
Abstract
Appendicitis is one of the most common conditions requiring acute surgery and can pose a threat to the lives of affected individuals. We performed a genome-wide association study of appendicitis in 7,276 Icelandic and 1,139 Dutch cases and large groups of controls. In a combined analysis of the Icelandic and Dutch data, we detected a single signal represented by an intergenic variant rs2129979 [G] close to the gene PITX2 associating with increased risk of appendicitis (OR = 1.15, P = 1.8 × 10−11). We only observe the association in patients diagnosed in adulthood. The marker is close to, but distinct from, a set of markers reported to associate with atrial fibrillation, which have been linked to PITX2. PITX2 has been implicated in determination of right-left symmetry during development. Anomalies in organ arrangement have been linked to increased prevalence of gastrointestinal and intra-abdominal complications, which may explain the effect of rs2129979 on appendicitis risk.
Similar content being viewed by others

Introduction
Appendicitis is a condition whereby the inner lining of the appendix becomes inflamed. Appendicitis has a relatively high prevalence; 8.6% of all males and 6.7% of all females will at some point develop appendicitis1. Rate of diagnosis peaks in childhood, although around half of cases are diagnosed in adulthood (Supplementary Table 1). The median age of diagnosis for all cases in the US is 211. Appendix perforation presents a significant risk in acute appendicitis, causing release of bacteria into the abdominal cavity and significantly worsening prognosis2. Obstruction of the appendiceal lumen appears to play an important role in the pathogenesis of appendicitis3, but the condition remains poorly understood4.
No sequence variants influencing risk of appendicitis have been reported. A family history of appendicitis greatly increases an individual’s risk, and twin studies suggest that genetics account for around 30% of appendicitis risk, with a strong sex-linked effect5, 6. We estimated the risk ratio among siblings (λS) of Icelandic patients with appendicitis (N = 8,160) to be 1.95 by cross-matching with the nationwide genealogy7 (Supplementary Table 2).
In order to discover sequence variants associating with risk of appendicitis, we performed a genome-wide association study (GWAS) of appendicitis, combining data from Icelandic and Dutch populations.
Results
We performed a GWAS on combined data from 7,267 Icelandic and 1,139 Dutch cases of appendicitis, and 327,134 Icelandic and 4,587 Dutch controls. The Icelandic cases were identified from hospital records indicating diseases of the appendix coded according to the International Classification of Diseases (ICD) between 1983 and 2015 (Supplementary Table 3), whereas the Dutch cases were self-reported. In Iceland, we test 32.5 million imputed markers identified through whole-genome sequencing of 15,220 Icelanders and subsequently imputed into chip-typed individuals through long-range haplotype phasing. Genotype probabilities were calculated for first and second-degree relatives of chip-typed individuals8. In the Netherlands we test 15,268,903 markers identified through whole-genome sequencing of 249 Dutch trios (GoNL). We combined the results of 11,290,636 markers found in both populations. When testing for association, we used weighted genome-wide significance thresholds depending on variant class9.
A sequence variant at 4q25 associates with appendicitis
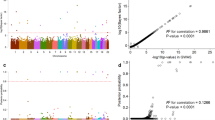
In the meta-analysis of Iceland and the Netherlands, we found a single genome-wide significant signal at 4q25 (Figs 1 and 2). The signal is represented by the common intergenic variant rs2129979 [G] (MAFICE = 29.29%; MAFNL = 27.66%) associating with an increased risk of appendicitis (OR = 1.15; 95% CI = 1.10, 1.20; P = 1.8 × 10−11) (Table 1). Three markers show high correlation with rs2129979 (r2 > 0.93) in Iceland and in the main 1000Genome populations10. The variant is the most significant marker in the Icelandic dataset (OR = 1.14; 95% CI = 1.09, 1.19; P = 3.5 × 10−9), and its effect in Holland is comparable to the one in Iceland (OR = 1.19; 95% CI = 1.07, 1.32; P = 1.1 × 10−3; Phet = 0.50). In addition, we called microsatellites and structural variants from the WGS set, but found no such markers that are highly correlated with or more significant than rs2129979. After conditional analysis, no further variants associated with appendicitis.

Manhattan plot for the combined Icelandic and Dutch appendicitis GWAS (N = 8,566). Only one region, 4q25, harbors a genome-wide significant signal. Variants are plotted by chromosomal position (x-axis) and -log10P values (y-axis). Individual Manhattan plots for the Icelandic and Dutch groups are shown in Supplementary Figures 4 and 5, respectively. Variants with P > 0.05 have been omitted.

Locus plot for combined Icelandic and Dutch data showing the association of rs2129979 and surrounding variants at 4q25 with appendicitis (N = 8,566). The leading variant is shown as a purple circle, and other variants are coloured according to correlation (r2) with the leading marker (legend at top-right). −log10P values are shown along the left y-axis, and correspond to the variants depicted in the plot. The right y-axis shows calculated recombination rates at the chromosomal location, plotted as a solid red line. PITX2 is located 177 kb upstream of rs2129979.
We assessed the genotypic effect of rs2129979 in Iceland under a full model limited to chip-typed individuals in Iceland (n = 151,677; Supplementary Table 4). The risk of appendicitis is significantly greater for homozygous carriers of the minor allele than for heterozygotes (P = 7.0 × 10−4). The genotypic effects of rs2129979 on appendicitis are consistent with an additive mode of inheritance.
We found that rs2129979[G] had a significant and positive correlation with age of appendicitis diagnosis among cases in Iceland (P = 3.9 × 10−5), but a significant association was not seen in the Dutch cohort (P = 0.43). The combined effect on age of diagnosis for both datasets is about two years later per copy of the rs2129979 G allele (P = 7.2 × 10−4). The median age of diagnosis for all cases in Iceland based on hospital record dates was 22 years, consistent with published data from the US1. When we divide Icelandic appendicitis patients into age-at-diagnosis quintiles, the appendicitis risk conferred by rs2129979 increased monotonically with age (from 1.03 to 1.30), and was only significant for the 3 strata corresponding to an adult age (Supplementary Tables 1 and 5, Fig. 3). When we stratified our case control test in Iceland for cases above or below the median age of diagnosis, no other signals besides rs2129979 were detected (Supplementary Figures 1 and 2).

Effect of rs2129979 on appendicitis risk in Iceland. Age quintiles are shown on the x-axis, and odds ratio (OR) on the y-axis. The cases (N = 7,297) were stratified by age at diagnosis and logistic regression was performed per quintile with the same group of controls (N = 307,292), adjusting for sex and county. Each grey dot represents the OR of rs2129979 for individuals in the age group in question, and the error bars represent the 95% confidence intervals of the OR.
No significant differences in variant frequency were found when the Icelandic appendicitis group was subdivided by sex, or presence of peritonitis (data not shown). We found no association of rs2129979 with any of 16 other phenotypes related to diseases of the colorectum, or infectious and inflammatory processes (Supplementary Tables 6 and 7).
Functional Annotation
The closest protein-coding gene to rs2129979 and its correlates is the homeobox transcription factor PITX2, located around 170 kb upstream from the variant. We examined eQTL data from adipose tissue and blood. We did not find rs2129979 to affect PITX2 expression in adipose tissue (P = 0.66; N = 684), and expression was too low in blood for an analysis to be conducted (N = 2,512). No variants affecting expression of PITX2 are reported by GTEx Portal11. Furthermore, rs2129979 did not affect expression of other nearby genes in our data or in GTEx.
The three variants are strongly correlated (r2 ≥ 0.93) with rs2129979 in the four main 1000Genome super-populations (AFR, AMR, EUR, ASN)10 (Supplementary Table 8), as well as in Iceland (r2 = 1.00) (Table 1). The four markers are all located within a 1.6 kb region. All four markers are reported to be within gastrointestinal enhancer histone marks specific to the fetal small intestine, and the three markers correlated to rs2129979 are also within enhancers specific to the fetal large intestine10.
Long range expression regulation has been found to occur to a large extent within so called topologically associated domains (TAD)12. rs2129979 and the three correlated appendicitis risk variants overlap a 1.5 Mb long TAD (chr4:110579395-112059395), within which PITX2 is the only protein-coding gene.
Relationship of appendicitis and other reported signals at 4q25
Two sequence variants (rs2200733 [T] and rs10033464 [T]) at the 4q25 locus have been reported to associate with increased risk of atrial fibrillation (AF) in the Icelandic population13 (Table 2). rs2200733 correlates modestly with rs2129979 (r2 = 0.32), and weakly with rs10033464 (r2 = 0.04). Although the markers are physically close, the association signals are fully distinct (Fig. 4, Supplementary Figure 3), suggesting two separate biological pathways. Two further AF signals have been reported in the region in studies of other populations: rs6843082 [G], and rs1448817 [G]14. The two variants are modestly correlated with rs2129979 in the Icelandic population (r2 = 0.11–0.38; Table 2). The distance between the reported AF markers and the signal represented by rs2129979 ranges from 236 bp to 80 kb. None of the four variants reported to associate with AF associate with risk of appendicitis in the Icelandic dataset after adjusting for the rs2129979 signal (all P > 1 × 10−3).

The associations of both rs2129979 (blue diamond) and rs6843082 (red diamond), and their correlated variants at 4q25, with appendicitis (above; N = 7,427) and atrial fibrillation (below; N = 13,471) in Iceland. Variants correlated to the leading variant appear in the same colour, with the degree of correlation represented by the colour saturation. Individual locus plots for the two phenotypes are shown in Supplementary Figures 6 and 7.
Discussion
We detected a significant association between four correlated sequence variants at 4q25 and appendicitis. The closest protein-coding gene is PITX2, encoding the transcription factor pituitary homeobox 2. The region containing PITX2 has previously been described in the context of atrial fibrillation13, 15, and several papers have provided speculation as to the direct functional involvement of PITX2 16, 17. PITX2 has, in addition, been conclusively demonstrated to be important in the determination of right-left symmetry in development (i.e. situs-specific morphogenesis)18. Mutations in PITX2 are the cause of several Mendelian diseases including Rieger syndrome, a morphogenesis disorder that can present with systemic anomalies that include imperforate anus and anal stenosis19. Many individuals with situs anomalies also display various intra-abdominal and/or gastrointestinal abnormalities20. PITX2 signaling has also been shown to regulate the development of the cecum in mice, an intestinal structure directly connected to the appendix21, 22, and to regulate gut looping and vascular development in the gut of various animal models23, 24.
The small intestine and colon are among the tissues in which PITX2 is expressed the most25. In the Roadmap data26, rs2129979 and its three correlates are assigned an enhancer state specifically in fetal small intestine, and all four overlap a H3K4me1 enhancer histone mark region in intestinal cell lines. Two of the variants are predicted27 to alter binding of transcription factors with tissue-specific expression in colon and/or small intestine: rs2129979 disrupts a binding site for EVI1 (encoded by the gene MECOM), and rs17042195 introduces an Early B-cell Factor (EBF) binding motif. This collection of evidence suggests that these variants affect appendicitis risk through modulation of tissue specific regulation of the PITX2 gene in intestinal tissues, and in a time-dependent fashion. We note that the second-closest protein-coding gene is ENPEP, but no clear links can be drawn between it and the effects of variants at 4q25 based on the literature.
It is unclear if rs2129979 or correlated variants at 4q25 directly increase the risk of appendicitis, the likelihood of being diagnosed with the condition, or patient prognosis and risk of complications. While the relationship between situs anomalies and appendicitis are unclear, we cannot exclude that an anatomical abnormality could affect the likelihood of appendicitis diagnosis.
The association of rs2129979 with appendicitis is limited to patients diagnosed above the median age of diagnosis. We note that the incidence of appendicitis is much lower among the adult population where we observe a higher risk conferred by rs2129979.
Our data do not indicate a direct relationship between the signals for atrial fibrillation and appendicitis at 4q25, although the reported appendicitis signal is close to previously reported atrial fibrillation signals. We speculate that both signals are encompassed within an important hub for tissue-specific regulatory elements close to 4q25.
We have discovered an association between a common sequence variant and risk of appendicitis at PITX2. We only detected association for cases diagnosed during adulthood, suggesting different pathogenesis for the disease in different age groups. Further studies are required, however, to elucidate biological mechanisms underlying this association.
Methods
Study subjects from Iceland
This study is based on whole-genome sequence data from the whole blood of 15,220 Icelanders participating in various disease projects at deCODE genetics. In addition, 151,677 Icelanders have been genotyped using Illumina SNP chips and genotype probabilities for untyped relatives has been calculated based on Icelandic genealogy.
All participating individuals who donated blood, or their guardians, provided written informed consent. The family history of participants donating blood was incorporated into the study by including the phenotypes of first- and second-degree relatives and integrating over their possible genotypes.
All sample identifiers were encrypted in accordance with the regulations of the Icelandic Data Protection Authority. Approval for the study was provided by the National Bioethics Committee (ref:VSNb2015100030/03.03). Personal identities of the participants and biological samples were encrypted by a third-party system approved and monitored by the Icelandic Data Protection Authority. The National Bioethics Committee approved the study, including the protocol, methodology and all documents presented to the participants, and all methods were performed in accordance with the relevant guidelines and regulations.
To identify appendicitis cases, we searched for patients with International Classification of Diseases (ICD10) codes, diagnosis codes K35-K38, indicative of appendicitis at Landspitali—The National University Hospital of Iceland in Reykjavik (LUH), a community hospital for half of Iceland’s population. The records spanned from 1983–2015. A total of 7,267 appendicitis cases were included in the association analysis; 175 of these were whole-genome sequenced, 3,372 were genotyped using various Illumina chips and imputed using long-range phased haplotypes, and genotype probabilities for 3,720 were imputed on the basis of information from genotyped close relatives. Controls comprised individuals recruited through different genetic research projects at deCODE. Individuals in the appendicitis cohort were excluded from the control group. Of the controls, 7,534 were whole-genome sequened, 134,153 were genotyped by chip, and 185,447 were imputed on the basis of the genotypes of close relatives. The total number of controls was 327,134.
The process used to whole-genome sequence the Icelandic population, and the subsequent imputation from which the data for this analysis were generated has been extensively described in a recent publication28.
Study subjects from the Netherlands
The Dutch cases consisted of 1,139 individuals with self-reported appendicitis and/or appendectomy. Of those, 659 cases were recruited in a project entitled “Nijmegen Biomedical Study”29. The remaining cases were recruited through previously described studies on bladder cancer (225 cases)30, prostate cancer (109 cases)31, breast cancer (109 cases)32 and ovarian cancer (37 cases)33. The 4,587 Dutch controls were individuals from the Nijmegen Biomedical Study that did not report having had appendicitis or appendectomy.
The study protocols of all the cancer studies and the Nijmegen Biomedical Study were approved by the Institutional Review Board of the Radboud University Medical Center and all study subjects gave written informed consent.
The Dutch study samples were genotyped using Omni-1 Quad-bead chips (Illumina, San Diego, CA, USA). Variants were excluded if they (i) had <94% yield, (ii) had <1% MAF, (iii) failed Hardy-Weinberg test (P < 1 × 10−6) or (iv) showed significant (P < 1 × 10−6) difference between genotype batches. Samples with <94% yield were excluded. The resulting genotypes were phased using SHAPEIT (v2.790)34, and used to impute un-genotyped variants using IMPUTE2 (v2.3.2)35. The Dutch study samples were imputed using The Genome of the Netherlands36 (GoNL) dataset generated by whole-genome sequencing of 249 Dutch trios (498 unrelated parents and 249 children).
Population structure
To study the population structure and the ancestry of samples in the Dutch cohort we used the ADMIXTURE (v 1.2)37 and EIGENSOFT (v 6.0.1)38 software. Samples were excluded if they were identified as ethnic outliers and to adjust for remaining population substructure 10 principle components were included as covariates in the subsequent association analysis.
Association testing
Logistic regression was used to test for association between variants and disease, assuming a multiplicative model, treating disease status as the response and expected genotype counts from imputation as covariates. For the Icelandic cohort this was done using software developed at deCODE genetics28, but the Dutch cohort was analyzed using the SNPTEST (v.2.5) software39. Testing was performed using the likelihood ratio statistic.
Meta-analysis
Variants in the GoNL imputation datasets were mapped to NCBI Build38 positions and matched to the variants in the Icelandic dataset based on allele variation. Results from the different study groups were combined using a Mantel-Haenszel model40 in which the groups were allowed to have different population frequencies for alleles and genotypes but were assumed to have a common OR. Heterogeneity was tested by comparing the null hypothesis of the effect being the same in all populations to the alternative hypothesis of each population having a different effect using a likelihood ratio test. I 2 lies between 0% and 100% and describes the proportion of total variation in study estimates that is due to heterogeneity.
References
Addiss, D. G., Shaffer, N., Fowler, B. S. & Tauxe, R. V. The epidemiology of appendicitis and appendectomy in the United States. Am J Epidemiol 132, 910–25 (1990).
Barrett, M. L., Hines, A. L. & Andrews, R. M. Trends in Rates of Perforated Appendix, 2001–2010: Statistical Brief #159. In Healthcare Cost and Utilization Project (HCUP) (Agency for Healthcare Research and Quality, 2013).
Pieper, R., Kager, L. & Tidefeldt, U. Obstruction of appendix vermiformis causing acute appendicitis. An experimental study in the rabbit. Acta Chir Scand 148, 63–72 (1982).
Bhangu, A., Soreide, K., Di Saverio, S., Assarsson, J. H. & Drake, F. T. Acute appendicitis: modern understanding of pathogenesis, diagnosis, and management. Lancet 386, 1278–87 (2015).
Ergul, E. Heredity and familial tendency of acute appendicitis. Scand J Surg 96, 290–2 (2007).
Sadr Azodi, O., Andren-Sandberg, A. & Larsson, H. Genetic and environmental influences on the risk of acute appendicitis in twins. Br J Surg 96, 1336–40 (2009).
Edvardsson, V. O., Palsson, R., Indridason, O. S., Thorvaldsson, S. & Stefansson, K. Familiality of kidney stone disease in Iceland. Scand J Urol Nephrol 43, 420–4 (2009).
Kong, A. et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat Genet 40, 1068–75 (2008).
Sveinbjornsson, G. et al. Weighting sequence variants based on their annotation increases power of whole-genome association studies. Nature Genetics 48, 314–317 (2016).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research 40, D930–D934 (2012).
GTEx Portal. Version 4.1, Build #201. Avaiable at: http://www.gtexportal.org (Accessed on 3 February 2017)
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–80 (2012).
Gudbjartsson, D. F. et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448, 353–7 (2007).
Mohanty, S. et al. Novel association of polymorphic genetic variants with predictors of outcome of catheter ablation in atrial fibrillation: new directions from a prospective study (DECAF). J Interv Card Electrophysiol 45, 7–17 (2016).
Ebana, Y. et al. Clinical utility and functional analysis of variants in atrial fibrillation-associated locus 4q25. J Cardiol (2017).
Ye, J. C. et al. A Functional Variant Associated with Atrial Fibrillation Regulates PITX2c Expression through TFAP2a. American Journal of Human Genetics 99, 1281–1291 (2016).
Kolek, M. J. et al. A Common Variant on Chromosome 4q25 is Associated With Prolonged PR Interval in Subjects With and Without Atrial Fibrillation. American Journal of Cardiology 113, 309–313 (2014).
Ryan, A. K. et al. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature 394, 545–51 (1998).
Perveen, R. et al. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest Ophthalmol Vis Sci 41, 2456–60 (2000).
Lee, S. E. et al. Situs anomalies and gastrointestinal abnormalities. J Pediatr Surg 41, 1237–42 (2006).
Al Alam, D. et al. FGF9-Pitx2-FGF10 signaling controls cecal formation in mice. Dev Biol 369, 340–8 (2012).
Nichol, P. F. & Saijoh, Y. Association For Academic Surgery Pitx2 is a Critical Early Regulatory Gene in Normal Cecal Development. Journal of Surgical Research 170, 107–111 (2011).
Mahadevan, A. et al. The left-right Pitx2 pathway drives organ-specific arterial and lymphatic development in the intestine. Dev Cell 31, 690–706 (2014).
Welsh, I. C. et al. Chromatin Architecture of the Pitx2 Locus Requires CTCF- and Pitx2-Dependent Asymmetry that Mirrors Embryonic Gut Laterality. Cell Reports 13, 337–349 (2015).
Carithers, L. J. et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank 13, 311–9 (2015).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–30 (2015).
Kheradpour, P. & Kellis, M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res 42, 2976–87 (2014).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 47, 435–44 (2015).
Wetzels, J. F., Kiemeney, L. A., Swinkels, D. W., Willems, H. L. & den Heijer, M. Age- and gender-specific reference values of estimated GFR in Caucasians: the Nijmegen Biomedical Study. Kidney Int 72, 632–7 (2007).
Rafnar, T. et al. European genome-wide association study identifies SLC14A1 as a new urinary bladder cancer susceptibility gene. Human Molecular Genetics 20, 4268–4281 (2011).
Gudmundsson, J. et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet 39, 977–83 (2007).
Stacey, S. N. et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39, 865–9 (2007).
Rafnar, T. et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet 43, 1104–7 (2011).
Delaneau, O., Howie, B., Cox, A. J., Zagury, J. F. & Marchini, J. Haplotype estimation using sequencing reads. Am J Hum Genet 93, 687–96 (2013).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009).
Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet 46, 818–25 (2014).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19, 1655–64 (2009).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–9 (2006).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39, 906–13 (2007).
Mantel, N. & Haenszel, W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 22, 719–48 (1959).
Author information
Authors and Affiliations
Contributions
R.P.K., S.B., D.F.G., U.T., T.R., P.S., and K.S. designed the study and interpreted the results. R.P.K., S.B., T.E.G., G.T., K.K.A., K.A., G.B.W., J.K.S., L.A.K., T.J., T.R., and P.S. carried out the subject ascertainment and recruitment. R.P.K., S.B., A.O., and P.S. performed the sequencing, genotyping and expression analyses. R.P.K., S.B., A.O., G.T., O.B.D., S.J., G.A.A., B.O.J., J.K.S., D.F.G., and P.S. performed the statistical and bioinformatics analyses. S.S., H.H., D.O.A., and G.T., worked to create the atrial fibrillation dataset. R.P.K., S.B., A.O., I.J., U.T., D.F.G., T.R., P.S., and K.S. drafted the manuscript. All authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Competing Interests
R.P.K., S.B., A.O., G.T., O.B.D., S.J., S.S., H.H., G.T., I.J., U.T., T.J., D.F.G., T.R., P.S., and K.S. who are affiliated with deCODE genetics/Amgen declare competing financial interests as employees. The remaining authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kristjansson, R.P., Benonisdottir, S., Oddsson, A. et al. Sequence variant at 4q25 near PITX2 associates with appendicitis. Sci Rep 7, 3119 (2017). https://doi.org/10.1038/s41598-017-03353-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03353-0
This article is cited by
-
Investigation of VGLL3 and sub-target genes in the aetiology of paediatric acute appendicitis: a prospective case–control study
Pediatric Surgery International (2023)
-
Elemental, fatty acid, and protein composition of appendicoliths
Scientific Reports (2022)
-
Genetic association and differential expression of PITX2 with acute appendicitis
Human Genetics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
