
Abstract
Malignant pleural mesothelioma (MPM) is an aggressive human cancer and miRNAs can play a key role for this disease. In order to broaden the knowledge in this field, the miRNA expression was investigated in a large series of MPM to discover new pathways helpful in diagnosis, prognosis and therapy. We employed nanoString nCounter system for miRNA profiling on 105 MPM samples and 10 healthy pleura. The analysis was followed by the validation of the most significantly deregulated miRNAs by RT-qPCR in an independent sample set. We identified 63 miRNAs deregulated in a statistically significant way. MiR-185, miR-197, and miR-299 were confirmed differentially expressed, after validation study. In addition, the results of the microarray analysis corroborated previous findings concerning miR-15b-5p, miR-126-3p, and miR-145-5p. Kaplan-Meier curves were used to explore the association between miRNA expression and overall survival (OS) and identified a 2-miRNA prognostic signature (Let-7c-5p and miR-151a-5p) related to hypoxia and energy metabolism respectively. In silico analyses with DIANA-microT-CDS highlighted 5 putative targets in common between two miRNAs. With the present work we showed that the pattern of miRNAs expression is highly deregulated in MPM and that a 2-miRNA signature can be a new useful tool for prognosis in MPM.
Similar content being viewed by others


Introduction
MicroRNAs (miRNAs) are highly conserved small non-coding RNA molecules, 20–25 nucleotides long, which play an important regulatory role at post-transcriptional level. Each miRNA has multiple targets, thus slight variations in their expression could affect the behavior of a large variety of genes. Their abnormal expression has been linked to multiple human diseases, including cancer1. MiRNAs can function as either tumour suppressors or oncogenes and this feature, together with their tissue-specific expression, has prompted the study of miRNAs expression levels as a possible important diagnostic and prognostic tool for several malignancies2. Moreover, miRNAs-driven genes/pathways deregulation gives countless potentially druggable targets for the development of innovative cures3.
In recent years a number of studies have profiled the miRNA content of Malignant Pleural Mesothelioma (MPM) cell lines and/or tumour tissues4. MPM is a lethal cancer with increasing worldwide incidence, often induced by asbestos exposure. To date, a few studies have evaluated the differential expression of miRNAs using different sample sources in the hope of improving MPM management. Recently, Truini et al. reviewed a wide range of publications regarding MPM miRNA expression data4 highlighting the heterogeneity of the results (sometimes contradictory) especially due to ‘technical factors’ such as: diversity of the histological subtypes examined, different sample sources, diversity of the control groups, statistical approaches and different high-throughput platform used. From the published literature it seems established that miRNAs deregulated in MPM target genes fundamental for the development and progression of the disease, such as CDKN2A, NF2, JUN, HGF, and PDGFA 4. As a matter of fact, most of these miRNAs mapped to chromosomal regions known to be altered in MPM, such as gain of 8q24 and deletion of 1p36, and 14q32.
To overcome some conflicting results, larger samples’ cohorts should be profiled and analysed to minimize false-positive findings. In order to broaden the knowledge on the miRNAs expression status in MPM, we profiled a large number of histologically confirmed FFPE MPM samples, using NanoString nCounter, a high throughput platform particularly suitable for miRNA profiling of FFPE samples. Validation of the most significantly deregulated miRNAs was done by the gold standard quantitative RT-qPCR on an independent group of MPM and controls tissues. Furthermore, using in silico approaches, we sought to better understand how the biology of MPM might be differentially impacted by the expression patterns of key miRNAs. Finally, miRNA expression was correlated to patient overall survival (OS). As a result, we built a 2-miRNA signature able to divide MPM patients in high and low risk that has allowed us to highlight the role of microenvironment and energy metabolism in MPM.
Results
Several miRNAs are deregulated in MPM
We used a microarray approach to analyse miRNAs profile of a series of 96 MPM patients compared to 10 normal pleura samples. Raw and normalised expression data are provided individually for each sample for each of the 800 miRNAs detected by the nanoString platform in Supplementary Table 1.
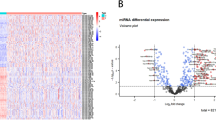
Limma analysis identified a total of 63 deregulated miRNAs represented in Fig. 1. Among them, 55 were down-regulated and 8 were up-regulated in MPM samples (detailed information are available in Supplementary Table 2). The expression levels of miRNAs were ranked according to BH values and the top five significant miRNAs were: miR-337-3p, miR-185-5p, miR-485-3p, miR-197-3p, and miR-299-5p, all down-regulated in MPM. These were selected for validation with RT-qPCR in an independent cohort of 16 MPM and 17 control tissues. The extent of differential expression was quantified by calculating the log2(fold change) (i.e., log2(FC)) using the normal pleural specimens as control samples. We observed a statistically significant down-regulation in MPM samples, after Bonferroni’s correction, for miR-197, miR-185 and miR-299 (see Table 1), whereas we could not validate the results for miR-337 and miR-485. Raw and normalised Ct (Threshold Cycle) values for each sample for each miRNA are provided in Supplementary Table 3.

Heat map representing hierarchical clustering of 63 statistically significantly deregulated miRNAs detected in the present work on 96 MPM and 10 control tissues. Rows: miRNAs; columns: samples; red: high expression; blue: low expression.
In addition, from our microarray analysis we could confirm previous findings from several high-throughput studies where mesothelial cell lines or normal mesothelial tissues were used as controls (Table 2). Interestingly, we found a statistically significant down-regulation of miR-126-3p, miR-15b-5p, and miR-145-5p, which showed to have a functional role in MPM according to in vitro and in vivo studies5,6,7. The log2 (FC) was −0.307 for miR-126 (p-value after BH correction = 0.014), −0.917 for miR-15b (0.00265), and −0.326 for miR-145 (0.014). Thus, overall, we confirmed the role played by miR-126, miR-15b and miR-145 and, in addition, we suggest a role for miR-185, miR-197, and miR-299 in MPM.
In silico analysis suggested targets and pathways affected by the deregulated miRNAs
The putative mRNAs targeted by the 6 mentioned miRNAs (i.e. miR-126, miR-15b, miR-145, miR-185, miR-197 and miR-299) were predicted with DIANA-microT-CDS in silico tool and the top 50 predicted targets according to miTG score are reported in Supplementary Table 4. Interestingly, the intersection of the 6 lists of putative targets (shown in the last sheet of Supplementary Table 4) highlighted 5 transcripts in common between two miRNAs (GOLGA1, GOLGB1, PTPN9, PSMD1, TNRC6A).
When miRNAs were analysed with miRPath tool, the union of predicted or experimentally validated targets showed an enrichment for pathways involved in cancer and signaling cascades such as Wnt or MAPK pathways. The top 5 pathways significantly enriched are reported in Table 3, together with the miRNAs and their target genes mapped in the pathway. On the other hand, the intersection of predicted or experimentally validated targets highlighted the presence of further transcripts targeted by two or more deregulated miRNAs: MPLZ1 (targeted by miR-145, -185, and -197), PRKAR2A (miR-185, -15b and -299), CCNE1 (miR-15b and miR-185), and VEGFA (miR-126 and miR-15b).
A 2-miRNA signature has an independent prognostic value for MPM
Finally, miRNAs from microarray analysis were also evaluated as potential prognostic biomarkers, considering only the 52 MPM patients for whom the survival data were available. We identified a 2-miRNA signature based on Let-7c-5p and miR-151a-5p expression levels. The OS and a risk score for each patient was calculated as explained in the materials and methods section. The OS rates were significantly lower in the patient group with the high-risk score (P = 0.004 by the log-rank test; Fig. 2A). We next sought to validate the risk score using the miRseq data from the TCGA (The Cancer Genome Atlas) mesothelioma dataset. When patients in validation cohort were stratified according to their risk score, the group with a low risk score had a significantly better prognosis (P = 0.021 for OS, log-rank test) than group with a high risk score (Fig. 2B). Similar results were obtained when assessing the 2-miRNA signature in association with prognosis in our cohort of 16 fresh frozen MPM (P = 0.038 for OS, log-rank test; Fig. 2C). Taken together, these results show that the classifier based on Let-7c-5p and miR-151a-5p expression levels has great potential as prognostic tool.

Kaplan-Meyer curves representing the correlation between 2-miRNA signature based on Let-7c-5p and miR-151a-5p expression levels and overall survival rates in MPM patients. High levels (grey line) of Let-7c-5p plus miR-151a-5p were associated with a significantly worse overall survival than low levels (black line) in patients recruited in the present study (A), in the TCGA mesothelioma dataset (B) and in an independent set of fresh frozen MPM (C) (P = 0.004, P = 0.021, and P = 0.038, respectively).
In order to ascertain whether the 2-microRNA signature indeed performs better than single miRNAs, we compared the performances of the 2-microRNA signature and the single miRNAs for OS by Akaike information criterion (AIC). The lowest AIC of the model is based on the two miRNAs taken together and it suggests its better performance than the single miRNAs in our cohort (307.1 vs 310.1 and 310.0) and in the TCGA cohort as well (402.3 vs 403.4 and 403).
The effect of risk score, gender, age and histotype on patient survival was further evaluated by multivariate Cox proportional hazard model on 52 MPM subjects from the microarray sample set. Histologic subtype and gender were not associated with survival in our sample set (P = 0.34 and P = 0.20, respectively), while a lower age at the diagnosis (<65 years old) was significantly associated with better prognosis (P = 0.001). In this model, risk score was an independent predictor of patient survival (P = 0.002).
Moreover, the effect of risk score and therapy on the prognosis was evaluated on 27 MPM subjects for whom the information regarding the received treatments was available. A non-symptomatic therapy (including surgery, chemotherapy or radiotherapy) was significantly associated with a better prognosis (P = 0.008), and the risk score was again an independent factor of patient survival (P = 0.027).
Discussion
MPM is a very aggressive and heterogeneous tumour8. Its biology is still unclear and further studies are needed in order to discover new pathways potentially helpful in diagnosis, prognosis and therapy. MiRNAs were shown to play a key role in the pathogenesis of MPM4.
Here, we aimed to broaden the knowledge about miRNAs in MPM investigating the presence of differentially expressed miRNAs using a microarray platform. This approach allowed the identification of six miRNAs (miR-15b, miR-126, miR-145, miR-185, miR-197, and miR-299), three of which (i.e. miR-185, miR-197 and miR-299) have never been linked, to the best of our knowledge, to MPM carcinogenesis. These miRNAs have been shown to target specific pathways involved in cancer outcome, progression or therapy. For instance, miR-185 was reported to be down-regulated in hepatocellular carcinoma and to inhibit tumour growth by targeting the DNMT1/PTEN/Akt pathway9, commonly activated in MPM10, 11. It was shown to suppress tumour proliferation in triple-negative breast cancer by directly targeting E2F6 and DNTM1 and by simultaneously upregulating BRCA112. A key anti-tumourigenic role of miR-185 was shown also in lung13 and gastric cancer14, modulating target mRNAs (i.e. AKT1 and TRIM29, respectively) involved in the carcinogenic process. Another study revealed a role of miR-197 as tumour suppressor in mediating apoptosis in multiple myeloma cells by targeting MCL1 15, while in non-small cell lung cancer it contributed, when down-regulated, to chemoresistance through the miR-197/CKS1B/STAT3-mediated PD-L1 network16. MiR-299 was found down-regulated in prostatic cancer cell lines compared to normal cells17 and to play a role in autophagy inhibition by suppressing Atg5 in neurons18.
Moreover, we could replicate some of the results obtained by other authors6, 7, 19,20,21,22,23 who attempted to define a set of miRNAs differentially expressed between MPM and non-MPM mesothelial tissues (Table 2). Although we found 18 miRNAs already linked with MPM, the fact that some of the results were not in agreement with what previously reported should not be surprising. In fact, there are several reasons that could explain the only partial overlap with the literature data, i.e. different sample sources (FFPE vs fresh frozen tissues), different high-throughput platforms, different normalization techniques and, last but not least, the intrinsic heterogeneity of MPM. Nonetheless, we confirmed some interesting findings about three miRNAs (i.e. miR-15b, miR-126, and miR-145) that were shown to play a biological role in MPM. MiR-15b was down-regulated in MPM tumour specimens and was shown to inhibit growth of MPM cell lines6. It was also shown to act as tumour suppressor in liver cancer24 and its reduced expression was associated with chemo-resistance25 and poor prognosis26. MiR-126 has been extensively studied in MPM, where it was shown to have a proper biological activity both in vitro and in vivo 5 and a potential value as biomarker22, 23, 27. Many targets of miR-126 were experimentally validated in other cancer types, including VEGF-A 28, ADAM9 29, Sox2 30, CRK, and PI3KR2 31. The involvement of miR-126 in the regulation of angiogenesis was shown also in hepatocellular carcinoma, where it acts as tumour suppressor by targeting LRP6 and PIK3R2 32. MiR-145 loss was reported to have pro-tumourigenic effects in MPM, via modulating clonogenicity, cell migration, and resistance to pemetrexed7. It was shown to be downregulated and to function as a tumour suppressor in other malignancies, namely nasopharyngeal carcinoma33, non-small cell lung cancer34, and bladder cancer35.
Altogether, the “miRNomic” approach allows highlighting a list of deregulated miRNAs, which could act as tumour suppressor or oncogene according to their target mRNAs. This strategy partially overcomes the limitation of the transcriptomics, which gives information about the dysregulated transcripts, but does not provide reliable data regarding their protein products, since mRNA levels often do not correlate with protein contents36. Indeed, miRNAs target analysis could help in the identification of a pattern of proteins possibly deregulated. Thus, following an in silico approach here we proposed novel targets for MPM, namely GOLGA1, GOLGB1, PTPN9, PSMD1, TNRC6A, MPLZ1, PRKAR2A, CCNE1 and VEGFA.
Once the protein targets will be validated as deregulated in MPM, novel therapeutic strategies could be undertaken using specific inhibitors. Following this concept, PSMD1 and VEGFA are among the most suitable candidate to be investigated. PSMD1 encodes one of the regulatory subunits of the proteasome complex and it was reported to be up-regulated in MPM37. It could suggest an unexplored therapeutic strategy for MPM, since the proteasome inhibitor Bortezomib has already shown to be effective in the treatment of multiple myeloma and mantle cell lymphoma38. VEGFA encodes the vascular endothelial growth factor A and it is reported to be up-regulated in mesothelioma39. The treatment with bevacizumab, a monoclonal antibody that blocks angiogenesis by inhibiting VEGF-A, in combination with pemetrexed plus cisplatin showed encouraging results in the therapy for MPM in a recent randomized phase III trial40.
A further aim of this study was to evaluate how miRNAs deregulation performs as a prognostic biomarker. Previous works highlighted the usefulness of miRNAs as prognostic biomarkers in MPM20, 23, 41, 42. While the earlier studies suggested a prognostic value for single miRNAs (miR-29c*41, miR-17-5p and miR-30c-5p20), subsequent works attempted to increase the accuracy of the survival analysis using a combination of multiple miRNAs (four-miRNA classifier23 or six-miRNA score42). In line with this approach, we proposed here a 2-miRNA signature (Let-7c-5p plus miR-151a-5p) relating to overall survival, whose prognostic value was also validated in the TCGA mesothelioma dataset and in an independent set of fresh frozen MPM samples. Our study showed some discrepancies compared to the literature data, where general poor overlap among studies could be at least partially ascribed to different microarray platforms (for example, miR-29c* and miR-652-3p probes were not included in the Nanostring platform), to subtle different statistical analyses in the calculation of the survival curves and, as acknowledged before, to the intrinsic heterogeneity of MPM. Nonetheless, our results were consistent with data provided recently about an eight-miRNA signature in bladder cancer, reporting an association between a high expression of Let-7c-5p and a poor outcome43. At a similar extent, high levels of miR-151 correlated with adverse effects on survival rate in prostate cancer44. Interestingly, both these miRNAs were reported to be associated with dysregulated metabolic conditions. In fact, Let-7c-5p plays a protective role against cerebral ischemia injury in mice45, while miR-151a-5p regulates energy metabolism reducing ATP production via mitochondrial dysfunction46. We have already stressed the role of mitochondrial abnormalities in MPM47, and we believe that hypoxia plays a crucial role in the resistance of MPM to conventional therapies. Let-7c-5p is reported to be involved in regulation of hypoxia48, 49, and can be considered members of the so-called “HypoxamiRs”, a group of miRs modulated by hypoxic conditions, involved in EMT and chemo-resistance and associated with worse prognosis50. Other evidences showed how miR-151-5p overexpression is linked with energy metabolism derangement and suggested a link with hypoxia51. This could explain their association with the overall survival that we observed here, since their high expression could promote the hypoxic microenvironment very well known to activate many signalling pathways involved in tumour initiation, progression and maintenance of MPM52, 53.
Although we provided overall reliable data about miRNA dysregulation in MPM, there are some limitations to the present study. First, we acknowledge a partial lack of clinical information for several patients since the sample collection was performed in different hospitals with different capabilities in retrieving old information from their records. Thus, the inclusion of more and more complete demographic details in the analyses for the identification of miRNA with diagnostic value could have improved the quality of the data provided. This limitation is particularly relevant in the prognostic calculations for the 2-microRNA signature, which were then done on relatively small sample numbers (n = 52, and information about relevant treatment was available only for 27 of them). Regarding the role of therapeutic treatments as prognostic variable, we acknowledge that our analysis was unable to discriminate among different treatments, since we simply grouped individuals in non-symptomatic vs symptomatic therapy in order to allow a sufficient statistical power in the multivariate analysis.
Despite this partial lack of clinical information, the baseline demographic features (age, gender and histological subtype) did not show any statistically significant difference between the cohorts with versus without survival data (see Supplementary Table 5 for further details), suggesting that the 2-miRNA prognostic signature could be applied to our entire sample set. Moreover, as explained in Table 4 and below in the materials and methods section, sufficient RNA could not be isolated from all samples, reducing the number of samples available for analysis. However, comparison of baseline characteristics of patients with and without RNA showed that there were no major differences between the respective groups (see Supplementary Table 5 for further details), thus the samples that we actually analysed were representative of a more general population.
Conclusion
In conclusion, with the present work we showed that the pattern of miRNAs expression is highly deregulated in MPM, as reported in many other tumour types. We also suggest that alterations in miRNA expression could modify cell pathways regulation and could allow the discovery of new druggable targets, following appropriate experimental validation. We also identified a 2-miRNA signature as useful biomarker for prognosis in MPM clearly addressing toward a role of metabolism in MPM aggressiveness. Patients stratified in high risk group, by our classifier, might benefit either of more targeted and aggressive cures or therapies targeting metabolic reprogramming in order to improve their outcome. Further studies on the miRNA target-genes are needed to better ascertain their fruitful exploitation as therapeutic targets or as diagnostic and prognostic biomarkers of MPM.
Materials and Methods
For this work, according to the Helsinki declaration, volunteers gave informed consent for the research. The study was approved by the institutional ethical committee of the University Hospital of Pisa. All the experimental protocols were carried out according to the relevant guidelines, as explained in details in the sections below.
Tissue collection
A consecutive series of 105 MPM patients provided corresponding 105 formalin-fixed paraffin-embedded MPM tissues. The MPM FFPE samples were biopsies collected from 2005 to 2009 in collaboration with the units of Thoracic Surgery and Occupational Medicine at the University Hospital of Pisa, together with the Hospital of Brescia, the Hospital of Venice (ASL 12), and the IRCCS AOU San Martino-IST in Genoa. All institutions providing the tissues followed the same protocol of sample collection and tissue preparation. The MPM diagnosis was issued from pathologists, following the standard clinical routine that was based on microscopic inspection and immuno-histochemical analyses of slides with antibodies to detect a panel of biomarkers (CK5/6, calretinin, vimentin, CK-Pan, EMA, TTF1, BerEP4, CEA). Pleural tissue specimens for the control sample set (n = 10) were collected in the University Hospital of Novara from individuals with chronic pleuritis (n = 6) and mesothelial hyperplasia (n = 4). Samples were selected for the microarray study only when RNA of sufficient quality and quantity was available after extraction from FFPE tissues. Nine MPM FFPE samples among the 105 available did not meet the quality criteria either in the RNA extraction or in the microarray run. Baseline characteristics of the 105 MPM patients (including the 96 samples used in the study) and the 10 non-MPM individuals are provided in Table 4. We used the full cohort of 96 RNA samples with known diagnosis of MPM to identify miRNAs with a potential diagnostic value, i.e. miRNAs that were differentially expressed between MPM and normal pleural samples. Among the 96 MPM patients used in this study, partial/total demographic information was available for 66 individuals. The survival time, calculated from the date of diagnosis, was available for 52 patients, who were then also employed to identify miRNAs with a potential prognostic value.
For the validation analysis, 22 MPM biopsies were collected during thoracoscopy in collaboration with the units of Thoracic Surgery and Occupational Medicine at the University Hospital of Pisa (Italy) before any treatment. About 70% of MPM patients had an ascertained positive history of exposure to asbestos, and approximately half of them had also a positive cigarette smoking history. Control tissues (20) were normal pleura from patients who underwent surgery for early-stage lung cancer (6 lung adenocarcinomas and 14 lung squamous cell carcinomas). Pleural specimens were collected far from the tumour site, eye-inspected by surgeons and analysed by pathologists in order to collect a small portion of pleura not containing evidence of lung cancer spread. All samples were fresh frozen, stored in the RNA later reagent (Qiagen, Hilden, Germany) and placed at −80 °C right after the collection. Following the same criteria of the microarray study, samples were selected for the validation study only when RNA of sufficient quality and quantity was available after extraction from fresh frozen tissues, and for this reason six and three RNA samples were excluded among the MPM and non-MPM subgroup, respectively. Baseline characteristics of the 42 MPM and non-MPM samples and the subset of 33 patients used in this validation study are provided in Table 5. The validation of the 2-miRNA signature was performed on the full set of 16 MPM with RNAs as the survival time was available for all of them.
Microarray Study
RNA extraction
Total RNAs were isolated from formalin-fixed paraffin-embedded tissues (FFPE) using Recover All kit (Thermo Fisher Scientific, Waltham, USA), following manufacturer’s protocol. About 100 ng of total RNA per samples was then processed with the Human miRNA Expression Assay from NanoString nCounter system (nanoString, Seattle, Washington, USA).
nanoString nCounter profile analysis
RNAs were processed by the nanoString nCounter system (nanoString, Seattle, Washington, USA) in the Nucleic Acid Shared Resource of The Ohio State University. The miRNA panel detects 800 endogenous miRNAs, five housekeeping transcripts [actin beta (NM_001101.2), beta-2 microglobulin (NM_004048.2), GAPDH (NM_002046.3), RPL19 (NM_000981.3), and RPLP0 (NM_001002.3)], six positive and eight negative controls (proprietary spike-in controls).
Data analysis, statistical methods, and figures
Raw expression data, which are proportional to copy number, were log-transformed and normalized by the quantile method after application of a manufacturer-supplied correction factor. Data were filtered to exclude features below the detection threshold (defined for each sample by a cutoff corresponding to ~twice standard deviation of negative control probes plus the means) in at least half of the samples. Using R/Bioconductor and the filtered dataset, linear models for microarray data analysis (limma) were employed with a contrast matrix for comparing normal vs tumour. Raw data that were above background (analysis not shown), as well as the corresponding quantile-normalized data, were also imported into MultiExperiment Viewer. Data processing and analysis were conducted using BRB-ArrayTools54, the MultiExperiment Viewer55, and R/BioConductor packages including limma56. The analysis of raw data was performed with nSlover Analysis Software provided by NanoString Technologies®. Data were collected as NanoString RCC files and provided as an Excel spreadsheet. P values were used to rank miRNAs of interest, and correction for multiple comparisons was done by the Benjamini-Hochberg (BH) method57.
Survival analysis was carried out using Kaplan–Meier curves on 52 MPM patients. To generate a risk score, we adopted a previously developed strategy58 that uses the Cox proportional hazards regression analysis to identify the miRNA associated with survival. MiRNAs whose expression was statistically significantly associated with survival at P ≤ 0.05 were selected by fitting Cox proportional hazards models to each miRNA in the training data. A Cox proportional hazards model was built using the principal components calculated from the statistically significant miRNA list. The OS and a risk score for each patient was calculated by multiplying the expression level of the 2 miRNAs by their corresponding Cox regression coefficient and summing the resulting values (risk score = sum of each Cox coefficient of miRNA x expression value of the corresponding miRNA). Thus, patients were dichotomized into groups at high or low risk using the 50th percentile (median) cutoff of the risk score as the threshold value. In order to validate our prognostic signature, the TCGA mesothelioma dataset (validation set/cohort, http://cancergenome.nih.gov/cancersselected/Mesothelioma) was used an independent set. MiRNA expression data for 83 tumours from patients with MPM were downloaded and the survival analysis was carried out applying directly the Cox coefficient derived by our cohort.
The performances of the 2-microRNA signature and single microRNAs for OS were compared by AIC, a measure of global fit that offers a relative estimate of the information lost when a given model is used to represent the process that generates the data, where low AIC values indicate better fit59.
The effect of other known prognostic factors on the survival of 52 MPM patients (histological subtype, gender and age) was also tested together with the risk score in a multivariate Cox proportional hazard model. Moreover, information about therapy was available for 27 patients with survival data, thus we performed another multivariate analysis considering the 2-miRNA risk score and the treatment (symptomatic therapy, n = 13, vs non-symptomatic therapy, n = 14) as prognostic variables.
Validation study
RNA isolation and cDNA synthesis
RNA from individual biopsies was purified by using Tri-Reagent method (Sigma Aldrich Corp. St Louis, MO, USA). In order to remove any contaminating genomic DNA, the extracted RNA was treated with DNase (Sigma Aldrich Corp. St Louis, MO, USA) and its concentration was determined by spectrophotometer (SmartSpec 3000, Bio-Rad Laboratories, Hercules, CA). The integrity and purity of total RNA were further verified by electrophoresis on ethidium bromide agarose gel, inspecting the 18 and 28 S ribosomal RNA bands. RNAs which did not meet the quality criteria (i.e. good concentration and integrity) were excluded from the analyses. The reverse transcription (RT) was performed using the TaqMan® MicroRNA Reverse Transcription Kit (Life Technologies, Monza, Italy) using stem-loop specific miRNA primers starting from 100 ng of total RNA.
Selection of reference miRNAs for RT-qPCR and RT-qPCR
In order to perform accurate RT-qPCR measurements, we tested a panel of possible reference miRNAs/short RNAs commonly used in miRNA expression analyses. The stability of each of them was measured by geNorm 60 and, according to the average M and the pair-wise variation values, U6, RNU48, RNU44 were found stable and were employed as reference miRNAs in all the experiments. Pre-designed TaqMan probes (Life Technologies, Monza, Italy) were employed. For the TaqMan assay, the reaction mixture consisted of 1.33 μl of cDNA template, 7.67 μl of deionized H2O, 1 μl of specific TaqMan Assay probe and primer mixture, and 10 μl of TaqMan® Universal Master Mix II NO UNG (Life Technologies, Monza, Italy). The thermal cycling conditions were: 10 min at 95 °C followed by 40 cycles made of 15 s at 95 °C and 60 s at 60 °C. TaqMan ID assays are reported in Supplementary Table 5. Each sample was run in triplicate, and the quality control of the derived expression values was performed according to MIQE guidelines61.
Statistical analyses
For validation analysis, miRNAs expression levels from MPM samples were compared to control tissue samples by applying the two-sided Mann–Whitney–Wilcoxon test. The adjustment for multiple testing was performed by using Bonferroni’s correction, thus the novel statistical significance threshold was 0.01. Data analysis was produced by the software Statgraphics Centurion XV (StatPoint, Inc.). Moreover, in order to confirm the 2-miRNA signature associated with prognosis, the OS and a risk score for each patient was calculated as previously explained for the microarray set of analyses.
In silico analyses
In silico prediction of the potential target mRNAs was performed for each interesting miRNA using DIANA-microT-CDS tool62, with threshold set at 0.6. DIANA-miRPath63 was employed to perform the over-representation analysis using the differentially expressed miRNAs as input list. DIANA-miRPath allows the identification of miRNA-target genes, either predicted (the “microT-CDS” mode) or experimentally validated (the “TarBase” mode). The option “Union Set”, that collects all genes targeted by at least one selected miRNAs, was applied for the over-representation analysis.
References
Croce, C. M. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10, 704–714 (2009).
Orellana, E. A. & Kasinski, A. L. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers (Basel) 7, 1388–1405 (2015).
Boccellino, M. et al. Gene interference strategies as a new tool for the treatment of prostate cancer. Endocrine 49, 588–605 (2015).
Truini, A. et al. Role of microRNAs in malignant mesothelioma. Cell Mol Life Sci 71, 2865–2878 (2014).
Tomasetti, M. et al. MicroRNA-126 suppresses mesothelioma malignancy by targeting IRS1 and interfering with the mitochondrial function. Antioxid Redox Signal 21, 2109–2125 (2014).
Reid, G. et al. Restoring expression of miR-16: a novel approach to therapy for malignant pleural mesothelioma. Ann Oncol 24, 3128–3135 (2013).
Cioce, M. et al. Protumorigenic effects of mir-145 loss in malignant pleural mesothelioma. Oncogene 33, 5319–5331 (2014).
Sun, X. et al. Molecular characterization of tumour heterogeneity and malignant mesothelioma cell differentiation by gene profiling. J Pathol 207, 91–101 (2005).
Qadir, X. V., Han, C., Lu, D., Zhang, J. & Wu, T. miR-185 inhibits hepatocellular carcinoma growth by targeting the DNMT1/PTEN/Akt pathway. Am J Pathol 184, 2355–2364 (2014).
Cedres, S. et al. Exploratory analysis of activation of PTEN-PI3K pathway and downstream proteins in malignant pleural mesothelioma (MPM). Lung Cancer 77, 192–198 (2012).
Zhou, S. et al. Multipoint targeting of the PI3K/mTOR pathway in mesothelioma. Br J Cancer 110, 2479–2488 (2014).
Tang, H. et al. miR-185 suppresses tumor proliferation by directly targeting E2F6 and DNMT1 and indirectly upregulating BRCA1 in triple-negative breast cancer. Mol Cancer Ther 13, 3185–3197 (2014).
Li, S. et al. MiR-185 acts as a tumor suppressor by targeting AKT1 in non-small cell lung cancer cells. Int J Clin Exp Pathol 8, 11854–11862 (2015).
Qiu, F., Xiong, J. P., Deng, J. & Xiang, X. J. TRIM29 functions as an oncogene in gastric cancer and is regulated by miR-185. Int J Clin Exp Pathol 8, 5053–5061 (2015).
Yang, Y. et al. miR-137 and miR-197 Induce Apoptosis and Suppress Tumorigenicity by Targeting MCL-1 in Multiple Myeloma. Clin Cancer Res 21, 2399–2411 (2015).
Fujita, Y. et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther 23, 717–27 (2015).
Formosa, A. et al. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene 33, 5173–5182 (2014).
Zhang, Y. et al. MiR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci Rep 6, 24566 (2016).
Guled, M. et al. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma -A miRNA microarray analysis. Genes Chromosomes Cancer 48, 615–623 (2009).
Busacca, S. et al. MicroRNA signature of malignant mesothelioma with potential diagnostic and prognostic implications. Am J Respir Cell Mol Biol 42, 312–319 (2010).
Balatti, V. et al. MicroRNAs dysregulation in human malignant pleural mesothelioma. J Thorac Oncol 6, 844–851 (2011).
Santarelli, L. et al. Association of MiR-126 with soluble mesothelin-related peptides, a marker for malignant mesothelioma. PLoS One 6, e18232 (2011).
Andersen, M. et al. Diagnostic potential of miR-126, miR-143, miR-145, and miR-652 in malignant pleural mesothelioma. J Mol Diagn 16, 418–430 (2014).
Zhang, Y., Huang, F., Wang, J., Peng, L. & Luo, H. MiR-15b mediates liver cancer cells proliferation through targeting BCL-2. Int J Clin Exp Pathol 8, 15677–15683 (2015).
Sun, L. et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 31, 432–445 (2012).
Sun, G. et al. Decreased Expression of miR-15b in Human Gliomas is Associated with Poor Prognosis. Cancer Biother Radiopharm 30, 169–173 (2015).
Tomasetti, M. et al. Clinical significance of circulating miR-126 quantification in malignant mesothelioma patients. Clin Biochem 45, 575–581 (2012).
Chen, H. et al. Reduced miR-126 expression facilitates angiogenesis of gastric cancer through its regulation on VEGF-A. Oncotarget 5, 11873–11885 (2014).
Wang, C. Z., Yuan, P. & Li, Y. miR-126 regulated breast cancer cell invasion by targeting ADAM9. Int J Clin Exp Pathol 8, 6547–6553 (2015).
Zhao, C., Li, Y., Zhang, M., Yang, Y. & Chang, L. miR-126 inhibits cell proliferation and induces cell apoptosis of hepatocellular carcinoma cells partially by targeting Sox2. Hum Cell 28, 91–99 (2015).
Liu, L. Y. et al. Mir-126 inhibits growth of SGC-7901 cells by synergistically targeting the oncogenes PI3KR2 and Crk, and the tumor suppressor PLK2. Int J Oncol 45, 1257–1265 (2014).
Du, C. et al. MiR-126-3p suppresses tumor metastasis and angiogenesis of hepatocellular carcinoma by targeting LRP6 and PIK3R2. J Transl Med 12, 259 (2014).
Wu, J. et al. MiR-145, a microRNA targeting ADAM17, inhibits the invasion and migration of nasopharyngeal carcinoma cells. Exp Cell Res 338, 232–238 (2015).
Ye, Z. et al. Low miR-145 silenced by DNA methylation promotes NSCLC cell proliferation, migration and invasion by targeting mucin 1. Cancer Biol Ther 16, 1071–1079 (2015).
Kou, B. et al. miR-145 inhibits invasion of bladder cancer cells by targeting PAK1. Urol Oncol 32, 846–854 (2014).
Rogers, S. et al. Investigating the correspondence between transcriptomic and proteomic expression profiles using coupled cluster models. Bioinformatics 24, 2894–2900 (2008).
Crispi, S. et al. Global gene expression profiling of human pleural mesotheliomas: identification of matrix metalloproteinase 14 (MMP-14) as potential tumour target. PLoS One 4, e7016 (2009).
McConkey, D. J. & Zhu, K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 11, 164–179 (2008).
Røe, O. D. et al. Genome-wide profile of pleural mesothelioma versus parietal and visceral pleura: the emerging gene portrait of the mesothelioma phenotype. PLoS One 4, e6554 (2009).
Zalcman, G. et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet 387, 1405–1414 (2016).
Pass, H. I. et al. hsa-miR-29c* is linked to the prognosis of malignant pleural mesothelioma. Cancer Res 70, 1916–1921 (2010).
Kirschner, M. B. et al. MiR-score: a novel 6-microRNA signature that predicts survival outcomes in patients with malignant pleural mesothelioma. Mol Oncol 9, 715–726 (2015).
Zhou, H. et al. A panel of eight-miRNA signature as a potential biomarker for predicting survival in bladder cancer. J Exp Clin Cancer Res 34, 53 (2015).
Chiyomaru, T. et al. Genistein suppresses prostate cancer growth through inhibition of oncogenic microRNA-151. PLoS One 7, e43812 (2012).
Ni, J. et al. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun 49, 75–85 (2015).
Zhou, R. et al. Mitochondria-related miR-151a-5p reduces cellular ATP production by targeting CYTB in asthenozoospermia. Sci Rep 5, 17743 (2015).
Manente, A. G. et al. Estrogen receptor β activation impairs mitochondrial oxidative metabolism and affects malignant mesothelioma cell growth in vitro and in vivo. Oncogenesis 2, e72 (2013).
Chen, Z. et al. Hypoxia-responsive miRNAs target argonaute 1 to promote angiogenesis. J Clin Invest 123, 1057–1067 (2013).
Zhao, C. & Popel, A. S. Computational Model of MicroRNA Control of HIF-VEGF Pathway: Insights into the Pathophysiology of Ischemic Vascular Disease and Cancer. PLoS Comput Biol 11, e1004612 (2015).
Greco, S. & Martelli, F. MicroRNAs in Hypoxia Response. Antioxid Redox Signal 21, 1164–1166 (2014).
Xu, P. et al. Variations of microRNAs in human placentas and plasma from preeclamptic pregnancy. Hypertension 63, 1276–1284 (2014).
Graziani, I. et al. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res 68, 9678–9685 (2008).
Goudarzi, H. et al. Enhancement of in vitro cell motility and invasiveness of human malignant pleural mesothelioma cells through the HIF-1α-MUC1 pathway. Cancer Lett 339, 82–92 (2013).
Simon, R. et al. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 3, 11–17 (2007).
Saeed, A. I. et al. TM4 microarray software suite. Methods Enzymol 411, 134–193 (2006).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015).
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N. & Golani, I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125, 279–284 (2001).
Gasparini, P. et al. microRNA expression profiling identifies a four microRNA signature as a novel diagnostic and prognostic biomarker in triple negative breast cancers. Oncotarget 5, 1174–1184 (2014).
Burnham, K. P. & Anderson, D. R. In Model Selection and Multimodel Inference (2nd ed), Ch. 2, 70–74 (Springer-Verlag, 2002).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, RESEARCH0034 (2002).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–622 (2009).
Paraskevopoulou, M.D. et al. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res 41 (Web Server issue), W169–173 (2013).
Vlachos, I. S. et al. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res 40, (Web Server issue), W498–4504 (2012).
Acknowledgements
This work was supported by GIME (Gruppo Italiano Mesotelioma) onlus and by Ministero della Salute-Bando Ricerca Finalizzata 2009 (RF-2009-1529895).
Author information
Authors and Affiliations
Contributions
M.L., M.M., A.T., A.T., B.M. and R.B. provided the FFPE samples and the related clinical features. A.B. managed, together with A.C. and R.F., the sample collection and RNA extraction. L.C. and P.G. performed the microarray study and the bioinformatics analysis. C.D.S. and O.M. managed the sample collection for the validation study and performed RNAs extraction, RT-qPCRs, in silico target prediction and pathway analyses. M.C. performed the RT-qPCRs of Let-7c-5p and miR-151a-5p on the MPM sample set (n = 16) for validation of the 2-miRNA signature, and the related statistical analyses were carried out by L.C. G.D.L. helped with the bioinformatics and the literature research, especially in relation to metabolism and hypoxia. F.G., S.L. and L.M. conceived the idea at the basis of the study. C.D.S. and O.M. wrote the manuscript, with the help of A.B., L.C. and G.D.L. F.G. helped to evaluate and edit the manuscript. S.L. and L.M. supervised the development of work, managed the data interpretation and revised the manuscript. C.D.S., O.M. and A.B. contributed equally to the manuscript. S.L. and L.M. were listed as joint senior authors. S.L. acted as corresponding author.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Santi, C., Melaiu, O., Bonotti, A. et al. Deregulation of miRNAs in malignant pleural mesothelioma is associated with prognosis and suggests an alteration of cell metabolism. Sci Rep 7, 3140 (2017). https://doi.org/10.1038/s41598-017-02694-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02694-0
This article is cited by
-
ATG5 as biomarker for early detection of malignant mesothelioma
BMC Research Notes (2023)
-
Temporally Altered miRNA Expression in a Piglet Model of Hypoxic Ischemic Brain Injury
Molecular Neurobiology (2020)
-
Exosomal miR-16-5p as a target for malignant mesothelioma
Scientific Reports (2019)
-
A perspective on the diagnostics, prognostics, and therapeutics of microRNAs of triple-negative breast cancer
Biophysical Reviews (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
