
Abstract
Asymmetric cell divisions are required for cellular diversity and defects can lead to altered daughter cell fates and numbers. In a genetic screen for C. elegans mutants with defects in dopaminergic head neuron specification or differentiation, we isolated a new allele of the transcription factor HAM-1 [HSN (Hermaphrodite-Specific Neurons) Abnormal Migration]. Loss of both HAM-1 and its target, the kinase PIG-1 [PAR-1(I)-like Gene], leads to abnormal dopaminergic head neuron numbers. We identified discrete genetic relationships between ham-1, pig-1 and apoptosis pathway genes in dopaminergic head neurons. We used an unbiased, quantitative mass spectrometry-based proteomics approach to characterise direct and indirect protein targets and pathways that mediate the effects of PIG-1 kinase loss in C. elegans embryos. Proteins showing changes in either abundance, or phosphorylation levels, between wild-type and pig-1 mutant embryos are predominantly connected with processes including cell cycle, asymmetric cell division, apoptosis and actomyosin-regulation. Several of these proteins play important roles in C. elegans development. Our data provide an in-depth characterisation of the C. elegans wild-type embryo proteome and phosphoproteome and can be explored via the Encyclopedia of Proteome Dynamics (EPD) – an open access, searchable online database.
Similar content being viewed by others


Introduction
Cellular diversity is based on the division of a cell into two unequal daughter cells; a process termed asymmetric cell division. Molecular mechanisms and key regulators of asymmetric cell division are conserved between vertebrates and invertebrates (for review see ref. 1). Thus, the nematode C. elegans with its largely invariant development has proven a valuable model to uncover pathways controlling asymmetric cell division. For example, studies into the first asymmetric cleavage of the C. elegans zygote led to the discovery of the polarity-regulating PAR (abnormal embryonic PARtitioning of cytoplasm) proteins, whose function is conserved in diverse animals (reviewed in ref. 2). In addition, a mechanistically conserved Wnt/β-catenin system resulting in unequal nuclear β-catenin levels in the two daughter cells has been described to act during several asymmetric divisions in C. elegans (reviewed in ref. 3). Finally, the transcription factor HAM-1 (HSN Abnormal Migration) and its target, the conserved kinase PIG-1 (PAR-1 (I)-like Gene), were both found to regulate asymmetric cell divisions resulting in an apoptotic daughter cell4,5,6. However, it is not known either whether HAM-1 and PIG-1 interact with the PAR proteins and/or the Wnt/β-catenin binary system, or how such potential interactions may be regulated.
Mutation of the transcription factor HAM-1 can result in survival of daughter cells destined for apoptotic cell death5,6,7. To the best of our knowledge, the kinase PIG-1 is the only known transcriptional target of HAM-1 involved in asymmetric cell divisions8, 9. Loss of ham-1 and pig-1 can transform the fate of daughter cells destined for apoptosis and result in them developing via the same neuronal fate as their surviving sister cells7, 8. Furthermore, mutation of either ham-1, or pig-1, alters the resulting sizes of daughter cells: thus, in ham-1 mutants, the normally smaller apoptotic daughter cell is larger than the sister cell. In pig-1 mutants and pig-1;ham-1 double mutants daughter cell sizes are now similar in size5, 8. In relation to these cell size phenotypes, it is interesting that HAM-1 and PIG-1 were suggested previously to regulate myosin polarisation and the positioning of the mitotic spindle9, 10, which are processes known to influence daughter cell sizes (reviewed in refs 1, 11). However, the molecular mechanisms and proteins mediating these phenotypes that result from mutation of HAM-1 and PIG-1 are unknown.
PIG-1 is a member of the polarity-regulating PAR-1/Kin1/SAD-1 family of serine/threonine kinases8 and may play an important role in the regulation of asymmetric cell divisions. Localised phosphorylation events are known to contribute to the unequal segregation of cell fate determinants1 and PIG-1 expression was found at centrosomes12 and at the cortex of adjacent cells13. The expression of PIG-1 is ubiquitous in early embryos and becomes progressively more restricted in older embryos and young larvae, in which PIG-1 is expressed in dividing cells8. In addition to its role in asymmetric cell divisions, C. elegans PIG-1 was shown to control caspase-independent cell death and cell shedding, a process in which cells are eliminated by being actively expelled from their surrounding tissue14, 15. During cell shedding and asymmetric cell divisions, PIG-1 is controlled by the PAR-4-STRD-1-MOP-25 complex12, 14. Furthermore, PAR-4/LKB1 was shown to act via PIG-1 to regulate myosin accumulation in the C. elegans one-cell embryo13. In summary, PAR-4 appears to be genetically upstream of PIG-1, but the downstream effectors of PIG-1 are unknown.
The mammalian orthologues of HAM-1 and PIG-1 are connected to cell cycle regulation and cell death. The HAM-1 orthologue, Storkhead box (STOX) protein 1, is related to forkhead box (FOX) transcription factors16, which have a conserved role in cell cycle-regulated gene expression (for review see ref. 17). Furthermore, STOX1 itself was found to promote mitotic entry18 and the proliferation of inner ear epithelial cells19. The PIG-1 orthologue MELK (Maternal Embryonic Leucine zipper Kinase, also known as pEg3 kinase and MPK38) is predominantly expressed in proliferating cell populations and was reported to influence tumour growth and aggressiveness (for review see ref. 20). Putative MELK targets support a role in the regulation of cell cycle progression, cell proliferation and cell death21,22,23,24,25,26,27,28,29,30,31; however, the global effects of PIG-1/MELK kinase loss have not been investigated.
The C. elegans lineages that produce dopaminergic neurons provide a valuable system to explore the interplay between cell specification – a process that happens during every asymmetric cell division – and terminal cell differentiation – a mechanism that occurs only after the last mitotic division. C. elegans hermaphrodites possess 8 dopaminergic neurons: 2 CEPD (CEPhalic sensilla Dorsal), 2 CEPV (CEPhalic sensilla Ventral) and 2 ADE (Anterior DEirid) neurons in the head and 2 PDE (Posterior DEirid) neurons in the midbody. These dopaminergic neurons are derived from distinct lineages and their final differentiation is driven by a combination of dopaminergic ‘terminal selectors’, which are transcription factors controlling the expression of cell-specific features32, 33. However, the potential mechanistic connections between the HAM-1/PIG-1, PAR protein and Wnt/β-catenin specification pathways and subsequent dopaminergic neuron differentiation have not been characterised.
To identify factors influencing dopaminergic neuron specification and differentiation, we screened for C. elegans mutants exhibiting defective dopaminergic head neurons. We isolated a new allele of ham-1 that causes an abnormal number of CEPD and ADE neurons and confirmed that the proposed downstream effector of ham-1, the kinase PIG-1, is also involved in the specification of CEPD and ADE neurons. We report epistasis between ham-1 and pig-1 and describe distinct genetic interactions with the apoptosis-defective mutant ced-4 (CEll Death abnormality) in the lineages leading to dopaminergic head neurons. Using a global and phospho-specific mass spectroscopy-based proteomics approach, we screened for proteins affected by PIG-1 kinase loss. We found that pig-1 mutation affects the abundance and phosphorylation status of key developmental proteins in the C. elegans embryo. In addition, we provide an in-depth characterisation of the phosphoproteome of C. elegans wild-type embryos. These results are accessible via the Encyclopaedia of Proteome Dynamics (EPD) − http://www.peptracker.com/epd/ − a searchable, open-access online database34 providing convenient tools for developmental biologists to explore and visualise these complex, large-scale proteomics datasets.
Results
ham-1 mutation causes distinct defects in dopaminergic head neurons
To find genes that affect dopaminergic neuron development, a mutagenised population of the BY200 strain that allows for visualisation of C. elegans dopaminergic neurons (Pdat-1::gfp)35 was screened for defective dopaminergic head neurons. One of the isolated mutants, gt1984, exhibited distinct phenotypes in dopaminergic head neurons: while the number of ventral cephalic sensilla (CEPV) neurons was unchanged, the number of dorsal cephalic sensilla (CEPD) neurons was decreased and the number of anterior deirids (ADE) neurons was increased (Fig. 1a,c). Based on the largely invariant cell lineages of C. elegans 36, 37, it can be derived that the respective ‘sister’ cells of CEPVL and CEPVR neurons die by apoptosis (Fig. 1d − left panels). In contrast, in the lineages of left and right CEPD and ADEL neurons it is an ‘aunt’ cell that becomes apoptotic (Fig. 1d − middle and right panels).

Distinct defects caused by ham-1 mutation and genetic interactions with the apoptosis pathway in the lineages leading to C. elegans dopaminergic head neurons. (a) Dopaminergic head neurons in BY200 wild-type animals and in ham-1(gt1984) mutants. The dorsal cephalic sensilla (CEPD) neurons are located more posteriorly than the ventral cephalic sensilla (CEPV) neurons. A misplaced CEPD neuron is marked with an asterisk. (b) ham-1 gene structure with winged helix box DNA-binding domain (in white) and positions of mutations in ham-1(gt1984) (in orange) and ham-1(n1438) (in grey). gt1984 comprises a 554 base pair (bp) deletion spanning from the ham-1 promoter into the first exon and also includes a splice site mutation at the start of the second exon. The 238 bp deletion of the n1438 allele is within the gt1984 deletion. (c) Number of dopaminergic head neurons in BY200 wild-type and ham-1 mutant animals. Error bars = SEM of 2−4 biological replicates with 50 animals each. (d) Cartoon depicting the last divisions in the cell lineages that produce C. elegans dopaminergic head neurons (dopaminergic neurons are indicated with a solid black circle): left and right ventral cephalic sensilla neurons (CEPVL/R), left and right dorsal cephalic sensilla neurons (CEPDL/R) and left and right anterior deirids neurons (ADEL/R). Cells undergoing developmental apoptosis are marked with a cross. Daughter cells resulting from an anteroposterior division are labelled with ‘a’ and ‘p’, respectively, while daughter cells resulting from a left-right division are labelled with ‘l’ and ‘r’, respectively, starting from the ‘AB’ precursor cell. (e) Number of dopaminergic head neurons in the ham-1 mutant, in the apoptosis pathway mutants ced-3 and ced-4 and in the ced-4;ham-1 double mutant. Error bars = SEM of 2−4 biological replicates with 50 animals each.
We mapped38, 39 gt1984 to the ham-1 (HSN Abnormal Migration) locus and found that the mutation comprises a deletion and a splice site mutation (Fig. 1b). Thus, gt1984 is expected to result in a loss-of-function of HAM-1, an orthologue of the Storkhead box protein 1 (STOX1) transcription factor6, 9 containing a winged helix DNA binding motif (Fig. 1b). We verified that the same dopaminergic head neuron defects occur in a second ham-1 mutant4 (Fig. 1c). While ham-1 mutants were found to exhibit dopaminergic neuron defects5, 40, genetic interactions in these lineages have not been described. We conclude that ham-1(gt1984) constitutes a new loss-of-function allele that differentially affects cell fate in dopaminergic head neuron lineages.
Apoptosis in C. elegans development is controlled by the cell death abnormality genes ced-3 and ced-4, which encode an executioner caspase and an Apaf1 (Apoptotic Peptidase-Activating Factor 1)-like molecule, respectively41. We tested if dopaminergic head neuron specification is affected by mutation of ced-3 and ced-4 alone. Blinded studies showed that the number of CEPV neurons - which possess an apoptotic sister cell – is increased in the ced-3 and ced-4 mutants; however, the number of CEPD and ADE neurons – which possess an apoptotic aunt cell – is unchanged (Fig. 1e). This observation is in line with studies showing that surviving cells in either the ced-3 or ced-4 mutants can adapt sister cell fates, but do not divide further to produce differentiated cells5, 41, 42. Moreover, we found that mutation of ham-1 in the apoptosis-deficient background did not result in a further increase in CEPV neuron numbers (Fig. 1e – left panel). Therefore, the prevention of cell death appears to be sufficient to transform the normally apoptotic sisters into additional CEPV neurons.
Cell fate transformations caused by mutation of ham-1 can be masked by cell death5,6,7. If this is true for the dopaminergic head neuron lineages, the lower number of CEPD neurons in the ham-1 mutant is expected to be reversed in an apoptosis-deficient mutant background. Indeed, we found that in the ced-4;ham-1 double mutant CEPD neuron numbers are either restored to wild-type levels, or are even higher (Fig. 1e – middle panel). However, the already increased number of ADE neurons in the ham-1 mutant was not altered by additional mutation of ced-4 (Fig. 1e – right panel). Hence, ham-1 leads to cell fate transformations in both CEPD and ADE neurons, however in CEPD neurons the transformed cells appear to die via apoptosis.
pig-1 genetically interacts with ham-1 to specify dopaminergic neurons
If PIG-1 is a general HAM-1 effector, pig-1 and ham-1 mutants are expected to show similar dopaminergic neuron defects. To determine pig-1 mutant phenotypes, we used pig-1(gm344), which likely represents a null allele (Fig. 2a). We found that both pig-1(gm344) single mutants and pig-1;ham-1 double mutants exhibited a wild-type number of CEPV neurons. Thus, CEPV neurons are not affected by loss of ham-1 and pig-1 (Fig. 2b – left panel). Furthermore, pig-1 mutants show a higher number of ADE neurons, similar to ham-1 mutants and pig-1;ham-1 double mutants (Fig. 2b – right panel). Hence, pig-1 and ham-1 appear to repress ADE cell fate via a similar mechanism. However, in contrast with ham-1 mutants, pig-1 mutants did not display an decreased number of CEPD neurons (Fig. 2b – middle panel and ref. 8) and mutation of pig-1 rescued the reduced number of CEPD neurons in the ham-1 mutant (Fig. 2b – middle panel). Therefore, pig-1 acts as a suppressor of neuronal fate in both CEPD and ADE lineages and is epistatic to ham-1 in CEPD neurons.

pig-1 is epistatic to ham-1 in the CEPD neuron lineage and genetically interacts with the apoptosis pathway. (a) pig-1 gene structure with position of the predicted kinase and kinase-associated domains (both in white) and the pig-1(gm344) 562 bp deletion (in green). In gm344, 400 bp of the pig-1 promoter and the first 132 bp of the kinase domain are deleted. (b) Number of dopaminergic head neurons in the ham-1, pig-1 and pig-1;ham-1 mutant. (c) Number of dopaminergic head neurons in the pig-1, ced-4 and ced-4;pig-1 mutant. (d) Number of dopaminergic head neurons in the double and triple mutants of ham-1, pig-1 and ced-4. Error bars = SEM of 2–4 biological replicates with 50 animals each.
We next tested genetic interactions between pig-1 and the ced-4 apoptosis gene in dopaminergic neuron lineages. We found that CEPD neuron numbers were higher in the ced-4;pig-1 double mutant, even though ced-4 and pig-1 single mutants did not exhibit a phenotype (Fig. 2c – middle panel). This synergism is likely caused by the unmasking of a cell fate conversion in the apoptosis-deficient background, as is the case for ham-1 and ced-4 in CEPD neurons (Fig. 1e). In contrast, the higher number of CEPV neurons in the ced-4 mutants was not further increased by mutation of pig-1 (Fig. 2c – left panel). Also, the higher number of ADE neurons in the pig-1 mutants was not further increased by mutation of ced-4 (Fig. 2c – right panel). These results show that only one of the three lineages leading to dopaminergic head neurons shows a synergistic genetic relationship between pig-1 and the apoptosis pathway as reported for other C. elegans lineages8. We further examined the phenotype of the ced-4;pig-1;ham-1 triple mutant and found that in the ADE neuron lineage – in contrast to the CEPV and CEPD neuron lineages – mutation of ced-4 did not lead to higher dopaminergic neuron numbers than in the ham-1;pig-1 mutant background (Fig. 2d). In summary, we report varying genetic interactions between ham-1, pig-1 and ced-4 in the distinct dopaminergic head neuron lineages.
Changes in protein abundance and signalling pathways in the pig-1 mutant
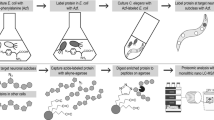
To identify proteins and pathways that are affected by loss of the PIG-1 kinase, we turned to an unbiased, mass spectrometry-based proteomics approach (Fig. 3a). We aimed to determine PIG-1 targets in ‘late’ C. elegans embryos, corresponding to the developmental time point at which the last cell divisions of the dopaminergic head neuron lineage take place. Thus, we compared the proteomes and phosphoproteomes of wild-type and pig-1 mutant embryos that were left to develop for 4 hours at 20 °C after isolation from a synchronised population of adult C. elegans hermaphrodites. Since PIG-1, which is an AMP-activated protein kinase-related protein, is likely to form part of a kinase cascade, we anticipate that both direct and indirect PIG-1 targets will be differentially phosphorylated in the pig-1 mutant. In addition, proteins participating in pathways affected by pig-1 mutation might also exhibit altered protein abundances. To facilitate interactive exploration of these large, complex datasets, the resulting protein abundance and protein phosphorylation data from the analysis of wild-type and pig-1 mutant embryos described below, were incorporated into the Encyclopedia of Proteome Dynamics (EPD) database (Fig. 3b).

Mass spectrometry-based proteomics analysis and data deposition at the Encyclopedia of Proteome Dynamics (EPD). (a) Proteomics pipeline with a phosphopeptide enrichment step using titanium dioxide (TiO2) (for experimental details please refer to Materials and Methods). 95% of each peptide fraction was used to enrich for phosphopeptides using TiO2 affinity chromatography43 before being analysed by LC-MS/MS. In addition, 5% of the peptide fractions were directly measured with LC-MS/MS to assess global protein abundances. HILIC = hydrophilic interaction chromatography, TiO2 = titanium dioxide, LC-MS/MS = liquid chromatography tandem mass spectrometry. (b) Overview of protein abundance and post-translational modification (PTM) data from wild-type and pig-1 mutant embryos available in the Encyclopedia of Proteome Dynamics (EPD).
In the 5% of each peptide fraction that was used to measure protein abundance, we detected 51,414 unique peptides for wild-type and 48,690 unique peptides for the pig-1 mutant, across the three biological replicates (Supplementary Table 1a). Peptides that were detected in at least two out of the three biological replicates were mapped to a total of 6,449 proteins for wild-type and 6,104 proteins for the pig-1 mutant, respectively. Of these proteins, 184 were found to be >2-fold upregulated and 113 > 2-fold downregulated in pig-1 mutant embryos, as compared with wild-type embryos, in at least two out of the three replicates (p-value < 0.05; false discovery rate (FDR) < 5% for both peptide and protein identification) (boxed in Fig. 4a) (Supplementary Table 2).

Global protein abundance changes in pig-1 mutant embryos. (a) Volcano plot (in log10 vs. log2 scale) depicting protein abundance changes in the pig-1 mutant embryo compared to wild-type embryos. The dashed orange and red lines indicate a p-value of 0.05 and 0.01 (1.3 and 2 in log10 scale), respectively. Boxed proteins were at least 2-fold down- or upregulated with a p-value of 0.05 and used for gene ontology term analysis. An interactive version of this volcano plot is available online via the EPD. To illustrate its functions, several proteins are highlighted and for one of them a tooltip with further available information is displayed. (b and c) Gene ontology term (GO) enrichment (biological process) of pig-1-variant proteins (boxed in the volcano plot). x and y-axis indicate semantic space used to group GO terms of related biological processes. Bubble sizes indicate the frequency of the GO term in the underlying C. elegans protein database (larger bubbles reflect more common terms) and bubble colour indicates statistical significance (the greener, the lower the p-value). (b) GO term analysis of proteins with at least 2-fold downregulated abundance in the pig-1 mutant (Supplementary Table 5a). (c) GO term analysis of proteins with at least 2-fold upregulated abundance in the pig-1 mutant (Supplementary Table 5b).
To investigate whether pig-1 mutation alters the abundance of proteins associated with specific biological processes, we analysed the Gene ontology (GO) term annotations associated with both the down- and upregulated proteins detected (Supplementary Table 5a,b). GO terms associated with downregulated proteins encompass myosin-regulation (‘striated muscle myosin thick filament assembly’, ‘muscle contraction’), locomotion (‘locomotion’, ‘inductive cell migration’) and metabolism (‘protein catabolic process’, ‘growth’, ‘cellular amino acid biosynthetic process’ and ‘metabolic process’) (Fig. 4b). An increase in abundance was observed for proteins linked to cell cycle regulation (‘DNA replication’, ‘cell cycle’, ‘cell division’, ‘mitotic spindle organisation’), embryo development, apoptosis (Fig. 4c) and microtubule regulation (Supplementary Table 5b). We conclude that pig-1 mutation leads to an overall decrease of proteins related to actomyosin processes, locomotion and metabolism and to an overall increase of proteins connected to the cell cycle, embryo development, apoptosis and microtubules.
We next examined selected proteins within these highlighted biological processes showing altered protein abundances in pig-1 mutant embryos. The selected proteins connected to either cell cycle regulation (Fig. 5a), or microtubule organisation (Fig. 5b), were more abundant in the pig-1 mutant than in wild-type embryos. In contrast, selected proteins connected to actomyosin regulation were downregulated: For example, we detected a >5-fold lower protein abundance (note the logarithmic scale in Fig. 5) of MLC-3 (myosin light chain), MYO-1 (myosin heavy chain structural genes), MYO-2, MYO-4, MYO-5 and MYO-6 (Fig. 5c).

Abundance changes of key pathway proteins in pig-1 mutant embryos. Proteins exhibiting a significantly different abundance (in log2 scale) in pig-1 mutant embryos as compared to wild-type embryos, in at least two out of the three biological replicates (p-value < 0.05, two-tailed t-test; FDR < 5%) (replicate values are indicated in black and the calculated average over the replicates is indicated in red). Proteins are grouped according to their functional association with. (a) cell cycle regulation, (b) microtubule regulation, (c) actomyosin regulation, (d) asymmetric cell division, (e) cell death or phagocytosis, (f) cell adhesion, or (g) vesicle trafficking.
Next, we examined protein abundance changes in pathways previously connected to pig-1 phenotypes. We found that proteins associated with asymmetric cell division, such as the centralspindlin complex-forming ZEN-4 (Zygotic epidermal ENclosure defective) and CYK-4 (CY to Kinesis defect) (reviewed in ref. 44), were more abundant in pig-1 mutant embryos (Fig. 5d). Further examples include the >3.5-fold higher abundances detected for GPR-2 (G Protein Regulator) and LIN-5 (abnormal cell LINeage), the orthologues of Drosophila PINS (Partner of INScuteable) and NuMA (Nuclear Mitotic Apparatus protein), respectively. GPR-2 and LIN-5 mediate the formation of differently sized daughter cells via microtubule-cortex interactions in the C. elegans one-cell embryo (for review see ref. 1). We also observed that Wnt signalling components, such as the Wnt receptor MOM-5 (MOre of MS) and DSH-2 (DiSHevelled related), showed higher abundance in pig-1 mutant embryos (Fig. 5d). Furthermore, as pig-1 genetically interacts with the apoptosis pathway (Fig. 2c, refs 8, 14, 15) and as pig-1 was suggested to promote the digestion of apoptotic cells14, we examined proteins related to cell death and corpse engulfment. We found that the apoptosis proteins CED-3 and CED-4 and the corpse engulfment proteins CED-2 and CED-12, all show increased abundance in pig-1 mutant embryos (Fig. 5e). We also detected altered abundances of several cell adhesion proteins in pig-1 mutant embryos (Fig. 5f). These data support the reported function of PIG-1 in cell detachment (‘shedding’) by interfering with the cell-surface expression of cell adhesion proteins in C. elegans caspase-deficient embryos14. PIG-1-mediated cell shedding was suggested to involve endocytosis14. Consistent with this proposal, we found that the vesicle trafficking protein ARF-1.2 (ADP-Ribosylation Factor-related), which is required for cell shedding14, was downregulated in pig-1 mutants, while the abundance of other vesicle trafficking proteins was upregulated (Fig. 5g).
Mutation of the PIG-1 kinase leads to phosphorylation changes in key development pathways
We hypothesised that both direct and indirect targets of the PIG-1 kinase may exhibit altered protein phosphorylation levels in pig-1 mutant embryos. To test this hypothesis, we compared phosphopeptide abundances in pig-1 mutant and wild-type embryos. Using a phosphopeptide enrichment step prior to liquid chromatography-mass spectroscopy analysis (Fig. 3a), we detected 7,247 phosphosites in wild-type embryos and 7,913 phosphosites in pig-1 mutant embryos across the three biological replicates (FDR < 5%, localisation probability ≥75%) (Fig. 6) (Supplementary Table 1b). To facilitate the convenient exploration of this complex phosphoproteomics dataset, we created an interactive, searchable version of the data summarised in Fig. 6, which is available online via the EPD (http://www.peptracker.com/epd/). The plot features a custom visualisation tool that allows every phosphorylation site we identified to be selected and traced through a set of cognate metadata. We provide information about the abundance of each selected protein and its phosphosites, the amino acid position of each phosphosite in the protein, statistical evaluation of biological replicates, as well as linking to the raw mass spectrometry files. For wild-type embryos, these phosphosites correspond to 1,925 phosphorylated proteins (localisation probability ≥75%) (Supplementary Table 1b, Supplementary Table 3). To the best of our knowledge, this is the first dataset describing global protein phosphorylation in C. elegans embryos.

Phosphosites detected in wild-type and pig-1 mutant embryos. (a) Detected protein post translational modification (PTM) phosphorylation sites in wild-type embryos. Each grey line represents a phosphorylation site and models its behaviour across multiple dimensions. The phosphorylation lines intersect the y-axes on the values that were detected for the specific site. The axis dimensions are ‘modified amino acid’ (threonine (T), serine (S) or tyrosine (Y)), ‘protein intensity’ (in log10 scale), ‘site intensity’ (in log10 scale), ‘PTM site probability’, ‘position in protein’ (in log10 scale), ‘score’ (of identification, the product of all peptide posterior error probabilities used for identification that is calculated in MaxQuant), and number of ‘replicates with data’. (b) Detected protein phosphorylation sites of two example proteins in pig-1 mutant embryos - the grey lines for background phosphorylation were removed in this graph. Both of these plots are available online as interactive visualisations via the EPD (http://www.peptracker.com/epd/). Firstly, the axes can be filtered such that only elements contained within the user-defined boxes are shown: in the depicted graphs an example filter of ≥0.75 was applied for the ‘PTM site probability’ axis. Secondly, proteins can be searched and their phosphorylation sites can be highlighted in colour: Here, this was done for the same two example proteins and the search bar is shown on top of one of the graphs. Thirdly, selection of a phosphorylation line will lead to the display of a tooltip box containing further information: In the graphs shown, this was done for three examples of phosphorylation sites.
To help evaluate the significance of differences in the phosphoproteomes of pig-1 mutant and wild-type embryos, we aimed to determine the stoichiometry of phosphorylation in relation to protein abundance. For this, we compared pig-1/wild-type phosphopeptide abundances to the corresponding pig-1/wild-type protein abundances (Fig. 7a). This showed that most proteins are located within two standard deviations of the diagonal of the plot, indicating that changes in phosphosite abundance tally with changes in protein abundance. Thus, these proteins are not significantly changing their stoichiometry of phosphorylation. However, proteins located more than two standard deviations from the mean were considered strong candidates for being differentially phosphorylated in the pig-1 mutant, as compared with wild-type, embryos (Supplementary Table 4a). In addition to the differentially phosphorylated proteins, we identified 220 proteins that were exclusively phosphorylated in wild-type embryos and 288 proteins that were only phosphorylated in pig-1 mutant embryos (Fig. 7b) (Supplementary Table 4b,c). The GO terms associated with proteins harbouring the respective up- and downregulated phosphosites were similar and connected to embryo development, the cell cycle and apoptosis (Fig. 7c,d). We note that this similarity in the biological processes connected with either increased, or decreased, levels of phosphorylation contrasts with our earlier finding that distinct processes were associated with the proteins showing either increased, or decreased abundance (Fig. 4b,c). In summary, we find that pig-1 mutation leads to differential phosphorylation of multiple proteins associated with key developmental processes.

Global phosphoproteome changes in pig-1 mutant embryos. (a) Phosphorylation site abundance ratio (pig-1/wild-type) compared to protein abundance ratio (pig-1/wild-type). The orange dotted line indicates where the phosphorylation site ratio is equal to the protein ratio. Points within the orange and red areas are within one and two standard deviations from equality, respectively. An interactive version of this plot is accessible online via the EPD. In this illustration, detected phosphorylation sites for three example proteins were marked and a tooltip containing further information is shown for one of these phosphorylation sites. (b) Number of phosphorylated proteins detected either in wild-type (red) or in pig-1 mutant embryos (blue) or in both genetic backgrounds (purple overlap)(localisation probability >75%). (c and d) Gene ontology (GO) term enrichment (biological process) of pig-1 variant phosphosites. x- and y-axis indicate semantic space used to group GO terms of related biological processes. Bubble sizes indicate the frequency of the GO term in the underlying C. elegans protein database (larger bubbles reflect more common terms) and bubble colour indicates statistical significance (the greener, the lower the p-value). (c) GO term analysis of downregulated phosphosites in the pig-1 mutant (related to Supplementary Table 5c). Phosphosites downregulated in pig-1 (outside of the 2 standard deviation range in (a)) were pooled with phosphosites that were only detected in wild-type embryos (red in (b)). (d) GO term analysis of phosphosites with upregulated abundance in the pig-1 mutant (Supplementary Table 5d). Phosphosites upregulated in pig-1 mutant embryos (outside of the 2 standard deviation range in (a)) were pooled with the phosphosites that were only detected in pig-1 mutant embryos (blue in b)).
Next, we examined the phosphorylation of selected proteins groups associated with changes in either protein abundance, or protein phosphorylation levels, in pig-1 mutant embryos, as determined by GO term analysis. As seen in the phosphosite GO term analysis (Fig. 7e,f), pig-1 mutation differentially affects phosphorylation of proteins mediating similar processes (Fig. 8). For example, phosphorylation of S80 in the cell division cycle protein CDC-25.2 was only detected in wild-type embryos, while phosphorylation of CDC-5L T377 was only detected in pig-1 mutants (Fig. 8a). Furthermore, phosphorylation of S58 and T530 in the DNA replication protein MCM-10 [yeast MCM (licensing factor) related] is only detected in pig-1 mutant embryos, while phosphorylation of ICP-1 (INCENP (inner centromere protein) homolog) is decreased at least 10-fold at 4 different sites (Fig. 8a) (note logarithmic scale in Fig. 8).

Phosphopeptide abundance changes of key pathway proteins in pig-1 mutant embryos. Ratio of phosphopeptide abundance and protein abundance (in log2 scale) in pig-1 mutants as compared to wild-type embryos (white diamonds). Phosphosites that were only detected in either pig-1 mutants (blue diamonds), or wild-type embryos (red diamonds), are shown above and below the y-axis, respectively. Proteins are grouped according to their functional association with (a) cell cycle regulation, (b) microtubule regulation, (c) actomyosin regulation, (d) asymmetric cell division, (e) cell death or phagocytosis, (f) cell adhesion, or (g) vesicle trafficking.
We found that pig-1 mutation affects the phosphorylation of proteins previously associated with pig-1 function. PIG-1 was shown to regulate the localisation of NMY-2 (Non-muscle MYosin)9, 10, 13 and NMY-2 itself affects the asymmetric distribution of several polarity proteins in the C. elegans one-cell embryo1. We found that in pig-1 mutant embryos, NMY-2 S211 and S1974 are ~8-fold and ~10-fold less phosphorylated (Fig. 8c). In addition, PIG-1 was shown to control myosin accumulation in a pathway parallel to ANI-1 [ANIllin (actin-binding protein)]13 and we found that ANI-1 S352 is over 8-fold and ANI-1 T486 over 40-fold less phosphorylated in pig-1 mutants (Fig. 8d). Furthermore, when investigating apoptotic corpse engulfment proteins, we found that CED-1 S1050 and CED-8 S411 are only phosphorylated in wild-type animals (Fig. 8e).
PIG-1 was suggested to affect the expression of cell adhesion proteins14 and we found that AJM-1 (Apical Junction Molecule) S766 and DLG-1 (Drosophila Discs LarGe homolog) S161 and S471 exhibit increased phosphorylation in pig-1 mutant embryos (Fig. 8f). DLG-1 is a MAGUK (membrane-associated guanylate kinase) protein that mediates a microtubule-dependent pathway important for the segregation of cell fate determinants in Drosophila (reviewed in ref. 1). Furthermore, the inappropriate cell-surface expression of cell-adhesion molecules in pig-1 mutants was proposed to be based on impaired endocytosis14, a process that appears to play an important role in asymmetric cell division1. We found increased phosphorylation for several endocytosis-associated proteins in pig-1 mutant embryos (Fig. 8g).
Lastly, we found that pig-1 mutation also affects the phosphorylation status of proteins connected to asymmetric cell division. PAR-3 (abnormal embryonic PARtitioning of cytoplasm) for example, the C. elegans orthologue of Drosophila Bazooka, exhibits decreased phosphorylation of S1032 (Fig. 8d). pig-1 mutation also leads to altered phosphorylation of MEX-5 (Muscle EXcess), MEX-6 and PLK-1 (POLO Kinase) (Fig. 8d), proteins that segregate into the anterior daughter cell and help to establish the correct division of the C. elegans one-cell embryo (reviewed refs 1, 45). Furthermore, we found that phosphorylation of DSH-1 S904 is only detected in pig-1 mutants (Fig. 8d). Dishevelled proteins are Wnt pathway components and normally enriched at the posterior part of an asymmetrically dividing cell3.
To provide the research community with convenient access to the results from this study, all the proteomics data describing protein and phosphopeptide abundance changes in the pig-1 mutant embryos, as well as phosphopeptide detection in C. elegans wild-type embryos, have been incorporated in the Encyclopaedia of Proteome Dynamics (EPD) − http://www.peptracker.com/epd/. The EPD currently includes the largest collection of annotated, searchable mass spectrometry-based proteomics data from studies in C. elegans, linked with other extensive data sets from studies on human cells and model organisms. In addition, the corresponding raw mass spectrometry data files are freely available via the PRIDE repository, a partner in the ProteomXchange Consortium (http://proteomecentral.proteomexchange.org/).
Discussion
We report that the transcription factor HAM-1 and the kinase PIG-1 have a role in the specification of C. elegans dopaminergic head neurons. ham-1 and pig-1 appear to be required for asymmetric cell divisions that result in an apoptotic daughter cell and either a neuron, or a neuronal precursor5, 6, 8, 9. In the case of dopaminergic head neurons, the last cell division in the CEPVL/R neuron lineages and the second last division for the CEPDL/R and ADEL/R neuron lineages, are the first ones to directly produce such an apoptotic daughter cell (Fig. 1d, http://wormweb.org/celllineage). We therefore consider it likely that ham-1 and pig-1 affect the asymmetric division of these neuroblasts. Genetic interactions in the CEPD lineage support that mutation of ham-1 and pig-1 might lead to sister-sister cell fate conversions, thereby turning a normally apoptotic aunt cell into a dopaminergic neuron precursor cell. When blocking cell death by mutation of the apoptosis pathway gene ced-4, these conversions are unmasked and result in higher neuron numbers. In addition, our experiments support that the genetic interaction between ham-1 and pig-1 depends on the exact neuronal lineage analysed. pig-1 was implicated as a transcriptional target and HAM-1 effector in the Q.a neuroblast9. Consistent with this, we find an increased number of ADE neurons in ham-1 and pig-1 single and double mutants. In contrast, the reduced number of CEPD neurons observed in ham-1 mutants is supressed by pig-1.
A key feature of this study was the use of a combination of quantitative, mass spectrometry-based proteomic and phosphoproteomic analyses to provide an in-depth, unbiased characterisation of the global changes that occur after genetic disruption of the PIG-1 kinase in C. elegans embryos. Direct targets of the PIG-1 kinase are expected to exhibit lower abundance of phosphorylated peptides in pig-1 mutants, as compared with wild-type embryos. However, by analysing overall protein phosphopeptide and protein abundance changes, we were also able to measure indirect effects of pig-1 inactivation. The extent of differential phosphorylation between wild-type and pig-1 mutant embryos suggests that the majority of detected phosphorylation sites affected may not depend on direct phosphorylation by PIG-1 kinase. We note that this is the first study to analyse the effects of a kinase mutation on the global phosphoproteome in C. elegans.
In addition to the comparison of wild-type and pig-1 mutant embryos described above, this study represents the first global phosphoproteome analysis reported for C. elegans embryos. We identify 5,051 phosphosites, corresponding to 1,639 phosphorylated proteins, in C. elegans wild-type embryos, which are detected in at least two out of the three biological replicates (FDR < 5%, localisation probability >75%) (Supplementary Table 1b). In addition, we detect 288 proteins that are only phosphorylated in pig-1 mutant embryos (Supplementary Table 4c). There are only a limited number of phosphoproteome studies in C. elegans, reporting the identification of ~6,80046, ~6,50047 and ~3,50048 phosphorylation sites, respectively, in adult animals. However, our dataset is the first to identify phosphosites in the embryo and thus represents a valuable resource for the investigation of C. elegans development. Comparison with the C. elegans dataset from PhosphoPep48, a protein phosphorylation database for model organisms, showed that ~50% of the phosphosites we detect in embryos are novel (Supplementary Table 6).
We found that mutation of the PIG-1 kinase preferentially affected proteins associated with the cell cycle and asymmetric cell division, supporting the idea that PIG-1 might regulate the timing of cell cycle events in the neuroblast and thereby influence neuroblast polarity8. We note that cell division, cell polarity and differentiation are all tightly coordinated during animal development (reviewed in refs 49, 50). In addition, our results indicate that PIG-1 influences proteins regulating myosin distribution and spindle positioning. These processes are known to influence asymmetric cell divisions (for review see refs 1, 11) and might thus provide clues as to why pig-1 mutants exhibit daughter cells of approximately equal size. Finally, microtubule-associated proteins are more abundant and differentially phosphorylated in pig-1 mutants. During asymmetric cell divisions, microtubules maintain the established cell polarity by mediating interactions between centrosomes and the cell cortex1, 11. Therefore, the affected candidate proteins offer a promising starting point for further research.
The mammalian PIG-1 homologue, MELK, has also been implicated in cell cycle regulation21, 27,28,29,30. For example, MELK was shown to phosphorylate the mitosis-promoting phosphatase CDC25B that promotes transition to mitosis21. Since MELK overexpression blocks the transition to mitosis21, 29, 51, CDC25B phosphorylation by MELK is assumed to be inhibitory and mutation of the MELK kinase is therefore expected to stimulate cell division. Moreover, MELK overexpression has been associated with tumour growth and aggressiveness (for review see ref. 20).
In summary, the data presented here provide important new insights into the regulatory processes and signalling events involving PIG-1 that control asymmetric cell division during neuronal differentiation in C. elegans. In addition, this first detailed phosphoproteomic analysis of C. elegans embryos, together with the new interactive data visualisation tools we have developed, provide a valuable resource for developmental biologists studying both C. elegans and other species.
Methods
C. elegans strains and maintenance
TG2435 Bristol, a backcrossed version of BY20035, was used as the wild-type strain. TG4200 (ham-1(gt1984) IV;vtIs1 V) was isolated following ethyl methanesulfonate (EMS)-mutagenesis and backcrossed six times. Worms were maintained at 20 °C on nematode growth medium (NGM) plates seeded with E. coli strain OP50.
Strains
BY200 vtIs[pdat-1::gfp;rol-6] V
TG2435 vtIs[pdat-1::gfp;rol-6] V
TG4200 ham-1(gt1984) IV;vtIs1 V
TG4201 ham-1(n1438) IV;vtIs1 V
TG4202 ced-3(n717);vtIs1 V
TG4203 ced-4(n1162);vtIs1 V
TG4204 ced-4(n1162);ham-1(gt1984) IV;vtIs1 V
TG4205 pig-1(gm344) IV;vtIs1 V
TG4206 pig-1(gm344) IV; ham-1(gt1984); vtIs1 V
TG4207 ced-4(n1162) III; pig-1(gm344) IV;vtIs1 V
TG4208 ced-4(n1162) III; pig-1(gm344) IV; ham-1(gt1984); vtIs1 V
Microscopy
For live imaging of neurons, L4 stage and young adult C. elegans hermaphrodites were placed in a drop of 20 mM levamisole on a slide containing an agar pad. A 1.5 mm cover slip was then placed on each slide and animals were imaged with a DeltaVision microscope. Quantification was performed blind with a Zeiss Axio Scope microscope.
Mass spectrometry
Protein extraction and digest
For protein extraction, twelve 9 cm plates containing a synchronised population of C. elegans gravid hermaphrodites (ca. 40,000 animals) were washed to remove already laid eggs. The unlaid eggs were collected by bleaching and let develop for 4 hours at 20 °C before adding 500 μL of lysis buffer (8 M urea in 100 mM triethyl ammonium bicarbonate (TEAB) pH 8.5). The C. elegans embryos were then flash-frozen in liquid nitrogen and stored at −20 °C. The samples were ultrasonicated using a Bioruptor (30 cycles: 30 sec on, 30 sec off), reduced using tris(2-carboxyethyl)phosphine (TCEP) (25 mM) for 30 minutes at room temperature, then alkylated in the dark for 30 minutes using 2-iodoacetamide (50 mM). Total protein was quantified using the EZQ assay (Life Technologies). The lysates were diluted with 100 mM TEAB 4-fold for the first digestion with endoprotease Lys-C (Wako), then further diluted 2.5-fold before a second digestion with trypsin. Lys-C and trypsin were used at an enzyme to substrate ratio of 1:50 (w/w). The digests were carried out overnight at 37 °C, then stopped by acidification with trifluoroacetic acid (TFA) to a final concentration of 1% (v/v).
Peptide desalting and solid phase extraction
Prior to fractionation, the protein digests were desalted using C18 Sep-Pak Cartridges (Waters). Cartridges were first activated with Acetonitrile (ACN) and equilibrated with 50% ACN in water according to the manufacturer’s protocol. The samples were loaded and washed 4 times with 500 μL water containing 0.1% TFA. The peptides were eluted into a fresh Eppendorf tube with 800 μL 50% ACN and then dried in vacuo.
Off-line HILIC fractionation
Hydrophilic interaction chromatography (HILIC) was performed on a Dionex UltiMate 3000 (Thermo Scientific) using a similar protocol to the method described previously52,53,54. The dried peptides were redissolved in 80% ACN incorporating 0.1% TFA. The peptides were resolved on TSK-gel amide 80- column (TOSOH) using an inverted organic gradient of solvent A (water, 0.1% TFA) and solvent B (ACN, 0.1% TFA). 24 fractions were collected in 96-well deep well plates.
Phosphopeptide enrichment
Phosphopeptide enrichment was carried out on the HILIC fractions with titanium IMAC (immobilised metal affinity chromatography) beads (MagReSyn) using the original protocol by Larsen et al.43 on an automated bead processing robot (KingFisher, Thermo Fisher Scientific) as described in Tape et al.55. The enriched phosphopeptide fractions were dried, redissolved in 5% formic acid and analysed in data-dependent mode on Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific).
Liquid chromatography electrospray tandem mass spectrometry analysis (LC-ES-MS/MS)
5% of the material was analysed using a Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with a Dionex ultra high-pressure liquid chromatography system (nano RSLC) without enrichment in order to quantify changes in protein expression levels. RP-LC was performed using a Dionex RSLC nano HPLC (Thermo Scientific). Peptides were injected onto a 75 μm × 2 cm PepMap-C18 pre-column and resolved on a 75 μM × 50 cm reverse phase-C18 EASY-Spray temperature-controlled integrated column-emitter (Thermo Fisher Scientific) using a four hour multistep gradient from 5% B to 35% B with a constant flow of 200 nL/min. The mobile phases were: 2% ACN incorporating 0.1% FA (Solvent A) and 80% ACN incorporating 0.1% FA (Solvent B). The spray was initiated by applying 2.5 kV to the EASY-Spray emitter and the data were acquired under the control of Xcalibur software in a data-dependent mode.
95% of the material was subject to phosphopeptide enrichment and analysed on a Q Exactive Plus using a two hour gradient.
Database searching and protein quantification
The raw mass spectrometry data were processed using the MaxQuant software suite (version 1.5.2.8). Proteins and peptides were identified using the UniProt C. elegans reference proteome database (Swiss-Prot and Trembl separately), using the Andromeda search engine56, 57 with the following search parameters: carbamidomethylation of cysteines was defined as a fixed modification, while methionine oxidation, acetylation of N-termini of proteins, conversion of glutamine to pyro-glutamate and phosphorylation on STY were set as variable modifications. The false discovery rate was set to 5% for proteins and peptides.
Mass spectrometry data analysis
The volcano plot depicting protein abundances was generated based on the label-free quantification intensity of proteins present in at least two of the three biological replicates in wild-type and mutant embryos. Statistical significance was determined with a two-tailed Student’s t-test with a randomisation setting fixed at 250 on the Perseus software platform58. For phosphorylation site analysis, data analysis was performed in Python using the scipy statistical package. Only phosphosites with a localisation site probability higher than 75% and a score higher than 40 were considered. Elements present in both wild-type and pig-1 mutant embryos were added to the phosphosite comparison analysis, while elements present in only one of the genotypes were separated into two genotype-specific output files and visualisations. In the comparison analysis, the mutant over wild-type phosphosite abundance and protein abundance ratios were calculated and converted into log2 format for visualisation purposes. In the genotype-specific analysis, a log10 transformation was applied to the protein intensity, phosphorylation site intensity and the position of the post-translational modification within the protein to render the visualisation more user-friendly. The visualisation is presented in D3.js and the data is stored in Cassandra.
Data depository
Data generated in this study is accessible via the Encyclopaedia of Proteome Dynamics (EPD)34 (http://www.peptracker.com/epd/) for convenient exploring and display. In addition, the raw mass spectrometry files as well as the MaxQuant output have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE59 partner repository with the dataset identifier PXD005752.
Gene ontology term analysis
Gene ontology (GO)-term enrichment analysis was performed with the DAVID functional annotation tool (https://david.ncifcrf.gov/)60, 61. The complete list of C. elegans proteins detected in mass spectrometry-based proteomics was used as background and the GO term subcategories ‘GOTERM_BP_DIRECT’ ‘GOTERM_CC_DIRECT’ and ‘GOTERM_MF_DIRECT’ selected for analysis. The results were analysed with REVIGO62 and plotted using the provided R script to remove GO term redundancy. In REVIGO, p-values were provided and the C. elegans database and the SimRel semantic similarity measure were used for analysis. A ‘medium’ REVIGO list size was selected.
References
Knoblich, Ja Asymmetric cell division: recent developments and their implications for tumour biology. Nat. Rev. Mol. Cell Biol. 11, 849–860 (2010).
Goldstein, B. & Macara, I. G. The PAR Proteins: Fundamental Players in Animal Cell Polarization. Dev. Cell 13, 609–622 (2007).
Bertrand, V. Β-Catenin-Driven Binary Cell Fate Decisions in Animal Development. Wiley Interdiscip. Rev. Dev. Biol. n/a–n/a, doi:10.1002/wdev.228 (2016).
Desai, C., Garriga, G., Mclntire, S. & Horvitz, H. A genetic pathway for the development of the Caenorhabditis elegans HSN motor neurons. Nature 336 (1988).
Guenther, C. & Garriga, G. Asymmetric distribution of the C. elegans HAM-1 protein in neuroblasts enables daughter cells to adopt distinct fates. Development 122, 3509–18 (1996).
Frank, C. A., Hawkins, N. C., Guenther, C., Horvitz, H. R. & Garriga, G. C. elegans HAM-1 positions the cleavage plane and regulates apoptosis in asymmetric neuroblast divisions. Dev. Biol. 284, 301–10 (2005).
Leung, A. et al. C. elegans HAM-1 functions in the nucleus to regulate asymmetric neuroblast division. Dev. Biol. 410, 56–69 (2015).
Cordes, S., Frank, C. A. & Garriga, G. The C. elegans MELK ortholog PIG-1 regulates cell size asymmetry and daughter cell fate in asymmetric neuroblast divisions. Development 133, 2747–56 (2006).
Feng, G. et al. Developmental stage-dependent transcriptional regulatory pathways control neuroblast lineage progression. Development 140, 3838–47 (2013).
Ou, G., Stuurman, N., D’Ambrosio, M. & Vale, R. D. Polarized myosin produces unequal-size daughters during asymmetric cell division. Science 330, 677–80 (2010).
Roubinet, C. & Cabernard, C. Control of asymmetric cell division. Curr. Opin. Cell Biol. 31, 84–91 (2014).
Chien, S.-C., Brinkmann, E.-M., Teuliere, J. & Garriga, G. Caenorhabditis elegans PIG-1/MELK acts in a conserved PAR-4/LKB1 polarity pathway to promote asymmetric neuroblast divisions. Genetics 193, 897–909 (2013).
Pacquelet, A., Uhart, P., Tassan, J. P. & Michaux, G. PAR-4 and anillin regulate myosin to coordinate spindle and furrow position during asymmetric division. 210, 1085–1099 (2015).
Denning, D. P., Hatch, V. & Horvitz, H. R. Programmed elimination of cells by caspase-independent cell extrusion in C. elegans. Nature 488, 226–230 (2012).
Hirose, T. & Horvitz, H. R. An Sp1 transcription factor coordinates caspase-dependent and -independent apoptotic pathways. Nature 500, 354–8 (2013).
van Dijk, M. et al. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat. Genet. 37, 514–9 (2005).
McInerny, C. J. Cell cycle regulated transcription: from yeast to cancer. F1000Research 5, 1–5 (2016).
van Abel, D., Abdul-Hamid, O., van Dijk, M. & Oudejans, C. B. M. Transcription factor STOX1A promotes mitotic entry by binding to the CCNB1 promotor. PLoS One 7, 1–8 (2012).
Nie, X. et al. Transcription factor STOX1 regulates proliferation of inner ear epithelial cells via the AKT pathway. Cell Prolif. 48, 209–220 (2015).
Ganguly, R. et al. MELK-a conserved kinase: functions, signaling, cancer, and controversy. Clin. Transl. Med. 4, 11 (2015).
Davezac, N., Baldin, V., Blot, J., Ducommun, B. & Tassan, J.-P. Human pEg3 kinase associates with and phosphorylates CDC25B phosphatase: a potential role for pEg3 in cell cycle regulation. Oncogene 21, 7630–41 (2002).
Seong, H. A., Jung, H. & Ha, H. Murine protein serine/threonine kinase 38 stimulates TGF-β signaling in a kinase-dependent manner via direct phosphorylation of smad proteins. J. Biol. Chem. 285, 30959–30970 (2010).
Seong, H. A., Jung, H., Manoharan, R. & Ha, H. PDK1 protein phosphorylation at Thr354 by murine protein serine-threonine kinase 38 contributes to negative regulation of PDK1 protein activity. J. Biol. Chem. 287, 20811–20822 (2012).
Seong, H. A. & Ha, H. Murine protein serine-threonine kinase 38 activates p53 function through Ser15 phosphorylation. J. Biol. Chem. 287, 20797–20810 (2012).
Jung, H., Seong, H. – & Ha, H. Murine Protein Serine/Threonine Kinase 38 Activates Apoptosis Signal-regulating Kinase 1 via Thr838 Phosphorylation. J. Biol. Chem. 283, 34541–34553 (2008).
Lin, M.-L., Park, J.-H., Nishidate, T., Nakamura, Y. & Katagiri, T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 9, R17 (2007).
Nakano, I. et al. Maternal embryonic leucine zipper kinase (MELK) regulates multipotent neural progenitor proliferation. J. Cell Biol. 170, 413–27 (2005).
Le Page, Y., Chartrain, I., Badouel, C. & Tassan, J.-P. A functional analysis of MELK in cell division reveals a transition in the mode of cytokinesis during Xenopus development. J. Cell Sci. 124, 958–68 (2011).
Gray, D. Maternal Embryonic Leucine Zipper Kinase/Murine Protein Serine-Threonine Kinase 38 Is a Promising Therapeutic Target for Multiple Cancers. Cancer Res. 65, 9751–9761 (2005).
Blot, J., Chartrain, I., Roghi, C., Philippe, M. & Tassan, J.-P. Cell Cycle Regulation of pEg3, a New Xenopus Protein Kinase of the KIN1/PAR-1/MARK Family. Dev. Biol. 241, 327–338 (2002).
Badouel, C., Chartrain, I., Blot, J. & Tassan, J. P. Maternal embryonic leucine zipper kinase is stabilized in mitosis by phosphorylation and is partially degraded upon mitotic exit. Exp. Cell Res. 316, 2166–2173 (2010).
Flames, N. & Hobert, O. Gene regulatory logic of dopamine neuron differentiation. Nature 458, 885–9 (2009).
Doitsidou, M. et al. A combinatorial regulatory signature controls terminal differentiation of the dopaminergic nervous system in C. elegans. Genes Dev. 27, 1391–405 (2013).
Larance, M., Ahmad, Y., Kirkwood, K. J., Ly, T. & Lamond, A. I. Global subcellular characterization of protein degradation using quantitative proteomics. Mol. Cell. Proteomics 12, 638–50 (2013).
Nass, R., Hall, D. H., Iii, D. M. M., Blakely, R. D. & Miller, D. M. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 99, 3264–9 (2002).
Sulston, J. E. & Horvitz, H. R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156 (1977).
Sulston, J. E., Schierenberg, E., White, J. G. & Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 (1983).
Doitsidou, M., Poole, R. J., Sarin, S., Bigelow, H. & Hobert, O. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5, e15435 (2010).
Minevich, G., Park, D. S., Blankenberg, D., Poole, R. J. & Hobert, O. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–69 (2012).
Doitsidou, M., Flames, N., Lee, A. C., Boyanov, A. & Hobert, O. Automated screening for mutants affecting specification in C. elegans. 5, 869–872 (2008).
Ellis, H. M. & Horvitz, H. R. Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829 (1986).
Avery, L. & Horvitz, H. R. A Cell That Dies during Wild-Type C. elegans Development Can Function as a Neuron in a ced-3 Mutant. Cell 51, 1071–1078 (1987).
Larsen, M. R., Thingholm, T. E., Jensen, O. N., Roepstorff, P. & Jørgensen, T. J. D. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886 (2005).
White, E. A. & Glotzer, M. Centralspindlin: At the heart of cytokinesis. Cytoskeleton 69, 882–892 (2012).
Noatynska, A. & Gotta, M. Cell polarity and asymmetric cell division: the C. elegans early embryo. Essays Biochem 53, 1–14 (2012).
Zielinska, D. F., Gnad, F., Jedrusik-Bode, M., Wiśniewski, J. R. & Mann, M. Caenorhabditis elegans has a phosphoproteome atypical for metazoans that is enriched in developmental and sex determination proteins. J. Proteome Res. 8, 4039–4049 (2009).
Rhoads, T. W. et al. NeuCode Labeling in Nematodes: Proteomic and Phosphoproteomic Impact of Ascaroside Treatment in Caenorhabditis elegans., doi:10.1074/mcp.M115.049684 (2015).
Bodenmiller, B. et al. PhosphoPep—a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 26, 1339–1340 (2008).
Ishidate, T., Elewa, A., Kim, S., Mello, C. C. & Shirayama, M. Divide and differentiate: CDK/Cyclins and the art of development. Cell Cycle 13, 1384–1391 (2014).
Noatynska, A., Tavernier, N., Gotta, M. & Pintard, L. Coordinating cell polarity and cell cycle progression: what can we learn from flies and worms? Open Biol. 3, 130083 (2013).
Vulsteke, V. et al. Inhibition of spliceosome assembly by the cell cycle-regulated protein kinase MELK and involvement of splicing factor NIPP1. J. Biol. Chem. 279, 8642–7 (2004).
Alpert, A. J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. 499, 177–196 (1990).
McNulty, D. E. & Annan, R. S. Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteomics 7, 971–980 (2008).
Bensaddek, D. et al. Micro-proteomics with iterative data analysis: Proteome analysis in C. elegans at the single worm level. Proteomics 16, 381–392 (2016).
Tape, C. J. et al. Reproducible automated phosphopeptide enrichment using magnetic TiO2 and Ti-IMAC. Anal. Chem. 86, 10296–10302 (2014).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–72 (2008).
Cox, J. et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Vizcaíno, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 6 (2011).
Acknowledgements
We thank Jens Januschke and Ralf Schnabel for useful discussions and commenting on the manuscript. Some strains were provided by the CGC (Caenorhabditis Genetics Centre), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). S.L.O. was supported by a PhD fellowship (Molecular and Cellular Biology programme) from the Wellcome Trust and by ISSF funding from the Wellcome Trust. D.B. is funded by the Human Induced Pluripotent Stem Cells Initiative (098503/E/12/Z). A.B.M. is funded by a Wellcome Trust programme grant to A.I.L. (073980/Z/03/B). This work was part funded by a Wellcome Trust Strategic award to A.I.L. (073980/Z/03/B) and by a Wellcome Trust Programme grant to A.G. (0909444/Z/09/Z) and supported by infrastructure funded by a Wellcome Trust Strategic award (097045/B/11/Z).
Author information
Authors and Affiliations
Contributions
S.L.O. and A.G. conceived, designed and interpreted experiments and wrote the paper. S.L.O. performed the C. elegans experiments and analysed the data. D.B. performed the mass spectrometry and analysed the data with MaxQuant. A.B.M. analysed and visualised data for the Encyclopedia of proteome dynamics. A.I.L. contributed tools and reagents and provided advice on mass spectrometry data analysis. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Offenburger, SL., Bensaddek, D., Murillo, A.B. et al. Comparative genetic, proteomic and phosphoproteomic analysis of C. elegans embryos with a focus on ham-1/STOX and pig-1/MELK in dopaminergic neuron development. Sci Rep 7, 4314 (2017). https://doi.org/10.1038/s41598-017-04375-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04375-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
