
Abstract
Alpha (α)-thalassaemia is one of the most prevalent hereditary blood disorders, commonly affecting Southeast Asian people, with the highest incidence (30–40%) being seen in northern Thailand. However, this high incidence was estimated without consideration of the variations between ethnic populations and the geographical location of the populations. To address this issue, a total of 688 samples from 13 different northern Thai ethnic groups (30 villages) categorized into three linguistic groups were genotyped for deletional alpha-thalassaemia (-α3.7, -α4.2, --SEA and --THAI) and/or non-deletional alpha-thalassaemia (αCS and αPS) via multiplex gap-PCR and dot-blot hybridization, respectively. Alpha+(-α3.7, -α4.2, αCS and αPS) and alpha°-thalassaemia (--SEA and --THAI) allele frequencies (with 95% Confidence Interval) were the highest in the Sino-Tibetan group [0.13 (0.08–0.18)] and the Tai-Kadai group [0.03 (0.02–0.05)], respectively. With regards to ethnicity, the varying allele frequency of α+ and α°-thalassaemia amongst a variety of ethnic groups was observed. The highest α+-thalassaemia allele frequency was found in the Paluang [0.21 (0.10–0.37)] while α°-thalassaemia allele frequency was the highest in the Yuan [0.04 (0.01–0.10)]. These detailed results of alpha thalassaemia allele frequency and genetic diversity amongst the northern Thai ethnic groups demonstrate the need for ethnicity based thalassaemia prevention programs.
Similar content being viewed by others

Introduction
Alpha (α)-thalassaemia is a group of hereditary blood disorders that are found with very high prevalence in tropical and subtropical regions, and in particular in the people of South and Southeast Asian countries. The overall incidence of alpha-thalassaemia in Thailand appears to be unusually high, especially in the northern part where approximately 30–40% of residents have been reported to be either carriers or homozygotes1.
The inheritance of α-thalassaemia is autosomal recessive and a person with α-thalassaemia genotype could be either a carrier or a patient. Carriers are healthy, and as such α-thalassaemia is continuously maintained over generations. However, couples who are both carriers are likely to give birth to a child with α-thalassaemia associated with clinical symptoms2, 3. The α-globin genes located on chromosome 16p13.3 are responsible for α-globin production4, 5. Each haploid chromosome contains two copies of α-globin genes giving a total of four allelic copies in combination with the other homologous chromosome.
Alpha-thalassaemia is characterized by anomalies of the α-globin genes leading to reduced α-globin chain production, and α-globin is one of the major constituents of the haemoglobin of red blood cells. The reduction of α-globin chains in α-thalassaemia is most frequently caused by large deletions (-α3.7, -α4.2, --SEA and --THAI), although non-deletional α-thalassaemia such as Hb Constant spring (αCS) and Hb Pakse (αPS) can occur1, 6,7,8,9. Presentation of α-thalassaemia is correlated with the number of α-globin genes affected. Loss of one (α+: -α3.7, -α4.2, αCS and αPS) or two (α°: --SEA and --THAI) α-globin gene/s on one chromosome generally presents as a silent carrier state, while loss of three (α+/α°) results in Hb H disease in which the pathology is primarily mediated by the relative excess of β-chains which can form tetramers of β-globin (β4) which can promote oxidative hemolysis. Loss of four α-globin genes (α°/α°) results in fatal Hb Barts’ hydrops fetalis syndrome. Where loss of three α-globin genes occurs through inheritance of a combination of deletional and non-deletional α-thalassaemia, presentation can be more severe than that which results from inheritance of deletional α-thalassaemia only, and consequently, the clinical symptom of an affected person with inherited α-thalassaemia alleles ranges from asymptomatic to blood transfusion-dependence to premature death of infants depending on the number of α-globin alleles affected8.
The morbidity and mortality of α-thalassaemia associated with significant clinical symptoms are therefore observed in haemoglobin H disease (Hb H, three missing functional α-globin alleles) and haemoglobin Bart’s hydrops fetalis syndrome (Hb Barts, a complete loss of functional α-globin alleles)10. In Thailand, due to the high prevalence of α-thalassaemia carriers, there is a significant number of patients with Hb H disease (7/1,000 newborns)11. More importantly, in northern Thailand, 0.33% of 52,625 fetuses were reported to be Hb Bart’s hydrops fetalis12. These confirm the necessity for accurate and effective management of α-thalassaemia in this part of the world.
Several α-thalassaemia surveys in the northern part of Thailand have demonstrated that there is a high (15–40%) prevalence of α-thalassaemia alleles in the northern Thai population13, 14. However, population sampling in most surveys was conducted on couples who went to hospitals for screening, so the prevalence observed was primarily determined from the overall population of the upper northern part of Thailand. Interestingly, a recent study that determined the prevalence of α-thalassaemia in a population-based study in the northern Thai population showed for the first time that the overall prevalence of α-thalassaemia in upper northern Thailand was 24% (33 of 141), and more importantly, the study highlighted the significantly different prevalence of α-thalassaemia amongst ethnic groups ranging from 0 to 50% of populations examined15. However, that study was limited by a low number of samples and sampling areas for some ethnic groups, and in particular, no hill-tribe groups belonging to the Sino-Tibetan and Hmong-Mien linguistic families were included in the study. To address these issues this study analysed a large cohort comprising of ethnic populations from numerous sampling areas throughout the northern part of Thailand including the northern minorities such as Shan, Karen and Htin. Thus, the objective of this study is to provide more comprehensive and meaningful data of common α-thalassaemia allele frequency in northern Thai people as well as in particular, in each ethnic population. This information will serve as a more practical basis for developing genetic counseling for the long-term effort to reduce the burden of Hb H and Hb Bart’s hydrops fetalis syndrome in the country.
Results
A total of 688 DNA samples from people belonging to 13 ethnic groups that are classified as part of three linguistic groups (Tai-Kadai, Austro-Asiatic and Sino-Tibetan) were analysed for four types of common deletional α-thalassaemias (-α3.7, -α4.2, --SEA, --THAI) by multiplex gap-PCR with nine specific primers for each type (Fig. 1a) and 350 of the total 688 samples were analysed for an additional two types of mutational α-thalassaemias (αCS and αPS). Of the six common α-thalassaemia screened for, three different deletions (-α3.7, -α4.2, --SEA) and one point mutation (αCS) were found in this cohort.

Six common α-thalassaemia types detected by multiplex-gap PCR and dot-blot hybridization techniques. (a) PCR products after alpha-globin gene analysis using the multiplex gap-PCR methodology, M = DNA marker, lane 1–4 = positive controls of alpha-globin heterozygotes which are --THAI/αα, --SEA/αα, -α4.2/αα and -α3.7/αα in order, lane 5 = negative control (αα/αα), lane 6 = unknown sample genotyped as -α3.7 homozygote, lanes 7–8 = unknown samples genotyped as normal and lanes 9–10 = unknown samples genotyped as --SEA heterozygotes (A cropped gel is shown). The full-length gel is presented in Supplementary Figure S1. (b) Dot-blot hybridization analysis of the Lue ethnic group. Samples TL-201 and TL-234 were genotyped as αCS heterozygotes. No samples were positive for αPS (A cropped blot is shown). The full-length blot is presented in Supplementary Figure S2.
The overall prevalence of the six common α-thalassaemia types assessed in this cohort of 13 ethnic groups is 19.51% (Table 1) with a frequency of 0.1008 (0.0788–0.1247) (Table 2). Almost all the α-thalassaemia detected in this study was heterozygous, except for one case of Hb H disease (-α3.7/--SEA) which was detected in one sample from the Yong ethnic group (Table 1).
The most prevalent deletional α-thalassaemia in the cohort examined was the -α3.7 deletion with an allele frequency of 0.0676 (0.0549–0.0822), followed by--SEA and -α4.2 at frequencies of 0.0203 (0.0136–0.0293) and 0.0029 (0.0008–0.0074), respectively (Table 2). The presence of non-deletional α-thalassaemia was investigated in 350 samples by a dot-blot hybridization method (Fig. 1b). The non-deletional αCS was detected at an allele frequency of 0.0100 (0.0040–0.0205), and interestingly, this allele was only found in the Tai-Kadai group (Yuan, Lue and Yong) and the mutation was not detected in the Austro-Asiatic groups. The highest α-thalassaemia allele frequency was observed in the Paluang ethnic group [0.2105 (0.0955–0.3732)] while the Lawa showed the lowest α-thalassaemia allele frequency (0.0000) (Table 2 and Fig. 2).

The allele frequency of common α-thalassaemia in each ethnic group. The bar graph represents α-thalassa emia allele frequency. At the bottom of the figure, the total allele frequency of common α-thalassaemia is shown regarding the analysis of the three linguistic groups (Tai-Kadai, Austro-Asiatic and Sino-Tibetan).
The Sino-Tibetan linguistic group carried the highest frequency of deletional α-thalassaemia [0.1262 (0.0841–0.1794)] followed by Tai-Kadai [0.1125 (0.0910–0.1455)] and the Austro-Asiatic linguistic group [0.0464 (0.0169–0.0963)] (Table 2 and Fig. 2). As noted, analysis of two linguistic groups (Tai-Kadai and Austro-Asiatic) showed that non-deletion α-thalassaemia was found only in the Tai-Kadai linguistic group, giving a frequency of 0.0100 (0.0040–0.0205) (Yong, Lue and Yuan).
The combined analysis of α+ and α°-thalassaemia allele frequency may overestimate the incidence of the disease in the population. More importantly, the allele frequency of α°-thalassaemia determines the burden of significant α-thalassaemia syndromes. Therefore, α+-thalassaemia was analysed separately from α°-thalassaemia allele frequency. The results showed that α+-thalassaemia allele frequency was the highest in the Sino-Tibetan group [0.1262 (0.0841–0.1794)] followed by the Tai-Kadai group [0.0845 (0.0630–0.1105)] and the Austro-Asiatic group [0.0387 (0.0124–0.0862)]. While α°-thalassaemia allele frequency was the highest in the Tai-Kadai group [0.0317 (0.0189–0.0496)] followed by the Austro-Asiatic group [0.0077 (0.0002–0.0415)]. No α°-thalassaemia allele frequency was observed in the Sino-Tibetan group. With regards to ethnicity, the varying allele frequency of α+-thalassaemia amongst a variety of ethnic groups ranging from the lowest (0.00) in the Lawa and the Htin, to the highest frequency [0.2105 (0.0955–0.3732)] in the Paluang. While α°-thalassaemia allele frequency was detected in 6 (Yuan, Shan, Lue, Yong, Blang and Htin) out of 13 ethnic groups, the Yuan was the highest [0.0417 (0.0115–0.1033)] (Table 2).
Discussion
Alpha thalassaemia is a global health problem that is a growing burden16, 17, particularly in Southeast Asian ethnic groups. The high prevalence (30%) of α- thalassaemia that has been previously reported in northern Thailand13, 14, 18, has been shown to vary from region to region and by ethnic group15. However, few studies describing the frequencies of α-thalassaemia in Thai ethnic groups have been conducted, and the data was limited by the small sample size and screening method9, 14 (Table 3). Our first survey undertaken using molecular analysis to identify α-thalassaemia amongst 8 ethnic groups had a small sample size15, but showed distinct variations between ethnic groups. Furthermore, the population in upper northern Thailand is comprised of a number of ethnic groups which can be categorized into three major linguistic groups. These are comprised of the Tai-Kadai group who are the majority of the present day northern Thai population, the Austro-Asiatic group who are recognized as the descendants of the prehistoric inhabitants of northern Thailand and mostly reside in remote areas, and the hill-tribes group which is comprised of ethnic groups that belong to the Sino-Tibetan and Hmong-Mien linguistic families. From this last group, the Karen ethnic group have the highest population number amongst the hill-tribes of northern Thailand19, 20. This is further complicated by the occurrence of diverse genetic backgrounds amongst northern ethnic groups21, 22, and therefore the overall incidence of α-thalassaemia from previous surveys might not represent the situation accurately. Thus it was of interest to conduct a larger survey to more accurately determine the real prevalence. Therefore in this study, a larger cohort comprising of individuals from 13 ethnic groups residing in northern Thailand was surveyed for six common α-thalassaemia types. The overall frequency of the six types of α-thalassaemia investigated in this study is 0.1008 (0.0788–0.1247) (Table 2), representing a prevalence of 19.51%) (Table 1). The prevalence data surveyed by this and our previous cohort15 are comparable, but lower than previous reports from the general Thai population that reported the prevalence of α-thalassaemia at 26.42% (28/106)9 (Table 3).
In accordance with our previous study15 and the findings of other studies14, 16, 18, the -α3.7 deletion is the most common α-thalassaemia present amongst Thais and Thai ethnic groups, followed by --SEA 6, 7. Interestingly, the heterozygous --SEA deletion is very common in the Tai-Kadai linguistic group. The data is also consistent with a study undertaken in the Yunnan province of Southwestern China which showed that the --SEA deletion type is the most common α-thalassaemia23 and supports evidence that the Tai-Kadai speaking people staying in Northern Thailand migrated there from the southwest of China24, 25. It also supports the genetic diversity of this abnormal gene between population groups.
Similarly, evidence for the genetic diversity of this gene was found with the αCS mutation. While the overall allele frequency of the αCS allele was 0.0100 (0.0040–0.0205) (Table 2), it was only found in the Tai-Kadai linguistic group (Yuan, Lue and Yong) and the mutation was not detected in the Austro-Asiatic groups. However, αCS is the most prevalent α-globin variant in the Southeast Asian population26 and while people with heterozygous αCS have an almost normal clinical presentation, when inherited in a compound heterozygous state along with α0-thalassaemia, a more severe presentation than deletional Hb H disease can occur26. In contrast to the native Thai population, we did not find the --THAI deletion or the Hb Pakse α-globin variant in this cohort. This latter observation is in accordance with an earlier study which showed that Hb Pakse was not found in the population residing in northern Thailand3.
With regards to the frequency of α-thalassaemia observed in each linguistic group, this study detected considerable variation amongst the different ethnic groups. The highest frequency of α-thalassaemia [0.1262 (0.0841–0.1794): α+ = 0.1262 (0.0841–0.1794), α0 = 0] was seen in the Sino-Tibetan (Karen) linguistic group and the -α3.7 deletion type was the only α-thalassaemia type existing in this group. Importantly, the Paluang, Karen and Shan ethnic groups showed a very high frequency of the -α3.7 deletion type. Since these three ethnic peoples live along the Thailand-Myanmar border27 which is a malaria endemic area28, the high frequency of the -α3.7 may reflect natural selection due to protection against severe malaria infection29. Moreover, the presence of the -α3.7 deletion in all three Karen ethnic groups (Skaw, Pwo and Padong) is at very similar levels, supporting the common origin of these ethnic groups, and showing that the Karen seem to have a homogenous genetic background. The frequency of α-thalassaemia is also high in the Tai-Kadai group [0.1125 (0.0910–0.1455): α+ = 0.0845 (0.0630–0.1105), α0 = 0.0317 (0.0189–0.0496)]. Interestingly, this linguistic group shows the highest frequency of heterozygous α-thalassaemia 1 (--SEA) which is characterized by deletion of two α-globin genes, and this was supported by the detection of one individual with Hb H disease in this group. In contrast to the other ethnic groups in the Tai-Kadai linguistic group, the Lue show significant gene diversity with 4 types of α-thalassaemia detected in this ethnic group. This is likely to be the result of a founder effect and/or inter-ethnic marriage between the Lue and other ethnic groups during their migration through Laos. The predominance of --SEA and αCS types in Tai-Kadai linguistic group elevates their risk of conceiving fetuses with Hb Bart’s hydrop fetalis or Hb H-CS disease. The lowest frequency was recorded in Austro-Asiatic linguistic group since no α-thalassaemia was detected in any of the 48 Lawa people investigated.
Conclusion
Our study presents the results of the screening of a large cohort representing 13 ethnic groups from northern Thailand for α-thalassaemia. As the prevalence of α-thalassaemia is relatively high and the majority of these groups are still unaware of their thalassaemia status, couples who are members of particular ethnic populations at risk for α°-thalassaemia (--SEA, --THAI) such as the Yuan, Shan, Lue and Yong should be recommended for haematological screening prior to planning for pregnancy to control the severe types of α-thalassaemia. Future studies might be directed to study the whole α-globin locus in order to determine whether novel α-globin gene abnormalities may exist that are unique to a particular ethnic group.
Materials and Methods
Study populations
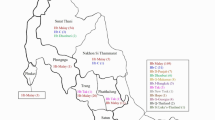
Northern Thailand has 18 officially recognized ethnic populations19, 20. For this survey samples were obtained from 13 ethnic groups from 30 villages distributed in five provinces of northern Thailand. The cohort comprised (a) 278 newly genotyped samples and (b) 269 subjects previously genotyped for hemoglobin E for whom α-thalassaemia genotype has not been reported30. In addition (c) α-thalassaemia genotypic data from 141 subjects as previously reported15 was included, giving a total 688 samples. The criteria for population sampling was as described elsewhere22, 24, 30, 31. Briefly, all volunteers enrolled in this study were healthy, over 20 years of age, unrelated, and recognized as a member of the study ethnic population for at least three generations with no admixture from other populations. The designed number of sample size enrolled in this study was 30 samples per ethnic group. Although some difficulties arose in obtaining appropriate number of samples from some ethnic groups such as the Padong Karen who practice endogamous marriage, the Palaung and Blang who have small population sizes and the Khuen who traditionally marry with people from other ethnic groups (interethnic marriage) giving offspring (admixed population) that cannot be recruited for this study, the sample sizes from such mentioned ethnic groups are still nearly in the power of calculation for population analyses as stated by Jobling et al., 2013 (20–50 individuals per populations are recommended)32. The location of sampling areas and details are shown in Table 4 and Fig. 3. All subjects from categories (a) to (c) were enrolled after informed consent. Ethical approval of all methods and experimental protocols according to the guidelines was follows: the Yong ethnic group (a) and all subjects of category (b) were approved by the Human Experimentation Committee, Research Institute for Health Sciences, Chiang Mai University, Thailand. All subjects of category (c) were approved by the Policy Review Board of the Pan Asia SNP consortium as described elsewhere33. It should be noted that both the Lue and the Htin ethnic population samples of category (a) were collected more than 10 years ago and therefore oral informed consent was implemented with the assistance of the head of each village.

Geographical map representation of sampling areas and distribution of the ethnic groups residing in northern Thailand. The red colour represents Tai-Kadai speaking ethnic groups, the blue colour represents the Austro-Asiatic speaking ethnic groups and orange indicates the Sino-Tibetan speaking ethnic groups. This figure was modified using the Photoshop program. The original source of this figure can be found at https://commons.wikimedia.org/wiki/File:Thailand_location_map.svg which is licensed under the “Creative Commons Attribution 3.0 Unported” that is free to share (to copy, distribute and transmit the work) and remix (to adapt the work).
DNA extraction
Five milliliters of peripheral blood from human subjects was collected after individual informed consent, and total genomic DNA was extracted using an inorganic salting out protocol as described elsewhere34. Quality and quantity of extracted genomic DNA from all samples were examined by 1% agarose gel electrophoresis and spectrophotometry (OD260/OD280). All samples were kept at -20 °C until use.
Multiplex gap-PCR analysis of the deletional alpha globin gene
The four most common deletional α-thalassaemias (-α3.7, -α4.2, --SEA and --THAI) in the Thai population were investigated in this study. All samples were genotyped for the four deletional types of α-thalassaemia by multiplex gap-polymerase chain reaction modified from Chong and colleagues35. Briefly, nine specific primers were used in PCR reaction, consisted of primers α2/3.7-F, 3.7-R, α2-R, 4.2-F, 4.2-R, SEA-F, SEA-R, THAI-F and THAI-R. Each PCR reaction was performed in a single tube for simultaneous amplification of different amplicons under an initial denaturation at 95 °C for 15 minutes and followed by thirty-five cycles of denaturation for 45 second at 98 °C, annealing at 60 °C for 1.30 minute, extension at 72 °C for 2.15 min with an additional final extension at 72oC for 5 min after the last cycle. PCR products were analysed by 1.5% agarose gel electrophoresis compared with positive controls as shown in Fig. 1a. To ensure the genotyping accuracy of the multiplex gap-PCR, every single round of PCR amplification of unknown samples was performed paralleled with the positive controls (Fig. 1a, lane 1, 2, 3 and 4 are --THAI/αα, --SEA/αα, -α4.2/αα and -α3.7/αα, respectively) and a negative control (lane 5 is genotyped as αα/αα). DNA samples from each unknown individual was genotyped at least in duplicate.
Dot-blot hybridization analysis of the mutational alpha globin gene
A total of 350 samples were screened for two common types of non-deletional α-thalassaemia (αCS and αPS). The dot-blot hybridization method was employed as described elsewhere36. The α-globin gene was amplified by PCR using primers αF and α2 R. The 331 bp-PCR products were validated by 1.5% agarose gel electrophoresis and were then subsequently hybridized with specific probes for αCS and αPS as well as a normal probe. The resulting genotype of each unknown sample was interpreted in parallel with controls, which consisted of a normal sample and homozygous αCS and αPS. A blue spot was interpreted as a positive signal (Fig. 1b). The genotyping quality of the dot blot hybridization of unknown samples was ensured by controls (Fig. 1b, positive controls are samples with known genotype of αCS homozygous and αPS homozygous while the negative control was αα/αα). Unknown samples were always tested in paralleled with controls, and analysis was conducted in duplicate.
Statistical Methods
All the allele frequencies were calculated using the Microsoft Excel program (version 2016, Microsoft Corporation, USA) with the function BinomLow and BinomHigh (add-ins) derived from JavaStat to compute the exact binomial confidence interval (95%). The bar graph was generated by the PRISM software (version 7.00, GraphPad Software, Inc. USA).
Data availability statement
The data sets generated and analysed during the current study are available within the paper.
References
Fucharoen, S. & Winichagoon, P. Haemoglobinopathies in Southeast Asia. The Indian J Med Res. 134, 498–506 (2011).
Kitsirisakul, B., Steger, H. F. & Sanguansermsri, T. Frequency of alpha-thalassemia-1 of the Southeast Asian-type among pregnant women in northern Thailand determined by PCR technique. Southeast Asian J Trop Med Public Health. 27, 362–363 (1996).
Lemmens-Zygulska, M. et al. Prevalence of alpha-thalassemias in northern Thailand. Hum Genet. 98, 345–347 (1996).
Higgs, D. et al. A review of the molecular genetics of the human a-globin gene cluster. Blood. 73, 1081–1104 (1989).
Kazazian, H. H. Jr. The thalassemia syndromes: molecular basis and prenatal diagnosis in 1990. Semin Hematol. 27, 209–228 (1990).
Tanphaichitr, V. S. et al. Studies of hemoglobin Bart and deletion of alpha-globin genes from cord blood in Thailand. Birth Defects Orig Artic Ser. 23, 15–21 (1987).
Laosombat, V. Thalassemia in children in Southern Thailand. J Med Assoc Thai. 69, 393–399 (1986).
Harteveld, C. L. & Higgs, D. R. α-thalassaemia. Orphanet J Rare Dis. 5, doi:10.1186/1750-1172-5-13 (2010).
Hundrieser, J., Sanguansermsri, T., Papp, T. & Flatz, G. Alpha-Thalassemia in Northern Thailand. Hum. Hered. 38, 211–215 (1988).
Laosombat, V. et al. Clinical features and molecular analysis in Thai patients with HbH disease. Ann Hematol. 88, 1185–1192 (2009).
Pansateinkul, B. A Group of Thai Hematologists (1990) Current situation and strategic plan for prevention and control of blood diseases in Thailand 1989–1990. 1st edn edn, (Num-Aksorn-Karnprim, 1990).
Tongsong, T. et al. Prenatal eradication of Hb Bart’s hydrops fetalis. J Reprod Med. 46, 18–22 (2001).
Fucharoen, S. & Winichagoon, P. New updating into hemoglobinopathies. Int J Lab Hematol. 34, 559–565 (2012).
Na-Nakorn, S. & Wasi, P. Alpha-thalassemia in northern Thailand. Am J Hum Genet. 22, 645–651 (1970).
Lithanatudom, P. et al. The prevalence of alpha-thalassemia amongst Tai and Mon-Khmer ethnic groups residing in northern Thailand: A population-based study. Hematology 21, 480–485 (2016).
Teawtrakul, N., Chansung, K., Sirijerachai, C., Wanitpongpun, C. & Thepsuthammarat, K. The impact and disease burden of thalassemia in Thailand: a population-based study in 2010. J Med Assoc Thai. 95, S211–216 (2012).
Piel, F. B. & Weatherall, D. J. The α-thalassemias. N Engl J Med. 371, 1908–1916 (2014).
Wanapirak, C., Muninthorn, W., Sanguansermsri, T., Dhananjayanonda, P. & Tongsong, T. Prevalence of thalassemia in pregnant women at Maharaj Nakorn Chiang Mai Hospital. J Med Assoc Thai. 87, 1415–1418 (2004).
Schliesinger, J. Ethnic Groups of Thailand: Non-Tai-Speaking Peoples 1st edition, 1–25 (White Lotus Co Ltd., 2000).
Schliesinger, J. Tai group of Thailand, Volume 1: Introduction and overview, 1st edition, 83–98 (White Lotus Co Ltd., 2001).
Besaggio, D. et al. Genetic variation in Northern Thailand Hill Tribes: origins and relationships with social structure and linguistic differences. BMC Evol Biol. 7, S12–S12 (2007).
Kutanan, W. et al. Genetic structure of the Mon-Khmer speaking groups and their affinity to the neighbouring Tai populations in Northern Thailand. BMC Genet. 12, doi:10.1186/1471-2156-12-56 (2011).
Zhang, J. et al. The spectrum of α-and β-thalassemia mutations in Yunnan Province of Southwestern China. Hemoglobin 36, 464–473 (2012).
Kampuansai, J. et al. Effect of migration patterns on maternal genetic structure: a case of Tai-Kadai migration from China to Thailand. J Hum Genet. 62, 223–228 (2016).
Qian, Y. P. et al. Mitochondrial DNA polymorphisms in Yunnan nationalities in China. J Hum Genet. 46, 211–220 (2001).
Hunt, D. M., Higgs, D. R., Winichagoon, P., Clegg, J. B. & Weatherall, D. J. Haemoglobin Constant Spring has an unstable α chain messenger RNA. British Br J Haematol. 51, 405–413 (1982).
Delang, C. O. Living at the Edge of Thai Society: The Karen in the Highlands of Northern Thailand. 1st edition, 2–3 (RoutledgeCurzon, 2003).
Parker, D. M. et al. Microgeography and molecular epidemiology of malaria at the Thailand-Myanmar border in the malaria pre-elimination phase. Malar J. 14, doi:10.1186/s12936-015-0712-5 (2015).
Enevold, A. et al. Associations between alpha+-thalassemia and Plasmodium falciparum malarial infection in northeastern Tanzania. J Infect Dis. 196, 451–459 (2007).
Lithanatudom, P. et al. Hemoglobin E Prevalence among Ethnic Groups Residing in Malaria-Endemic Areas of Northern Thailand and Its Lack of Association with Plasmodium falciparum Invasion In Vitro. PLoS One. 11, doi:10.1371/journal.pone.0148079 (2016).
Kampuansai, J. et al. Mitochondrial DNA variation of Tai speaking peoples in Northern Thailand. ScienceAsia 33, 443–448 (2007).
Jobling, M., Hollox, E., Kivisild, T. & Tyler-Smith, C. Human evolutionary genetics. 2nd edition, 328–329 (Garland Science Publishing, 2013).
The HUGO Pan-Asian SNP Consortium. Mapping Human Genetic Diversity in Asia. Science 326, 1541–1545 (2009).
Seielstad, M., Bekele, E., Ibrahim, M., Toure, A. & Traore, M. A view of modern human origins from Y chromosome microsatellite variation. Genome, Res. 9, 558–567 (1999).
Chong, S. S., Boehm, C. D., Higgs, D. R. & Cutting, G. R. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 95, 360–362 (2000).
Pichanun, D. et al. Molecular screening of the Hbs Constant Spring (codon 142, TAA>CAA, alpha2) and Pakse (codon 142, TAA>TAT, alpha2) mutations in Thailand. Hemoglobin 34, 582–586 (2010).
Acknowledgements
The authors would like to express their sincere thanks to all volunteers and village heads for their contribution. We are also grateful to the CMU Junior Research Fellowship program (2015) and Prof. Sakorn Pornprasert for his all valuable assistance.
Author information
Authors and Affiliations
Contributions
M.K., T.M., M.W. and P.L. performed the research. P.L., M.K. and K.L. analysed the data and drafted the manuscript. J.K., D.K. and P.L. were responsible for sample collections. S.F., S.S., D.R.S. and P.L. designed the research study. All authors contributed to revising the manuscript and all authors approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kulaphisit, M., Kampuansai, J., Leecharoenkiat, K. et al. A comprehensive ethnic-based analysis of alpha thalassaemia allelle frequency in northern Thailand. Sci Rep 7, 4690 (2017). https://doi.org/10.1038/s41598-017-04957-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04957-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}