
Abstract
In the photosynthetic apparatus of plants a high proportion of LHCII protein is needed to integrate 50% non-bilayer lipid MGDG into the lamellar thylakoid membrane, but whether and how the stability of the protein is also affected is not known. Here we use single-molecule force spectroscopy to map the stability of LHCII against mechanical unfolding along the polypeptide chain as a function of oligomerization state and lipid composition. Comparing unfolding forces between monomeric and trimeric LHCII demonstrates that the stability does not increase significantly upon trimerization but can mainly be correlated with specific contact sites between adjacent monomers. In contrast, unfolding of trimeric complexes in membranes composed of different thylakoid lipids reveals that the non-bilayer lipid MGDG substantially increases the mechanical stability of LHCII in many segments of the protein compared to other lipids such as DGDG or POPG. We attribute these findings to steric matching of conically formed MGDG and the hourglass shape of trimeric LHCII, thereby extending the role of non-bilayer lipids to the structural stabilization of membrane proteins in addition to the modulation of their folding, conformation and function.
Similar content being viewed by others


Introduction
The major light-harvesting complex (LHCII) found in the chloroplasts of green plants contains more than half of the chlorophylls (Chl) and is the most abundant membrane protein on earth. LHCII plays a key role in photosynthesis by collecting sunlight and efficiently transferring excitation energy to the reaction centers. At the same time, LHCII serves to protect the photosynthetic apparatus from damage of excessive light and to organize the grana structures of the thylakoid membrane1. The 25 kDa LHCII apoprotein contains three transmembrane and two amphiphilic helices, non-covalently binding 8 Chl a, 6 Chl b, 4 carotenoid and 2 lipid molecules in a densely packed arrangement as revealed by crystal structure analysis at 2.5 Å resolution2, 3. LHCII monomers are assembled into trimeric complexes by the end of the greening process of the thylakoid membrane4, although trimerization has been proposed to be reversible upon illumination5. Trimerization prevents LHCII from proteolysis under high light conditions6 and increases its thermal stability7. Inspection of the LHCII crystal structure allowed to localize the contact sites between the monomers2, in part confirming earlier biochemical data8, 9. However, these findings alone provide little information about how much individual structural features contribute to the stability of the LHCII trimer or monomer.
LHCII is embedded in the thylakoid membrane, a membrane with an unusual architecture and composition. The thylakoid membrane contains 80% uncharged glycolipids (30% digalactosyl diacylglycerol (DGDG) and 50% monogalactosyl diacylglycerol (MGDG)), and 20% negatively charged lipids (10% each of phosphatidyldiacylglycerol (PG) and sulfoquinovosyl diacylglycerol (SQDG))10. Although the alkyl chains of DGDG are highly unsaturated (mainly 18:3, 18:3), DGDG still has a cylindrical shape due to sterical compensation by its bulky head group containing 2 galactose rings. By contrast, MGDG comprises only 1 galactose moiety and therefore adopts a conical shape with a tendency to form a hexagonal phase11. On the one hand, DGDG as well as the fraction of negatively charged thylakoid lipids, modelled here by 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), form a lamellar phase (Fig. 1a,b). On the other hand, MGDG is a non-bilayer lipid, and even the thylakoid membrane with a MGDG fraction of 50% is unable to adopt a lamellar structure unless proteins are inserted into the membrane12. The role of the high content of MGDG in the thylakoid is still unclear. Proposed contributions of MGDG include structural aspects like the mediation of spontaneous curvature, the balance of excess membrane area, and specific tasks in protein functionality13,14,15,16,17. The lipid composition of chloroplasts, especially the balance of non-bilayer and bilayer lipids, is very sensitive to various kinds of external stress and a main pathway for plants to deal with changing environmental cues18,19,20,21.

Mechanical unfolding of LHCII in membranes assembled from thylakoid lipids. (a) Chemical structures of model phospholipid POPG and thylakoid glycolipids DGDG and MGDG. (b) Lipid geometries corresponding to (a) and lipid compositions of the membranes applied for unfolding experiments (1–3). The degree of saturation in the alkyl chain region and the size of the head group determine the shape of lipids. Hence, POPG (blue) and DGDG (orange) adopt a cylindrical shape, forming lamellar membranes (1,2). Due to its conical shape the non-bilayer lipid MGDG (red) causes alterations in the lateral pressure profile upon incorporation into lamellar membranes23, 24; here: lipid ratio of DGDG / MGDG = 2:1 (3). (c) Schematic representation of SMFS with LHCII embedded in lipid bilayers with compositions according to (b). Site-directed unfolding of LHCII is achieved by covalently attaching a gold-coated AFM tip to a cysteine motif (yellow) at the amino-terminus N of the polypeptide. Retraction of the tip applies a pulling force F to LHCII monomers, which get extracted from the membrane out of their trimeric assembly (transparent presentation), thereby inducing stepwise unfolding of the protein; gray: polypeptide, dark grey: pigments (obtained with Chimera and PDB ID code 2BHW). (d) Exemplary force-distance curve recorded during the unfolding process described in (c). Each force peak, representing an unfolding event of LHCII, is fitted with the WLC model (yellow) which provides information about the positions of the stabilized domains (contour lengths) and the forces needed to overcome these barriers along the LHCII polypeptide.
Another intriguing feature of the thylakoid membrane is its high protein density. 70–80% of the membrane area is occupied by proteins, with LHCII comprising more than 70% of the proteins in the grana structures22. LHCII in particular was found to be essential for the integration of the non-bilayer lipid MGDG in high quantities into the lamellar thylakoid membrane12. Conversely, the presence of non-bilayer lipids in lamellar membranes is known to cause alterations in the lateral pressure profile23, 24, which in turn can influence the structure and function of transmembrane proteins like LHCII25,26,27. Based on energy transfer experiments in liposomes it has been hypothesized that lateral pressure exerted by MGDG facilitates the structural association of LHCII and photosystem II28. Spectroscopic measurements revealed that the structure of LHCII is sensitive to the molecular environment surrounding the protein29, 30, thus one might expect MGDG to have a notable impact on the structure and stability of LHCII. However, the presence of MGDG in liposomes has not been found to influence the thermal stability of LHCII trimers significantly31. In this context, new methodical approaches are required to shed light on a potential effect of lateral pressure stabilizing LHCII.
Single-molecule force spectroscopy (SMFS) is an excellent technique to study the stability of individual domains of a transmembrane protein by measuring the force needed to unfold them, while pulling them out of the membrane32,33,34,35,36,37,38,39,40,41,42. Whereas thermal denaturation experiments address the overall protein structure, SMFS provides information about subdomains by sequentially unfolding them. SMFS has also been demonstrated to be a suitable technique to characterize lipid-protein interactions36, 37. Recently, Scheuring and coworkers were able to apply SMFS to bacterial LH2 elucidating the free energy of oligomerization38. In the present work, SMFS has been used to study the mechanical stability of LHCII inserted in supported membranes as a function of lipid composition and oligomerization state. This allowed us to analyze the relationship between the structural stability of the protein and the physicochemical properties of its membrane environment, with particular emphasis on lateral pressure caused by MGDG (Fig. 1b,c). Furthermore, we investigated both trimeric and monomeric LHCII in order to localize the regions specifically contributing to trimer stability. Our findings indicate that MGDG matches the hourglass shape of LHCII thereby providing stability against unfolding.
Results
Mechanical unfolding of LHCII
For atomic force microscopy (AFM) based unfolding experiments, recombinant LHCII was reconstituted into preformed liposomes, which were spread on a flat mica surface. Topographical AFM images of the supported lipid bilayers revealed connected bilayer patches (Supplementary Figure 1). The glycolipids (pure DGDG or DGDG/MGDG mixtures) showed a surface coverage of approximately 30%, while POPG had an increased surface coverage of 60%. Proteins were visible as elevated objects either ~1 nm or ~1.5 nm higher than the bilayer, approximately corresponding to lumenal and stromal protrusions of LHCII, respectively3, with diameters on the order of 10 to 100 nm. Apparent differences in protein organization between different lipid compositions were not observed. A Cys3 cluster near the N-terminus of the LHCII apoprotein allowed stable and selective attachment to gold-coated AFM cantilever tips suitable for SMFS experiments (Fig. 1c). Insertion of recombinant LHCII in artificial membranes presumably results in random up-down orientation; however, the N-proximal Cys3 attachment site permits to select those complexes for unfolding that expose their stromal surface32. The cantilever tip was brought into contact with the membrane surface and was then retracted to unfold the peptide chain as indicated by the saw tooth signature, characteristic for protein unfolding (Fig. 1d), with each force peak representing unfolding of an individual segment of the polypeptide chain34, 35. Only force-distance curves showing unfolding of the complete peptide chain were taken into account for further analysis as indicated by a force peak in a 10 nm interval around the contour length of the LHCII monomer estimated to be 88 nm. Each individual unfolding peak was fitted using the worm-like chain (WLC) model assuming a persistence length of 0.4 nm43, 44 to assess the contour length l C of the unfolded segment and the corresponding unfolding forces (Fig. 1d).
Frequent observation of a particular unfolding peak is usually linked to stable structural elements of the mechanically challenged protein that need not necessarily be identical with secondary-structure components33. Interestingly, the histogram of all contour lengths obtained by the WLC fits (Fig. 2a, shown as histograms on the x-axes) revealed a very broad distribution with unfolding events almost evenly spread over the full length of the peptide chain. In contrast to unfolding experiments on numerous other proteins distinct peaks were absent (as illustrated in Supplementary Figure 2)32, 36, 37, 39,40,41,42. The broad distribution might be attributed to the complex structure of LHCII forming a superhelix in combination with a high number of bound auxiliary pigments (see Fig. 1c)2, 3, 45. In contrast to the contour length histograms, the corresponding unfolding force histograms are less broad and show a clear preference for unfolding forces around 50 pN dependent on the lipid matrix and the oligomerization state (Fig. 2a, shown as histograms on the y-axes).

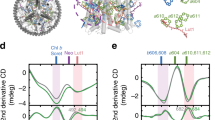
Unfolding forces of LHCII depend on trimerization and the lipid matrix. (a) Multivariate kernel density estimate of the unfolding force as a function of the contour length for LHCII monomers in POPG (top right) and LHCII trimers in POPG (top left), pure DGDG (bottom left) and a mixture of DGDG and MGDG (bottom right). The dots mark the most likely unfolding forces for a given contour length, while the diameter of the dots represents their relative probability of at least 40% in relation to the most probable unfolding event. The histograms (grey) at the axes show the individual underlying distributions of unfolding forces and contour lengths. (b) Most likely unfolding force as a function of the contour length (obtained from the dots in (a)) as a function of oligomerization state (top) and lipid mixture (bottom). Helices (H1-5) are indicated as shaded gray areas. The color shaded areas indicate the standard error of the most likely unfolding force. The trimer shows enhanced stabilization compared to the monomer in distinct domains of the protein, indicated by numbers 1 to 5. Both glycolipids provide additional stability for trimeric LHCII at helix 4, while the addition of MGDG strongly increases the unfolding forces in many segments along the LHCII polypeptide.
The common procedure for analysis of unfolding force curves is to map recurring unfolding peaks to stable elements and categorize every detected unfolding event accordingly. Due to the broad distribution of detected contour lengths such an analysis would be very unreliable and prone to false assignment of peaks to structural elements. Therefore, in order to circumvent categorization of unfolding events to structural elements, we estimated the probability to find a certain unfolding force at a given contour length as obtained from the WLC fits by using a multivariate kernel density estimate (Fig. 2a). Analysis of this “stability map” then allowed us to relate the mechanical response of the polypeptide to the stabilization of structural elements depending on different intrinsic and environmental conditions (see below). In order to compare unfolding forces between different experiments we considered the most likely unfolding force as a function of the contour length (colored dots in Fig. 2a,b). To find the most prominent contributions to protein stability, only unfolding events with a likelihood of at least 40% of the most probable unfolding event were selected. This also sorts out unfolding events with unfolding forces above 100 pN, which we attribute to simultaneous unfolding of more than one monomer attached to the cantilever in parallel.
Impact of LHCII trimerization
LHCII monomers inserted in PG-containing membranes tend to trimerize spontaneously46; therefore, in order to address the impact of trimerization on the mechanical stability of LHCII, a LHCII variant was used with two point mutations in its trimerization motif (positions 16 and 17) that render the protein incapable of trimerization, as described earlier8. For comparing LHCII monomers and trimers, the complexes were inserted into membranes consisting of POPG exclusively. The distribution of unfolding forces (Fig. 2a, shown as histograms on the y-axes) reveals that the most frequent unfolding force is increased by around 10 pN from monomers to trimers, demonstrating a slightly stabilizing effect of trimerization on LHCII. This is in agreement with previous thermal denaturation experiments revealing an increased stability of the trimer7.
Comparing the most likely unfolding force at each contour length (colored dots in Fig. 2a top left, top right) for monomeric LHCII and a single LHCII polypeptide unfolded out of the trimer reveals selective stabilization in the trimer. In some contour length intervals unfolding forces in the trimer are similar to those in the monomer, whereas in other parts of the polypeptide -although most of the disparities are not significant unless stated otherwise- the forces are higher for trimers than for monomers (Fig. 2b top). While the N-terminal hydrophilic domain at contour lengths of l C ≈ 10 nm shows no detectable unfolding events for the monomer, there is a high probability to observe unfolding events for trimeric LHCII (Fig. 2b top, indicated by 1). Along those lines, unfolding forces of the trimer were generally higher from l C ≈ 27 nm to l C ≈ 34 nm (∆F ≈ 8 pN), at l C ≈ 47 nm (∆F ≈ 7 pN), l C ≈ 77 nm (∆F ≈ 12 pN), and for the C-terminus at l C > 82 nm (∆F ≈ 8 pN, same figure, indicated by 2, 3, 4 and 5, respectively). Interestingly, in two segments of the protein, unfolding forces were slightly higher for the monomer than for the trimer (from l C ≈ 15 nm to l C ≈ 25 nm and from l C ≈ 65 nm to l C ≈ 70 nm).
Stabilization of the trimer compared to the monomer, as indicated by higher unfolding forces, can be linked to interactions in distinct protein domains facilitating LHCII trimerization. Both the N-terminus and the C-terminus have previously been described to be important for trimerization based on mutation analysis8, 9 and structural data2, which is mirrored here in the significant stabilization at l C ≈ 10 nm (peak 1) and the stabilization at l C > 82 nm (peak 5). The key trimerization motif has been identified as WYGPDR at aa17–aa22 (l C = 6–8 nm)8. However, detaching the cantilever tip from the membrane surface causes adhesion peaks at the beginning of each force-distance curve that may obscure unfolding events in the very proximal segment of the N-terminus. Hence, the precision in determining contour lengths in this domain is not sufficient to be sure that the peak at l C ≈ 10 nm is in fact due to the trimerization motif.
A critical factor for trimerization is the binding of pigments and lipids. Liu et al. pointed out an important role of helix 3 by coordinating several Chl b molecules, mediating hydrophobic interactions between adjacent monomers2. Here, we may observe a contribution to trimer stability by one chlorophyll in particular: Chl8 (nomenclature according to Standfuß et al.3) is bound to two different monomers via coordination of the central Mg2+ by His212 (l C = 77 nm, corresponding to peak 4 within helix 5) of one monomer and hydrophobic interaction of the phytol tail with Trp128 (l C = 47 nm, corresponding to peak 3 at the beginning of helix 3) of the other monomer (Supplementary Figure 3). In addition, Trp222 (l C = 81 nm) has been described to be a sensitive site for trimer formation and stability9, 45, which may explain an enhanced stabilizing effect at peak 4 compared to the other regions.
Liu et al. identified Chl4 and Chl5 bound via Glu65 (l C = 24 nm) and His68 (l C = 25 nm), respectively, to be crucial for the trimer structure2. However, unfolding forces of trimeric and monomeric LHCII show little differences at these contour lengths. Strong intramolecular interactions, like the salt bridge at the helix cross between helix 1 at Arg70 (l C = 25 nm) and helix 345, may be dominating in this segment, obscuring the effect of neighboring monomers as discussed by Sapra et al. for bacteriorhodopsin assemblies47.
There is no straightforward assignment of the significant stabilization between l C ≈ 27 nm and l C ≈ 34 nm (region 2) to the structure of LHCII. This contour length interval corresponds to the lumenal end of the transmembrane helix 1 and the amphiphilic helix 2. However, Liu et al. found a highly reduced degree of trimerization after an exchange of W97 to alanine (l C = 35 nm) at the beginning of helix 248, representing a binding site for lutein 245. This pigment has been suggested to assume a trimer specific conformation7, indicating that lutein 2 and hence W97 might contribute to the stability of trimeric LHCII to a certain extent as reflected by our data.
It seems surprising that we observed one position at the N-terminal domain close to helix 1 and one at the beginning of helix 4 with higher unfolding forces for the monomer than for the trimer. The PG molecule bound to LHCII via Tyr44 (l C = 16 nm) and Lys182 (l C = 66 nm)3 has been identified to be a major factor in trimerization49, 50. Moreover, there are indications that the monomeric complex alone is not able to bind PG50. While PG facilitates trimer formation, electrostatic repulsion between the negatively charged head groups of the bound lipid and the membrane lipids is energetically unfavorable and might destabilize the trimeric protein at these positions in a POPG matrix.
Effect of lipid matrix
To assess the impact of the lipid matrix on the mechanical stability of LHCII we performed unfolding experiments with trimeric LHCII in three different lipid matrices: pure POPG, pure DGDG and a mixture of DGDG and MGDG (2:1). Lipid mixtures containing more than roughly 30% MGDG no longer form lipid vesicles51, therefore we abstained from using higher percentages of MGDG, expecting that if the lateral membrane pressure has an effect on LHCII, this is seen at 30% MGDG. Since neither of the bilayer lipids POPG and DGDG affects the lateral pressure profile of a lamellar membrane, differences in unfolding forces between these lipid environments can primarily be attributed to the charge of the lipid head groups (Fig. 1a,b). Alterations in protein stability upon addition of 30% non-bilayer lipid MGDG to DGDG membranes provide information about the impact of the modified lateral pressure profile24, while the charge of the membrane and the chemical properties of the head groups remain unaffected.
The distribution of unfolding forces varies only little between the two cylindrical lipids POPG and DGDG (Fig. 2a top left, bottom left; shown on the y-axes). When considering the most likely unfolding force for every contour length, the N-terminus around l C ≈ 10 nm and the region from the end of the stromal loop to the beginning of helix 4 around l C ≈ 60 nm are slightly more stable (∆F ≈ 10 pN) in DGDG compared to POPG (Fig. 2b bottom). The lumenal side of helix 4 from l C ≈ 70 nm to l C ≈ 75 nm is greatly stabilized (∆F ≈ 25 pN) in both glycolipid matrices (DGDG and DGDG + MGDG). This narrow peak points to a protein domain interacting with lipid head groups on the lumenal side of helix 4.
Adding MGDG to the DGDG membrane significantly raises the overall occurrence of unfolding forces above 70 pN (Fig. 2a bottom left, bottom right; shown on the y-axes) and strongly enhances the unfolding forces in several segments of the protein (Fig. 2b bottom). Unfolding forces are increased by up to 20 pN from l C ≈ 6 nm to l C ≈ 12 nm close to the N-terminus, from l C ≈ 24 nm to l C ≈ 32 nm within helix 1, from l C ≈ 34 nm to l C ≈ 46 nm comprising helix 2, the lumenal loop and part of helix 3, and from l C ≈ 50 nm to l C ≈ 54 nm at the stromal side of helix 3. In contrast to the protein stabilization due to trimer formation, the stabilized regions along the peptide chain due to the surrounding lipids are rather broad extending over up to 30 amino acids, indicating a regional stabilization rather than local interactions between individual amino acids.
Discussion
Unfolding monomers and trimers reveals a connection between structure and stability of LHCII, despite the structural intricacy of the pigment-protein complex
Site-directed and complete unfolding of recombinant LHCII by SMFS was facilitated by linking the cantilever to an N-proximally introduced Cys3 cluster32. In contrast to other transmembrane proteins studied by SMFS32, 36, 37, 39,40,41,42, unfolding of different LHCII peptides does not always follow the same unfolding trajectory as indicated by the notable absence of clear peaks in the unfolding contour length histograms (Fig. 2a; see also Supplementary Figure 2). Since the broad distribution of contour lengths could in principle also arise from careless data evaluation or experimental errors, a number of quality control measures were taken to minimize the impact of those effects. To this end, the protein was site-specifically linked to the cantilever at the amino-terminus, with the aim to reduce the impact of nonspecific adhesion and random pulling from the stromal or the lumenal side during the unfolding experiments. Notably we do not observe any symmetry in the most frequent unfolding forces along the LHCII polypeptide, which would indicate stochastic pulling from either the N- or the C-terminus (Fig. 2b). Nonspecific attachment of the cantilever to the stromal or lumenal loop leads to force-distance curves with unfolding events at tip-sample separations much shorter than 78 nm, which were excluded from analysis. Similarly, force-distance curves lacking the characteristic WLC shape of protein unfolding were not analyzed. Additionally, the AFM setup used here has low instrumental noise with a standard deviation of 4 pN due to rigorous screening against external acoustic sources.
Therefore, we ascribe our findings to the intricate structure of the protein. The two tilted helices 1 and 4 are interlocked by two salt bridges, forming a super-secondary structure3, 45 (see Fig. 1c). In addition, the high number of pigments coordinated by LHCII (18 per monomer) leads to added links between different segments of the polypeptide, each capable to stabilize the protein structure. Considering more than 20 known pigment binding sites45 and the contour length of the LHCII monomer of 88 nm, the average distance along the peptide chain between two pigment binding sites is around 4 nm. Assuming an experimental error for the determination of the contour length of an unfolded segment of 2 nm (as a lower bound), it becomes clear that adjacent pigment binding sites cannot be resolved with the current experimental capabilities according to this simple calculation.
By analyzing the most likely unfolding force at every position along the polypeptide chain we avoided to focus on selected structural elements in the protein originating from the frequency of contour lengths only. We used this approach to analyze unfolding curves of the LHCII trimer and an LHCII point mutant incapable of trimerization, revealing localized differences in unfolding forces between monomeric and trimeric LHCII which can be directly linked to the protein structure. Although not at all positions along the contour of the protein statistically secured, we find this approach for data analysis reasonable as the disparities in unfolding forces due to trimerization can be largely correlated with previously published structural and biochemical data. We therefore relied on the method to further explore how the stability of LHCII is modulated by the lipid matrix surrounding the protein. In contrast to the rather subtle impact of oligomerization on the unfolding forces of LHCII we found that the lipid matrix has a substantial influence on protein stability as discussed below.
Lateral pressure by MGDG greatly stabilizes trimeric LHCII due to its hourglass shape
The regions of the LHCII trimer that are stabilized in DGDG compared to POPG membranes are mainly located in the extra-membrane domains of the protein in plane with the lipid head groups, suggesting a destabilizing impact of the negatively charged phosphate group and/or stabilizing effects by the galactose moieties (Fig. 2b bottom). The N-terminal region from l C ≈ 11 nm to l C ≈ 13 nm is rich in polar amino acids (Ser29, Ser32, Ser34, Tyr35 and Thr37) capable of forming hydrogen bonds with the sugar residues, which may lead to an increased stability by both glycolipids (MGDG and DGDG) in this segment of the polypeptide. Negatively charged amino acids (Asp162, Asp168, Asp169 and Glu171) in the stromal loop from l C ≈ 59 nm to l C ≈ 62 nm may generate repulsive ionic interactions with phosphate head groups and thus destabilize the protein at the end of the stromal hydrophilic domain in a POPG matrix. In addition, repulsive forces due to the bound PG-molecule at l C ≈ 66 nm may extend the destabilization of this segment to the stromal side of helix 4, similar to the statement made above for the comparison of the monomer and the trimer3. The greatly enhanced stabilization of LHCII against unfolding by both glycolipid mixtures at the lumenal side of helix 4 around l C ≈ 75 nm probably arises from hydrogen bonds between the corresponding polar amino acids (Gln197, Thr201 and Lys203) and the galactose rings of either DGDG or MGDG, as the presence of positively charged Lys203 renders a repulsive ionic interaction with the POPG head group highly unlikely. However, besides the vast chemical differences between POPG and DGDG, especially considering the different charges, disparities in unfolding force are minuscule.
By contrast, large differences are seen in unfolding forces between the DGDG/MGDG mixture and pure DGDG, despite the similarities between DGDG and MGDG with regard to charge and functional groups. This rules out specific or electrostatic interactions as a reason for the mechanical stabilization. The stabilizing effect of MGDG is not restricted across the membrane plane since it includes transmembrane as well as extra-membrane domains of LHCII (shown as schematic representation in Fig. 3a). A view on the stromal and lumenal faces of LHCII reveals that the stabilization is confined to the periphery of the trimer where it is in direct contact with the lipid matrix (Fig. 3b). We therefore attribute the stabilization to steric interactions between the hourglass shape of trimeric LHCII and the conical shape of MGDG (Fig. 3c), pointing to bulk physical properties of the lipid since it is not part of the LHCII structure2. Our results are in accordance with recent simulations demonstrating that concavely formed proteins are energetically favored in membrane domains exhibiting negative curvature stress52, which is a measure for alterations in the lateral membrane pressure profile induced by non-bilayer lipids such as MGDG24, 26. Yang et al. monitored the dissociation of LHCII trimers into monomers as a function of thylakoid lipid composition, showing only a slight effect of MGDG31. This is in agreement with our data as the impact of MGDG is particularly intense in the periphery of LHCII (Fig. 3b), whereas trimerization is mainly based on pigment mediated interactions in the hydrophobic core2.

Lateral pressure by MGDG stabilizes LHCII in a structure dependent manner. (a) Schematic presentation of the LHCII monomer structure with degree of stabilization by MGDG (red color gradient) derived from Fig. 2b. Stabilization comprises domains in the transmembrane (helices H1 and H3) and extra-membrane domains (N-terminus, H2 and lumenal loop) of the protein; small arrows indicate contour length positions along the LHCII polypeptide. (b) Hotspots of MGDG mediated stabilization (red) in the crystal structure of LHCII from lumenal and stromal view corresponding to the highest intensities in (a) and l C ± 1 nm of the peak centers in Fig. 2b respectively. The effect of MGDG is the strongest in the periphery of the protein. Within a trimer, the monomers are represented by the surface structure, the tertiary structure (helices H1-H5 indicated) with and without pigments; gray or white: polypeptide, dark gray: pigments. (c) Steric interplay between trimeric LHCII and MGDG. Stabilization of LHCII is accomplished as the concave-like surface structure of trimeric LHCII (emphasized with dashed lines) matches the conical shape of MGDG (red); colors (gray: polypeptide, dark gray: pigments) highlight one LHCII monomer within the trimeric assembly. Structures obtained with Chimera and PDB ID code 2BHW.
A lot of attention has been paid to how non-bilayer lipids, particularly phosphatidylethanolamine (PE) representing up to 38% of the inner mitochondrial membrane lipids53, modulate the folding, conformation and functionality of integral membrane proteins26, 27 (see also references therein). However, only little is known about their impact on the stability of transmembrane proteins, even though complementary shapes of transmembrane proteins and lipids appear to be a widespread phenomenon. The concavely formed ion channel gramicidin and the non-bilayer lipid lysophosphatidylcholine as well as the potassium channel KcsA and PE are prominent examples for the proposed steric interplay54, 55. Unfolding of LHCII via SMFS allowed us to observe a stabilizing effect of complementary lipid and protein shapes (as illustrated in Fig. 3c), thereby displaying the impact of the lateral pressure profile over the whole membrane cross section (Fig. 3a). While LHCII is needed to stabilize MGDG in a lamellar phase, our data clearly show that vice versa MGDG increases the mechanical stability of LHCII. Our results will contribute to the ongoing discussion of the role on non-bilayer lipids in many biological membranes.
Methods
Preparation of recombinant LHCII
The LHCII variants used in this study were derivatives of Lhcb1*2 (AB80) gene from pea (Pisum sativum)56 containing a C-terminal hexahistidine (His6) tag and having the native cysteine at position 79 replaced with serine. In order to achieve site-specific unfolding a cysteine motif (Cys-Cys-Cys) was added at the N-terminus of the protein by using the Phusion site-directed mutagenesis kit (Thermo Scientific). This version also served as a template to construct the LHCII variant WY16,17AV8 by using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies), which was applied for the monomer studies. The genes were inserted in the pDS12-RBSII vector and overexpressed overnight in Escherichia coli strain JM101, followed by isolation of the apoprotein according to established protocols57. The apoprotein was reconstituted to monomeric LHCII with total pigment extract from pea thylakoids58 by the detergent exchange method59. His6 tag mediated trimerization of the monomers was performed on Ni2+-sepharose columns60, followed by purification via ultracentrifugation through a sucrose density gradient as described earlier61, but using a different buffer (500 mM sucrose, 10 mM Tris-HCl pH 7.5, 5 mM TCEP, 0.05% (w/v) Triton X-100). In case of the variant WY16,17AV the trimerization step was omitted and the reconstitution mix was directly loaded on the sucrose gradient containing a higher concentration of Triton X-100 (0.1%). The bands corresponding to LHCII trimers or monomers (in case of WY16,17AV) respectively were collected and checked for correct assembly via CD spectroscopy on a J-810 spectropolarimeter (Jasco) in the visible range, with trimeric LHCII exhibiting a characteristic negative peak at 474 nm61. For insertion into preformed liposomes the buffer was exchanged to AFM buffer (50 mM NaCl, 10 mM Tris-HCl pH 7.5, 5 mM TCEP) using Amicon 30-kDa centrifugal filters, while the suspension still contained Triton X-100.
Reconstitution of LHCII into liposomes
Lipid aliquots of pure POPG (purchased from Avanti Polar Lipids), pure DGDG or a mixture of DGDG and MGDG (each lipid purchased from Lipid Products) at a molar ratio of 2:1 were dissolved in chloroform. The chloroform was slowly removed in a rotary evaporator by which a thin lipid film was formed on the inner wall of a glass flask. After complete removal of the solvent under high vacuum at 40 °C for 1 h, the lipid film was hydrated by rigorous mixing (3 × 30 s at 60 °C, MS2 minishaker, IKA) in AFM buffer at a total lipid concentration of 1.5 mM to produce preformed liposomes. For MGDG containing samples the mixing step was repeated several times until no more lipid traces could be detected by eye on the bottom of the flask. Sonication in a tip sonicator (Vibra cell, Sonics & Materials) for 4 min, followed by 3 freeze-thaw cycles, yielded large unilamellar vesicles. The vesicle suspension was then extruded 21 times (LipoFast-Basic, Avestin) through a polycarbonate membrane (pore diameter 100 nm) and mixed with Triton X-100 to a final concentration of 0.05% (w/v). Reconstitution of recombinant LHCII into preformed liposomes was performed according to established protocols31, but with some modifications: the protein suspension was added dropwise under continuous mixing at 4 °C in the dark to a molar lipid/protein ratio of 500–1000, and subsequently polystyrene beads (Bio-Beads SM-2; Bio-RAD) at 30 mg/ml were added. In order to remove Triton X-100 the mixture was incubated overnight under constant rotating at 4 °C in the dark, and the supernatant was removed to a new tube containing fresh Bio-Beads, followed by incubation for 1 h. The last step was repeated, and finally the supernatant was collected. The inserted LHCII trimers or monomers were checked for integrity on a FluoroMax-2 spectrometer (Horiba Scientific) as described earlier60.
SMFS experiments
Prior to the SMFS experiments, proteoliposomes were extruded 21 times (LipoFast-Basic, Avestin) through a polycarbonate membrane (pore diameter 100 nm). CaCl2 solution (100 mM) was added to a final concentration of 5 mM and the suspension (60 µl) was incubated for 10 min on freshly cleaved mica, leading to surface supported bilayers containing LHCII proteins (Supplementary Figure 1). Nonadsorbed vesicles were removed by intensive rinsing (4 × 1 ml) with AFM buffer. SMFS measurements were conducted in AFM buffer at room temperature using a commercial atomic force microscope (MFP-3D Infinity, Oxford Instruments Asylum Research) and gold-coated silicon nitride cantilevers (Biolever, Olympus). Each cantilever was calibrated with the thermal noise method yielding spring constants around 6 pN/nm. The cantilever was moved toward the surface at 500 nm/s until a load force of 100 pN was reached. The tip was kept in contact with the membrane for 1 s to allow for binding of the Cys3-motif at the N-terminus of LHCII to the gold-coated cantilever tip. The cantilever was then retracted at 1000 nm/s to unfold the peptide, giving rise to a force-distance curve. For each sample only a single force-distance curve was collected within an area of 1 µm2 and each area of the supported bilayer was only scanned once. Roughly 10% of the curves showed force peaks corresponding to unfolding events of the protein, of which ~7% exhibited a force peak corresponding to a contour length of 88 ± 10 nm indicating full unfolding of the LHCII polypeptide. Only the latter were selected for further analysis, thus more than 25,000 force-distance curves were recorded for each experimental condition to obtain a sufficient number of curves showing full unfolding (monomer in POPG: n = 193 out of ~30,000 curves; trimer in POPG: n = 328 out of ~50,000 curves; trimer in DGDG: n = 262 out of ~50,000 curves; trimer in DGDG/MGDG: n = 195 out of ~25,000 curves). The SMFS measurements were repeated at least 3 times for each experimental condition.
SMFS data analysis
Only force-distance curves showing the saw tooth signature characteristic for protein unfolding with an unfolding peak corresponding to a contour length of 88 ± 10 nm were selected for further analysis. Each individual unfolding peak was fitted using a WLC model43, 44:
with the force \(F(x)\,\) at tip-sample separation \(\,x\), the Boltzmann constant \({k}_{B}\), the persistence length \(b=4\,\AA \), the contour length of the unfolded peptide \({l}_{C}\,\) and the temperature \(T=298\,\text{K}\). Additionally, the peak force \({F}_{max}\) of each unfolding peak was recorded. To estimate the distribution of the value pairs of \({l}_{C}\,\) and \({F}_{max}\) for all recorded unfolding events, we calculated a multivariate kernel density estimate using the approximated experimental uncertainty \(\sigma ({l}_{C})=2\,\mathrm{nm}\) and \(\sigma (F)=12\,\mathrm{pN}\) as kernel bandwidths, as estimated from the standard deviation of the measured force during the baseline of the force-distance curve, i.e. recorded far away from the surface. The uncertainty of the contour length was calculated from the uncertainty of the measured force and the spring constant. This gives us an unfolding force distribution at any given contour length. The maxima of these distributions give us the most likely unfolding force \({F}^{\ast }\) as a function of the contour length. The most likely unfolding force was evaluated at discrete contour lengths with a fixed distance between two points Δl C = 1.25 nm. As an estimate of the uncertainty of the most likely unfolding force we calculated the standard error of the most likely unfolding force \(\sigma ({F}^{\ast })\) for each contour length \({l}_{C}\,\) as \(\sigma ({F}^{\ast })=\sqrt{\frac{{\rm{Var}}({F}_{max})}{N}}\), using the variance \({\rm{Var}}({F}_{max})\) of all measured unfolding forces \({F}_{max}\) in the interval \({l}_{C}\pm \frac{1}{2}{\rm{\Delta }}{l}_{C}\) and the number of data points in that interval \(N\).
Structure representation
The LHCII structure (PDB ID 2BHW)3 representation was created using the UCSF chimera package62.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Horton, P., Wentworth, M. & Ruban, A. V. Control of the light harvesting function of chloroplast membranes: the LHCII-aggregation model for non-photochemical quenching. FEBS Lett 579, 4201–4206 (2005).
Liu, Z. et al. Crystal structure of spinach major light harvesting complex at 2.72 A resolution. Nature 428, 287–292 (2004).
Standfuss, J., van Scheltinga, A. C. T., Lamborghini, M. & Kühlbrandt, W. Mechanisms of photoprotection and nonphotochemical quenching in pea lightharvesting complex at 2.5 A° resolution. EMBO J 24, 919–928 (2005).
Dreyfuss, B. W. & Thornber, J. P. Assembly of the light-harvesting complexes (LHCs) of Photosystem II. Plant Physiol 106, 829–839 (1994).
Garab, G. et al. Light-induced trimer to monomer transition in the main light-harvesting antenna complex of plants: thermo-optic mechanism. Biochemistry 41, 15121–15129 (2002).
Yang, D.-H., Paulsen, H. & Andersson, B. The N-terminal domain of the light-harvesting chlorophyll a/b-binding protein complex (LHCII) is essential for its acclimative proteolysis. FEBS Lett 466, 385–388 (2000).
Wentworth, M., Ruban, A. V. & Horton, P. The functional significance of the monomeric and trimeric States of the photosystem II light harvesting complexes. Biochemistry 43, 501–509 (2004).
Hobe, S., Förster, S., Klingler, J. & Paulsen, H. N-proximal sequence motif in light-harvesting chlorophyll a/b-cinding protein is essential for the trimerization of light-harvesting chlorophyll a/b complex. Biochemistry 34, 10224–10228 (1995).
Kuttkat, A., Hartmann, A., Hobe, S. & Paulsen, H. The C-terminal domain of light-harvesting chlorophyll-a/b-binding protein is involved in the stabilisation of trimeric light-harvesting complex. Eur J Biochem 242, 288–292 (1996).
Douce, R. & Joyard, J. Biochemistry and function of the plastid envelope. Annu Rev Cell Biol 6, 173–216 (1990).
Hoyo, J., Guaus, E. & Torrent-Burgués, J. Monogalactosyldiacylglycerol and digalactosyldiacylglycerol role, physical states, applications and biomimetic monolayer films. Eur Phys J E 39 (2016).
Simidjiev, I. et al. Self-assembly of large, ordered lamellae from non-bilayer lipids and integral membrane proteins in vitro. Proc Natl Acad Sci USA 97, 1473–1476 (2000).
Jarvis, P. et al. Galactolipid deficiency and abnormal chloroplast development in the Arabidopsis MGD synthase 1 mutant. Proc Natl Acad Sci USA 97, 8175–8179 (2000).
Pali, T., Garab, G., Horvath, L. I. & Kota, Z. Functional significance of the lipid-protein interface in photosynthetic membranes. Cell Mol Life Sci 60, 1591–1606 (2003).
Deme, B., Cataye, C., Block, M. A., Marechal, E. & Jouhet, J. Contribution of galactoglycerolipids to the 3-dimensional architecture of thylakoids. FASEB J 28, 3373–3383 (2014).
Loll, B., Kern, J., Saenger, W., Zouni, A. & Biesiadka, J. Lipids in photosystem II: interactions with protein and cofactors. Biochim Biophys Acta 1767, 509–519 (2007).
Jones, M. R. Lipids in photosynthetic reaction centres: structural roles and functional holes. Prog Lipid Res 46, 56–87 (2007).
Laval-Martin, D. & Troton, D. Chilling-resistance of photosynthetic performances in diuron-adapted Euglena gracilis. Plant Sci 72, 213–222 (1990).
Harwood, J. L. Involvement of chloroplast lipids in the reaction of plants submitted to stress. Lipids in Photosynthesis: Structure, Function and Genetics (Kluwer Academic Publishers, Dordrecht, The Netherlands), 290–300 (1998).
Gigon, A., Matos, A.-R., Laffray, D., Zuily-Fodil, Y. & Pham-Thi, A.-T. Effect of drought stress on lipid metabolism in the leaves of Arabidopsis thaliana (Ecotype Columbia). Ann Bot 94, 345–351 (2004).
Dankov, K. G. et al. LHCII organization and thylakoid lipids affect the sensitivity of the photosynthetic apparatus to high-light treatment. Plant Physiol Bioch 49, 629–635 (2011).
Kirchhoff, H., Tremmel, I., Haase, W. & Kubitscheck, U. Supramolecular photosystem II organization in grana thylakoid membranes: evidence for a structured arrangement. Biochemistry 43, 9204–9213 (2004).
Ding, W., Palaiokostas, M., Wang, W. & Orsi, M. Effects of lipid composition on bilayer membranes quantified by all-atom molecular dynamics. J Phys Chem B 119, 15263–15274 (2015).
Baczynski, K., Markiewicz, M. & Pasenkiewicz-Gierula, M. A computer model of a polyunsaturated monogalactolipid bilayer. Biochimie 118, 129–140 (2015).
de Kruijff, B. Lipids beyond the bilayer. Nature 386, 129–130 (1997).
van den Brink-van der Laan, E., Antoinette Killian, J. & de Kruijff, B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim Biophys Acta 1666, 275–288 (2004).
Brown, M. F. Curvature forces in membrane lipid–protein interactions. Biochemistry 51, 9782–9795 (2012).
Zhou, F. et al. Effect of monogalactosyldiacylglycerol on the interaction between photosystem II core complex and its antenna complexes in liposomes of thylakoid lipids. Photosynth Res 99, 185–193 (2009).
Pandit, A. et al. Assembly of the major light-harvesting complex II in lipid nanodiscs. Biophys J 101, 2507–2515 (2011).
Akhtar, P. et al. Pigment interactions in light-harvesting complex II in different molecular environments. J Biol Chem 290, 4877–4886 (2015).
Yang, C., Boggasch, S., Haase, W. & Paulsen, H. Thermal stability of trimeric light-harvesting chlorophyll a/b complex (LHCIIb) in liposomes of thylakoid lipids. Biochim Biophys Acta 1757, 1642–1648 (2006).
Oesterhelt, F. et al. Unfolding pathways of individual bacteriorhodopsins. Science 288, 143–146 (2000).
Kedrov, A., Janovjak, H., Sapra, K. T. & Müller, D. J. Deciphering molecular interactions of native membrane proteins by single-molecule force spectroscopy. Annu Rev Biophys Biomol Struct 36, 233–260 (2007).
Engel, A. & Gaub, H. E. Structure and mechanics of membrane proteins. Annu Rev Biochem 77, 127–148 (2008).
Bippes, C. A. & Müller, D. J. High-resolution atomic force microscopy and spectroscopy of native membrane proteins. Rep Prog Phys 74, 86601 (2011).
Zocher, M., Zhang, C., Rasmussen, S. G. F., Kobilka, B. K. & Müller, D. J. Cholesterol increases kinetic, energetic, and mechanical stability of the human 2-adrenergic receptor. Proc Natl Acad Sci USA 109, E3463–E3472 (2012).
Serdiuk, T., Sugihara, J., Mari, S. A., Kaback, H. R. & Müller, D. J. Observing a lipid-dependent alteration in single lactose permeases. Structure 23, 754–761 (2015).
Liu, L.-N., Duquesne, K., Oesterhelt, F., Sturgis, J. N. & Scheuring, S. Forces guiding assembly of light-harvesting complex 2 in native membranes. Proc Natl Acad Sci USA 108, 9455–9459 (2011).
Ge, L. et al. Molecular plasticity of the human voltage-dependent anion channel embedded into a membrane. Structure 24, 585–594 (2016).
Maity, S. et al. Conformational rearrangements in the transmembrane domain of CNGA1 channels revealed by single-molecule force spectroscopy. Nat Commun 6, 1–15 (2015).
Damaghi, M. et al. Dual energy landscape: the functional state of the β-barrel outer membrane protein G molds its unfolding energy landscape. Proteomics 10, 4151–4162 (2010).
Bippes, C. A. et al. Peptide transporter DtpA has two alternate conformations, one of which is promoted by inhibitor binding. Proc Natl Acad Sci USA 110, E3978–E3986 (2013).
Bustamante, C., Marko, J. F., Siggia, E. D. & Smith, S. Entropic elasticity of λ-phage DNA. Science 265, 1599–1600 (1994).
Rief, M., Gautel, M., Oesterhelt, F., Fernandez, J. M. & Gaub, H. E. Reversible unfolding of individual titin immunoglobin domains by AFM. Science 276, 1109–1112 (1997).
Barros, T. & Kühlbrandt, W. Crystallisation, structure and function of plant light-harvesting complex II. Biochim Biophys Acta 1787, 753–772 (2009).
Werwie, M., Xu, X., Haase, M., Basché, T. & Paulsen, H. Bio serves nano: biological light-harvesting complex as energy donor for semiconductor quantum dots. Langmuir 28, 5810–5818 (2012).
Sapra, K. T., Besir, H., Oesterhelt, D. & Müller, D. J. Characterizing molecular interactions in different characterizing molecular interactions in different bacteriorhodopsin assemblies by single-molecule force spectroscopy. J Mol Biol 355, 640–650 (2006).
Liu, C., Rao, Y., Zhang, L. & Yang, C. Identification of the roles of individual amino acid residues of the helix E of the major antenna of photosystem II (LHCII) by alanine scanning mutagenesis. J Biochem 156, 203–210 (2014).
Trémolières, A., Dubacq, J.-P., Ambard-Bretteville, F. & Rémy, R. Lipid composition of chlorophyl-protein complexes. Specific enrichment in trans-hexadecenoic acid of an oligomeric form of light-harvesting chlorophyll a/b protein. FEBS Lett 130, 27–31 (1981).
Nußberger, S., Dörr, K., Wang, D. N. & Kühlbrandt, W. Lipid-protein interactions in crystals of plant light-harvesting complex. J Mol Biol 234, 347–356 (1993).
Sprague, S. G. & Staehelin, A. Effects of reconstitution method on the structural organization of isolated chloroplast membrane lipids. Biochim Biophys Acta 777, 306–322 (1984).
Frewein, M., Kollmitzer, B., Heftberger, P. & Pabst, G. Lateral pressure-mediated protein partitioning into liquid-ordered/liquid-disordered domains. Soft Matter 12, 3189–3195 (2016).
Daum, G. & Vace, J. E. Import of lipids into mitochondria. Prog Lipid Res 36, 103–130 (1997).
Kelkar, D. A. & Chattopadhyay, A. The gramicidin ion channel: a model membrane protein. Biochim Biophys Acta 1768, 2011–2025 (2007).
van den Brink-van der Laan, E., Chupin, V., Killian, J. A. & de Kruijff, B. Stability of KcsA tetramer depends on membrane lateral pressure. Biochemistry 43, 4240–4250 (2004).
Cashmore, A. R. Structure and expression of a pea nuclear gene encoding chlorophyll a/b-binding polypeptide. Proc Natl Acad Sci USA 81, 2960–2964 (1984).
Paulsen, H., Rümler, U. & Rüdiger, W. Reconstitution of pigment-containing complexes from light-harvesting chlorophyll a/b-binding protein overexpressed in Escherichia coli. Planta 181, 204–211 (1990).
Booth, P. J. & Paulsen, H. Assembly of light-harvesting chlorophyll a/b complex in vitro. Time-resolved fluorescence measurements. Biochemistry 35, 5103–5108 (1996).
Paulsen, H., Finkenzeller, B. & Kühlein, N. Pigments induce folding of light-harvesting chlorophyll a/b-binding protein. Eur J Biochem 215, 809–816 (1993).
Yang, C., Horn, R. & Paulsen, H. The light-Harvesting chlorophyll a/b complex can be reconstituted in vitro from its completely unfolded apoprotein. Biochemistry 42, 4527–4533 (2003).
Hobe, S., Prytulla, S., Kühlbrandt, W. & Paulsen, H. Trimerization and crystallization of reconstituted light-harvesting chlorophyll a/b complex. EMBO J 13, 3423–3429 (1994).
Pettersen, E. F. et al. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004).
Author information
Authors and Affiliations
Contributions
D.S., H.W., A.J. and H.P. conceived the experiments. D.S. and H.W. performed and analyzed the experiments. D.S. and H.W. wrote the manuscript. A.J. and H.P. edited the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seiwert, D., Witt, H., Janshoff, A. et al. The non-bilayer lipid MGDG stabilizes the major light-harvesting complex (LHCII) against unfolding. Sci Rep 7, 5158 (2017). https://doi.org/10.1038/s41598-017-05328-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05328-7
This article is cited by
-
Cryo-EM structures of LHCII in photo-active and photo-protecting states reveal allosteric regulation of light harvesting and excess energy dissipation
Nature Plants (2023)
-
Accumulation of geranylgeranylated chlorophylls in the pigment-protein complexes of Arabidopsis thaliana acclimated to green light: effects on the organization of light-harvesting complex II and photosystem II functions
Photosynthesis Research (2021)
-
Interactions Determining the Structural Integrity of the Trimer of Plant Light Harvesting Complex in Lipid Membranes
The Journal of Membrane Biology (2021)
-
Modulation of non-bilayer lipid phases and the structure and functions of thylakoid membranes: effects on the water-soluble enzyme violaxanthin de-epoxidase
Scientific Reports (2020)
-
Dynamical and allosteric regulation of photoprotection in light harvesting complex II
Science China Chemistry (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
