of energy substrate on account of nutrient deprivation and enhanced energy demand in competition, the generation of fatty acids (FAs), glycerols and other lysolipids which were pivotal substrates for energy production and for harmful compounds detoxification27. The up-regulation of lipolysis production glycerophosphocholine (GPC) and its derivatives in our work, which may contribute to stress remediation of mycelia in the interaction zone as an osmolyte28, 46, was associated with host’s responses. Besides, the role of carnitine in fatty acid oxidation and oxidative stress protection has been recognized in fungi25, 47. The generation of acetyl CoA during fatty acids β-oxidation in the peroxisome could enter mitochondria as carnitine esters for TCA metabolism47, 48. The function of carnitine to buffer CoA pool can also supply energy for the enhanced catabolism of fatty acids in organism25, 48. Moreover, carnitine also serve as a defensive molecular under stressful condition. Based on carnitine involved metabolic pathways, the synergistic up-regulation of carnitine and its conjugates in all three interactions maybe accompanied by the nutrient competition and damages of oxidative stress during fungal interspecific interactions.
We also noticed that some metabolite inductions such as ACC and T-6-P were distinct significant for one or two interactions, not for all three (Supplementary Table S4). Previous studies revealed that ethylene can be produced not only in plant but also in fungi29, 49, 50. As ethylene in plant was involved in the response to biotic and abiotic stresses from environmental stimuli29, 50, the role of ethylene in fungi might be similar to that in plant. In this study, the synergistic up-regulation of ethylene precursor ACC was detected in the interactions of TvPo and TvDs, the strongly induced ethylene pathway in at least two of the interactions possibly implied that the abiotic and oxidative stresses were stimulated in the fungal antagonistic interaction.
Trehalose is an integral compound of various glycolipids. It not only can serve as a source of energy in organism51, but also plays defensive role of protecting cells from oxidative damage caused by ROS, which can be overproduced during fungal combative interaction6, 35. UDP-sugars and T-6-P, which were two important metabolites in the widely known synthetic pathway of trehalose, were significantly increased in two interactions of TvPo and TvDs, but showed no obvious change in PoDs. The result might be caused by the differential regulation mechanisms of trehalose-6-phosphate phosphatase (T6PP) in the trehalose pathway for the various pair-wise fungal interactions34, 52. By contrast, trehalose was decreased in all three interaction zones, suggesting fungus probably consumed its own trehalose or glucose hydrolyzed from trehalose to supply energy and carbohydrate source when growth and nutrient limiting occurred under stressful condition11, 52. The similar change of trehalose has been reported in a previous study, which showed decreased level of trehalose in white rot fungus Phanerochaete chrysosporium under a toxic stress response53. This result was also probably the main reason for the significant decrease of N-acetylglucosamine in all three interaction zones (Table 1). N-acetylglucosamine is the essential component of fungal cell wall chitin54 and plays an vital role in regulating the intracellular signaling proteins55. Therefore, we supposed that the attack or defense response leaded to cell wall degradation during interspecific combative interaction, and the degradation product N-acetylglucosamine could be utilized by competitors as carbon and nitrogen source.
In summary, our metabolomics analysis revealed that intercellular metabolites differentially exist in the interspecific interaction zones relative to their isolates among three white rot fungi. We also noticed that there were common in vivo metabolic reactions in white rot fungi in responses to stressful condition. The intracellular metabolic changes were correlated with the interactions between different white rot fungi, which probably affected the cell wall synthesis, osmolyte production and carbon/energy regulation in the interaction zones. Importantly, this study provided important information on the understanding of defense mechanism when different fungal mycelia were physically interacted with each other. The work on identification of differential metabolites provided insight into the role of specific metabolites in the interaction zone. These results can be further used for correlation analysis with transcriptomics and proteomics to deeply understand the metabolic processes during fungal combative interaction.
Materials and Methods
Strains and culture
Strains of Trametes versicolor (Tv), Pleurotus ostreatus (Po) were from the Biological Resource Center, NITE (NBRC) and Dichomitus squalens (Ds) was from the Deutsche Sammlung von Microorganismen and Zellkulturen (DSMZ). All the three white rot fungi were cultured on potato dextrose agar (PDA) slants and stored at 4 °C. Before use, the stored fungi were inoculated onto the newly prepared PDA plates at 28 °C. Cultures were routinely cultured every 7 days.
Mono-and paired-culture conditions
For mono-cultures, a 7 mm agar plug of a fungal pre-culture was inoculated in the center of a 90 mm petri dish containing 30 mL of Sc agar media (10 g/liter glucose,1.5/liter L-asparagine, 0.12 mg/liter Thiaminiumdichloride, 0.46 g /liter KH2PO4, 1 g/liter K2HPO4, 0.5 g/liter MgSO4·7 H2O, 2 mL trace elements, 20 g/liter Agar). The petri dishes were incubated at 28 °C for 9 days. Similarly, pair-culture experiments were inoculated with two 7 mm agar plugs from different fungal pre-culture on opposite sides of a petri dish containing Sc agar media(Fig. 1), there were three combinations(TvDs, TvPo, PoDs) in pair-culture and the petri dishes were also incubated at 28 °C for 9 days. Each mono- and pair-cultures were set five biological repeats.
Metabolite extraction and analysis
Mycelium from the interaction zones (~2 mm wide) of three fungal pair-cultures (TvDs, TvPo, PoDs) and isolates of three mono-cultures (Tv, Po, Ds) were excised with a razor blade and then freeze dried −80 °C. Each sample received was accessioned into the Metabolon Laboratory Information Management System (LIMS) and was assigned by the LIMS a unique identifier that was associated with the original source identifier only. This identifier was used to track all sample handling, tasks, results, etc. The samples (and all derived aliquots) were tracked by the LIMS system. All portions of any sample were automatically assigned their own unique identifiers by the LIMS when a new task was created; the relationship of these samples was also tracked. All samples were maintained at −80 °C until processed. Each sample group were set five biological repeats.
Interaction and isolate samples were prepared using the automated MicroLab STAR® system from Hamilton Company to measure metabolome. A recovery standard was added prior to the first step in the extraction process for QC purposes. To remove protein, dissociate small molecules bound to protein or trapped in the precipitated protein matrix, and to recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 min (Glen Mills GenoGrinder 2000) followed by centrifugation. The resulting extract was divided into four fractions: one for analysis by Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS) with positive ion mode electrospray ionization, one for analysis by UPLC-MS/MS with negative ion mode electrospray ionization, one for analysis by Gas Chromatography-Mass Spectroscopy (GC-MS), and one sample was reserved for backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. Each sample was then frozen and dried under vacuum, then prepared for the appropriate instrument.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
The LC-MS portion of the platform was based on a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo-Finnigan LTQ mass spectrometer operated at nominal mass resolution, which consisted of an electrospray ionization (ESI) source and linear ion-trap (LIT) mass analyzer. The sample extract was dried then reconstituted in acidic or basic LC-compatible solvents, each of which contained 12 or more injection standards at fixed concentrations. One aliquot was analyzed using acidic positive ion-optimized conditions and the other using basic negative ion-optimized conditions in two independent injections using separate dedicated columns (Waters UPLC BEH C18-2.1 × 100 mm, 1.7 µm). Extracts reconstituted in acidic conditions were gradient eluted using water and methanol containing 0.1% formic acid, while the basic extracts, which also used water/methanol, contained 6.5 mM ammonium bicarbonate. The MS analysis alternated between MS and data-dependent MS/MS scans using dynamic exclusion and the scan range was from 80–1000 m/z. Raw data files are archived and extracted as described below.
Gas Chromatography-Mass Spectroscopy (GC-MS)
The samples destined for analysis by GC-MS were dried under vacuum for a minimum of 18 h prior to being derivatized under dried nitrogen using bistrimethyl-silyltrifluoroacetamide. Derivatized samples were separated on a 5% diphenyl/95% dimethyl polysiloxane fused silica column (20 m × 0.18 mm ID; 0.18 um film thickness) with helium as carrier gas and a temperature ramp from 64° to 340 °C in a 17.5 min period. Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization (EI) and operated at unit mass resolving power. The scan range was from 50–750 m/z. Raw data files are archived and extracted as described below.
Data extraction and compound identification
Raw data was extracted, peak-identified and QC processed using Metabolon’s hardware and software. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Metabolon maintains a library based on authenticated standards that contains the retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data) on all molecules present in the library. Furthermore, biochemical identifications are based on three criteria: retention index within a narrow RI window of the proposed identification, accurate mass match to the library +/−0.005 amu, and the MS/MS forward and reverse scores between the experimental data and authentic standards. The MS/MS scores are based on a comparison of the ions present in the experimental spectrum to the ions present in the library spectrum. While there may be similarities between these molecules based on one of these factors, the use of all three data points can be utilized to distinguish and differential metabolites. More than 3300 commercially available purified standard compounds have been acquired and registered into LIMS for distribution to both the LC-MS and GC-MS platforms for determination of their analytical characteristics. Additional mass spectral entries have been created for structurally unnamed metabolites, which have been identified by virtue of their recurrent nature (both chromatographic and mass spectral).These compounds have the potential to be identified by future acquisition of a matching purified standard or by classical structural analysis.
Data analysis
Data were normalized for internal consistency by processing a constant weight per volume of extraction solvent for each sample. Data were scaled to the median value for each compound, then missing values were imputed with the minimum detected value for that compound (both RAW and SCALED IMPUTED data tables are provided in Supplementary Table S1 and S2). Moreover, a variety of curation procedures were carried out to ensure that a high quality data set was made available for statistical analysis and data interpretation. The QC and curation processes were designed to ensure accurate and consistent identification of true chemical entities, and to remove those representing system artifacts, mis-assignments, and background noise. Metabolon data analysts use proprietary visualization and interpretation software to confirm the consistency of peak identification among the various samples. Library matches for each compound were checked for each sample and corrected if necessary.
References
Boddy, L. Interspecific combative interactions between wood-decaying basidiomycetes[J]. FEMS Microbiol Ecol. 31, 185–94 (2000).
Wells, J. M. & Boddy, L. Interspecific carbon exchange and cost of interactions between basidiomycete mycelia in soil and wood[J]. Functional ecology. 16, 153–61 (2002).
Heilmann-Clausen, J. & Boddy, L. Inhibition and stimulation effects in communities of wood decay fungi: exudates from colonized wood influence growth by other species[J]. Microbial ecology. 49(3), 399–406 (2005).
Hu, H. L. et al. Improved enzyme production by co-cultivation of Aspergillus niger and Aspergillus oryzae and with other fungi[J]. International Biodeterioration & Biodegradation. 65(1), 248–52 (2011).
Evans, J. A., Eyre, C. A., Rogers, H. J., Boddy, L. & Müller, C. T. Changes in volatile production during interspecific interactions between four wood rotting fungi growing in artificial media[J]. Fungal Ecology. 1(2–3), 57–68 (2008).
Silar, P. Peroxide accumulation and cell death in filamentous fungi induced by contact with a contestant[J]. Mycological research. 109(2), 137–49 (2005).
Li, Q. et al. Oxidative stress in fungal fermentation processes: the roles of alternative respiration[J]. Biotechnology letters. 33(3), 457–67 (2011).
Ferreira Gregorio, A. P., Da Silva, I. R., Sedarati, M. R. & Hedger, J. N. Changes in production of lignin degrading enzymes during interactions between mycelia of the tropical decomposer basidiomycetes Marasmiellus troyanus and Marasmius pallescens[J]. Mycological research. 110(Pt 2), 161–8 (2006).
Rasmussen, S., Parsons, A. J., Fraser, K., Xue, H. & Newman, J. A. Metabolic profiles of Lolium perenne are differentially affected by nitrogen supply, carbohydrate content, and fungal endophyte infection[J]. Plant physiology. 146(3), 1440–53 (2008).
Misra, B. B., de Armas, E. & Chen, S. Differential metabolomic responses of PAMP-triggered immunity and effector-triggered immunity in Arabidopsis suspension cells[J]. Metabolomics. 12(4) (2016).
Luo, F. et al. Differential metabolic responses of Beauveria bassiana cultured in pupae extracts, root exudates and its interactions with insect and plant[J]. Journal of invertebrate pathology. 130, 154–64 (2015).
Tweeddale, H., Notley-McRobb, L. & Ferenci, T. Assessing the effect of reactive oxygen species on Escherichia coli using a metabolome approach[J]. Redox report: communications in free radical research 4(5), 237–41 (1999).
Gonzalez-Menendez, V. et al. Multicomponent Analysis of the Differential Induction of Secondary Metabolite Profiles in Fungal Endophytes[J]. Molecules. 21(2) (2016).
Farag, M. A., Huhman, D. V., Dixon, R. A. & Sumner, L. W. Metabolomics reveals novel pathways and differential mechanistic and elicitor-specific responses in phenylpropanoid and isoflavonoid biosynthesis in Medicago truncatula cell cultures[J]. Plant physiology. 146(2), 387–402 (2008).
Chatterjee, S., Kuang, Y., Splivallo, R., Chatterjee, P. & Karlovsky, P. Interactions among filamentous fungi Aspergillus niger, Fusarium verticillioides and Clonostachys rosea: fungal biomass, diversity of secreted metabolites and fumonisin production[J]. BMC microbiology. 16(1), 83 (2016).
Peiris, D. et al. Metabolite profiles of interacting mycelial fronts differ for pairings of the wood decay basidiomycete fungus, Stereum hirsutum with its competitors Coprinus micaceus and Coprinus disseminatus[J]. Metabolomics. 4(1), 52–62 (2007).
Bertrand, S. et al. Detection of metabolite induction in fungal co-cultures on solid media by high-throughput differential ultra-high pressure liquid chromatography-time-of-flight mass spectrometry fingerprinting[J]. Journal of chromatography A. 1292, 219–28 (2013).
Glauser, G. et al. Differential Analysis of Mycoalexins in Confrontation Zones of Grapevine Fungal Pathogens by Ultrahigh Pressure Liquid Chromatography/Time-of-Flight Mass Spectrometry and Capillary Nuclear Magnetic Resonance[J]. Journal of agricultural and food chemistry 57, 1127–34 (2009).
Rodriguez Estrada, A. E., Hegeman, A., Kistler, H. C. & May, G. In vitro interactions between Fusarium verticillioides and Ustilago maydis through real-time PCR and metabolic profiling[J]. Fungal genetics and biology: FG & B. 48(9), 874–85 (2011).
Cooney, J. M., Lauren, D. R. & Di, M. M. Impact of competitive fungi on Trichothecene production by Fusarium graminearum[J]. Journal of Agricultural & Food Chemistry. 49(1), 522 (2001).
Bertrand, S. et al. De novo production of metabolites by fungal co-culture of Trichophyton rubrum and Bionectria ochroleuca[J]. Journal of natural products. 76(6), 1157–65 (2013).
Losada, L., Ajayi, O., Frisvad, J. C., Yu, J. & Nierman, W. C. Effect of competition on the production and activity of secondary metabolites in Aspergillus species[J]. Medical mycology. 47(Suppl 1), S88–96 (2009).
Bremer, J. Carnitine-Metabolism and Functions[J]. Physiological Reviews. 63(4), 1421–49 (1983).
Bhambra, G. K., Wang, Z. Y., Soanes, D. M., Wakley, G. E. & Talbot, N. J. Peroxisomal carnitine acetyl transferase is required for elaboration of penetration hyphae during plant infection by Magnaporthe grisea[J]. Molecular microbiology. 61(1), 46–60 (2006).
Steiber, A., Kerner, J. & Hoppel, C. L. Carnitine: a nutritional, biosynthetic, and functional perspective[J]. Molecular aspects of medicine. 25(5-6), 455–73 (2004).
Mahmoud, A. G. Potential Role of Phospholipases in Virulence and Fungal Pathogenesis[J]. Clinical Microbiology Reviews. 13(1), 122–43 (2000).
Zechner, R., Kienesberger, P. C., Haemmerle, G., Zimmermann, R. & Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores[J]. Journal of lipid research. 50(1), 3–21 (2009).
Zablocki, K., Miller, S. P. F., Garcea-perez, A. & Burg, M. B. Accumulation of glycerophosphocholine (GPC) by renal cells: Osmotic regulation of GPC:choline phosphodiesterase[J]. Proc Nati Acad Sci. 88, 7820–4 (1991).
Wang, K. L. C., Li, H. & Ecker, J. R. Ethylene biosynthesis and signaling networks[J]. The Plant Cell. S131–S151 (2002).
Garcia-Sanchez, M. et al. Defence response of tomato seedlings to oxidative stress induced by phenolic compounds from dry olive mill residue[J]. Chemosphere. 89(6), 708–16 (2012).
Ujor, V. C., Monti, M., Peiris, D. G., Clements, M. O. & Hedger, J. N. The mycelial response of the white-rot fungus, Schizophyllum commune to the biocontrol agent, Trichoderma viride[J]. Fungal biology. 116(2), 332–41 (2012).
Wen, J. Q., Huang, F., Liang, W. S. & Liang, H. G. Increase of HCN and β-cyanoalanine synthase activity during ageing of potato tuber slices[J]. Plant Science. 125, 147–51 (1997).
Chamber, J. K., Macdonald, L. E., Sarau, H. M., Ames, R. S. & Freeman, K. A G Protein-coupled Receptor for UDP-glucose[J]. The journal of biological chemistry 275(15), pp. 10767–71 (2000).
Paul, M. Trehalose 6-phosphate[J]. Current opinion in plant biology. 10(3), 303–9 (2007).
Herdeiro, R. S., Pereira, M. D., Panek, A. D. & Eleutherio, E. C. Trehalose protects Saccharomyces cerevisiae from lipid peroxidation during oxidative stress[J]. Biochimica et biophysica acta. 1760(3), 340–6 (2006).
Diwaniyan, S., Sharma, K. K. & Kuhad, R. C. Laccase from an alkalitolerant basidiomycetes Crinipellis sp. RCK-1: production optimization by response surface methodology[J]. Journal of basic microbiology 52(4), 397–407 (2012).
Mayer, A. M. & Staples, R. C. Laccase: new functions for an old enzyme[J]. Phytochemistry. 60, 551–65 (2002).
Piscitelli, A. et al. Induction and Transcriptional Regulation of Laccases in Fungi[J]. Current Genomics. 12, 104–12 (2011).
Zhu, X. & Williamson, P. R. Role of laccase in the biology and virulence of Cryptococcus neoformans[J]. FEMS yeast research. 5(1), 1–10 (2004).
Cho, N. S., Wilkolazka, A. J., Staszczak, M., Cho, H. Y. & Ohga, S. The Role of Laccase from White Rot Fungi to Stress Conditions[J]. Journal- faculty of agriculture Kyushu University. 54, 81–3 (2009).
Flores, C., Vidal, C., Trejo-Hernandez, M. R., Galindo, E. & Serrano-Carreon, L. Selection of Trichoderma strains capable of increasing laccase production by Pleurotus ostreatus and Agaricus bisporus in dual cultures[J]. Journal of applied microbiology. 106(1), 249–57 (2009).
Chi, Y., Hatakka, A. & Maijala, P. Can co-culturing of two white-rot fungi increase lignin degradation and the production of lignin-degrading enzymes?[J]. International Biodeterioration & Biodegradation. 59(1), 32–9 (2007).
Baldrian, P. Increase of laccase activity during interspecific interactions of white-rot fungi[J]. FEMS microbiology ecology. 50(3), 245–53 (2004).
Hiscox, J., Baldrian, P., Rogers, H. J. & Boddy, L. Changes in oxidative enzyme activity during interspecific mycelial interactions involving the white-rot fungus Trametes versicolor[J]. Fungal genetics and biology: FG & B. 47(6), 562–71 (2010).
Kuhar, F., Castiglia, V. & Levin, L. Enhancement of laccase production and malachite green decolorization by co-culturing Ganoderma lucidum and Trametes versicolor in solid-state fermentation[J]. International Biodeterioration & Biodegradation. 104, 238–43 (2015).
Burg, M. B. & Ferraris, J. D. Intracellular organic osmolytes: function and regulation[J]. J Biol Chem. 283(12), 7309–13 (2008).
Franken, J., Kroppenstedt, S., Swiegers, J. H. & Bauer, F. F. Carnitine and carnitine acetyltransferases in the yeast Saccharomyces cerevisiae: a role for carnitine in stress protection[J]. Current genetics. 53(6), 347–60 (2008).
Strijbis, K. & Distel, B. Intracellular acetyl unit transport in fungal carbon metabolism[J]. Eukaryotic cell. 9(12), 1809–15 (2010).
Graham, J. H. Ethylene production by ectomycorrhizal fungi, Fusariuin oxysporum f. sp. pini, and by aseptically synthesized ectomycorrhizae and Fusarium-infected Douglas-fir roots[J]. Can J microbiol. 26, 1340–7 (1980).
Cristescu, S. M., De Martinis, D., te Lintel Hekkert, S., Parker, D. H. & Harren, F. J. M. Ethylene Production by Botrytis cinerea In Vitro and in Tomatoes[J]. Applied and environmental microbiology. 68(11), 5342–50 (2002).
Elbein, A. D., Pan, Y. T., Pastuszak, I. & Carroll, D. New insights on trehalose: a multifunctional molecule[J]. Glycobiology. 13(4), 17R–27R (2003).
Puttikamonkul, S. et al. Trehalose 6-phosphate phosphatase is required for cell wall integrity and fungal virulence but not trehalose biosynthesis in the human fungal pathogen Aspergillus fumigatus[J]. Molecular microbiology. 77(4), 891–911 (2010).
Matsuzaki, F., Shimizu, M. & Wariishi, H. Proteomic and Metabolomic Analyses of the White-Rot Fungus Phanerochaete chrysosporium Exposed to Exogenous Benzoic Acid[J]. Journal of Proteome Research 7, 2342–50 (2008).
Lenardon, M. D., Munro, C. A. & Gow, N. A. Chitin synthesis and fungal pathogenesis[J]. Current opinion in microbiology. 13(4), 416–23 (2010).
Konopka, J. B. N-acetylglucosamine (GlcNAc) functions in cell signaling[J]. Scientifica. 2012 (2012).
Acknowledgements
This study was funded by the National Natural Foundation of China (31400063), Fundamental Research Funds for the Central Universities (No. XDJK2011B009; XDJK2017B030), Research Funds of Scientific Platform and Base Construction (No. cstc2014pt-sy0017), Chongqing Research Program of Social undertaking and livelihood security (No. cstc2016shmszx1176) and The Recruitment Program for Foreign Experts (No. WQ20125500073).
Author information
Authors and Affiliations
Contributions
F.L., Z.Z., L.L. and N.L. performed the experiments and wrote the manuscript, and Y.I., D.X. and N.L. designed the experiments and revised the manuscript. All authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, F., Zhong, Z., Liu, L. et al. Metabolomic differential analysis of interspecific interactions among white rot fungi Trametes versicolor, Dichomitus squalens and Pleurotus ostreatus . Sci Rep 7, 5265 (2017). https://doi.org/10.1038/s41598-017-05669-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05669-3
This article is cited by
-
The regular pattern of metabolite changes in mushroom Inonotus hispidus in different growth periods and exploration of their indicator compounds
Scientific Reports (2022)
-
Melanin production and laccase mediated oxidative stress alleviation during fungal-fungal interaction among basidiomycete fungi
IMA Fungus (2021)
-
Enhanced lignin biodegradation by consortium of white rot fungi: microbial synergistic effects and product mapping
Biotechnology for Biofuels (2021)
-
Critical thresholds of 1-Octen-3-ol shape inter-species Aspergillus interactions modulating the growth and secondary metabolism
Scientific Reports (2020)
-
Comparative transcriptomics and transcriptional regulation analysis of enhanced laccase production induced by co-culture of Pleurotus eryngii var. ferulae with Rhodotorula mucilaginosa
Applied Microbiology and Biotechnology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
