
Abstract
A new acylphloroglucinol with a novel architecture including an unprecedented dearomatic dibenzofuran core, named callistemenonone A (1), was isolated from the leaves of Callistemon viminalis (Myrtaceae). The structure was fully characterized on the basis of extensive spectroscopic analysis, including UV, HRESIMS, as well as 1D and 2D NMR spectral data (HSQC, HMBC, and ROESY). The deduced structure represents the first example of a natural dibenzofuran with two phenyl moieties coupling through tertiary hydroxy and ketal carbons. A plausible biogenetic pathway involving oxidative coupling and dearomatization as key steps is proposed to account for the biosynthesis of this novel class of dibenzofuran. Moreover, antimicrobial assays, in conjunction with the time-killing and biophysical studies, revealed that 1 exerted potent bactericidal activity against a panel of methicillin resistant pathogenic microbes with a unique mechanism.
Similar content being viewed by others
Introduction
The genus Callistemon belongs to the family Myrtaceae, and is comprised of over 30 species with most being woody, aromatic, and ornamental trees or shrubs endemic to Australia1. Callistemon viminalis (Sol. ex Gaertn,) G. Don (commonly known as red bottlebrush) is an Australian native green shrub with leaves and stems reputed to possess a variety of biological effects, including antidiabetic2, antimicrobial, anti-inflammatory, antistaphylococal, antithrombin3,4,5,6, and nematicidal, larvicidal, as well as pupicidal activities7. It was introduced into China some decades ago. Presently, it is widely cultivated in southern China as an ornamental tree and is adopted into Chinese folk medicine to treat colds and arthralgia8.
Plants in the genus Callistemon are known for their enriched phloroglucinol content, which is responsible for a variety of the biological activities of these herbal medicines4, 5. Previous phytochemical studies on different parts of C. viminalis showed the presence of flavonoids9, triterpenoids9, tannins, and phloroglucinol derivatives10, 11. In a continuation of our efforts to discover new antibacterial constituents from the plants of family Myrtaceae12,13,14,15,16,17, the ethanolic extract of C. viminalis was found to exhibit in vitro antibacterial activity with a MIC value of 100 μg/mL against MRSA JCSC 2172. Antibactewrial activity guided isolation has now led to the isolation of a new potent (MIC 20 μg/mL, anti-MRSA JCSC 2172) acylphloroglucinol with an unprecedented dearomatic dibenzofuran skeleton, designated as callistemenonone A (1) (Fig. 1). Herein, the isolation, structural elucidation, bioassay, and antibacterial mechanism of 1 are described.

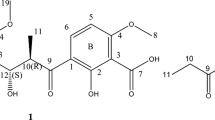
Structure of callistemenonone A (1).
Results and Discussion
C. viminalis leaves were extracted at room temperature with 95% EtOH, and the resulting pooled crude extract was concentrated under reduced pressure and then partitioned to afford n-hexane-soluble, EtOAc-soluble, and aqueous fractions. The n-hexane and EtOAc-soluble fraction were combined and subjected to repeated silica gel column chromatography to obtain 1 as a yellowish oil.
Compound 1 exhibited a deprotonated molecular ion at m/z 461.1819 (calcd m/z 461.1812) in the negative high-resolution electrospray ionization mass spectroscopy (HRESIMS) spectrum (HRESIMS and 1D and 2D spectra see supplementary information), consistent with the molecular formula C24H29O9, indicating nine indices of hydrogen deficiency. In good agreement with the molecular formula, inspection of the 13C NMR (Table 1) and HSQC spectra identified a total of 24 carbons, including four carbonyl (δ C 191.1, 191.9, 200.1, 204.1), eight quaternary (δ C 168.7, 162.0, 160.3, 110.3, 108.4, 105.2, 101.7, 82.8), three methylene (δ C 51.7, 45.6, 42.2), three methine (δ C 94.1, 26.5, 26.0), four methyl (δ C 22.8, 22.6, 22.4, 22.3), and two methoxy (δ C 55.9, 56.1) carbons. The 1H NMR data (Table 1) showed characteristic resonance signals for four methyl groups at δ H 0.98 (H-10), 1.02 (H-11), 0.75 (H-10′), and 0.80 (H-11′), as well as two methoxy groups at δ H 3.85 (3-OMe) and 3.59 (3′-OMe).
The 1H–1H COSY spectrum revealed the presence of two spin-coupling fragments (Fig. 2), namely, a (C-8/C-9/C-10/C-11) and b (C-8′/C-9′/C-10′/C-11′). The key HMBC cross-peaks (Fig. 2) observed in the coupling fragment a were H3-10 to C-8, C-9, and C-11; H3-11 to C-8, C-9, and C-10; H-9 to C-7; and H-8 to C-6. These correlations confirmed the presence of an isovaleryl group. Moreover, the HMBC correlations of the 5-OH to C-4, and C-6, of H-4 to C-2, and C-6, as well as of the 3-OMe to C-3 indicated the presence of a penta-substituted benzene moiety. Furthermore, the HMBC cross peak between H-8 to C-6 suggested a connection of the isovaleryl group to C-6 of the benzene moiety, which led to the construction of a typical phloroglucinol unit I, which was characteristically found in the genus Callistemon 18, 19. Similarly, the HMBC correlations of H3-10′ to C-8′, C-9′, and C-11′; of H3-11′ to C-8′, C-9′, and C-10′; of H-9′ to C-7′, and of H-8′ to C-6′, together with key HMBC correlations of H-4′ to C-3′, of C-2′ and C-6′, as well as of the 3′-OMe to C-3′, confirmed the other sub-structure portion as II (Fig. 2).

The key 1H-1H COSY and HMBC correlations (H → C) of 1.
Although the HMBC experiment failed to provide direct and conclusive evidence for the linkage between the structural fragments I and II due to the absence of a suitable correlative proton, assembly of the two moieties was achieved through a connection of C-1-O-C-3′ which led to the presence of a furan ring and, together with portions I and II, accounted for the nine double bond equivalents. Compound 1 might be an equimolar mixture of the two enantiomers based on its zero optical rotation value10. The relative configuration of 1 was deduced from an analysis of the ROESY correlations (Fig. 3). The key ROE correlations between the 2′-OH/H-4a′ and the 3′-OMe/H-4′a indicated that the 2′-OH and 3′-OMe were cofacial. Therefore, the structure was determined as 1, and given the trivial name callistemenonone A, representing the first member of a new family of phloroglucinol derivatives with an unprecedented dearomatic dibenzofuran core combining an α,β-triketone with a phloroglucinol unit.

The key ROESY correlations (H → H) of 1.
A biotransformation pathway (Fig. 4) was proposed to account for the biogenesis of callistemenonone A (1). Briefly, isovaleryl-CoA (2) and malonyl-CoA (3) would undergo PKS (polyketide synthase)-catalyzed condensation to yield the acylphloroglucinol 4, which has previously been reported from plants of the Myrtaceae family11, 20, 21. Oxidative coupling leads to the putative key precursor 6 through the radical intermediate 5. Further oxidative dearomatization of 6 would generate the intermediate 7, leading to construction of the unique tertiary hydroxyl carbon and the formation of the dibenzofuran architecture in callistemenonone A (1) through Michael addition.

Proposed biogenesis of callistemenonone A (1).
The antimicrobial activity of 1 was evaluated towards a panel of Gram-positive bacteria, including Staphylococcus aureus strain (CMCC 26003), Bacillus cereus strain (CMCC 63302) and several strains of methicillin-resistant S. aureus MRSA JCSC 2172, MRSA JCSC 4469, and MRSA JCSC 4744 using standard MIC method and following the CLSI guidelines. As a result, compound 1 was shown to possess potent antibacterial activity against these Gram-positive bacteria with MICs and MBCs ranging from 5 to 80 μg/mL as presented in Table 2. The potency is 3–60 fold less than that the positive control substance vancomycin (Van). Callistemenonone A (1) was inactive towards the Gram-negative bacteria Escherichia coli (ATCC 8739), presumably due to its relative inability to penetrate the permeability barrier presented by the outer membrane and/or the action of efflux transporters.
In order to understand the actions of callistemenonone A (1) at the morphologic level and to shed light on the mechanism underlying the antimicrobial activity against the clinical MRSA isolate JCSC 2172, a mechanistic investigation was initiated combining morphological, biochemical, and biophysical studies. As a result, time-kill kinetic experiments dosing with 1 × MIC revealed that 1 exhibited rapid, in vitro bactericidal activity (killing > 90% within 4 h) and complete bactericidal effect, with a sharp reduction in CFU of 99.99%, after 24 h exposure (Fig. 5A). However, in contrast to vancomycin, concentration-dependent experiment of 1 with 8 × MIC did not illustrate a dose-response effect.

Antibacterial action of callistemenonone A (1). (A) Time-kill assay of MRSA (JCSC 2172) using 1 and vancomycin as control; (B) 1-induced membrane depolarization of MRSA with different concentrations in the presence of DiSC3-5, DMSO was used as the untreated control with no effect on the depolarization, 8 × MIC concentration of 1 can not induce membrane depolarization of E. coli; (C) SYTOX green assay of MRSA with 1 (8 × MIC = 160 μg/mL) and melittin (10 μg/mL) as positive control; (D) SYTOX green assay after treating MRSA with different concentrations of 1 for 4 h and melittin as positive control. MIC values: callistemenonone A = 20 μg/mL, melittin = 1.25 μg/mL.
Further biophysical studies using fluorescence probes for membrane potential were performed to determine whether 1 caused cytoplasm membrane depolarization22,23,24. As shown in Fig. 5B, after treating the MRSA JCSC 2172 cells with serial concentrations of 1 (0.5 × MIC, 1 × MIC, 2 × MIC, 4 × MIC, 8 × MIC), a sharp increase in the fluorescence was observed, suggesting that the favorable bactericidal action of 1 could be attributed to a significant dissipation of the membrane potential due to depolarization. However, the SYTOX green assay indicated that the bactericidal action of 1 was independent of membrane disruption25, 26, because there was no obvious increase of fluorescence detected when the MRSA suspension was treated even with 8 × MIC of 1 for up to 4 h. In contrast, the positive control melittin caused an unambiguous increase in fluorescence within 60 s (Fig. 5C) and in the whole concentrations range from 0.5 to 8 MIC after 4 h (Fig. 5D). These results were a strong indication that the bactericidal action of 1 was independent of membrane disruption.
Moreover, the aforementioned results were good in agreement with the morphological study using scanning electron microscopy, wherein 1 failed to disrupt the cytoplasmic membrane structure of MRSA JCSC 2172 cells following treatment with 8 × MIC of 1 for 4 h. As shown in Fig. 6, the morphology of the treated cells remained intact with smooth spheres comparable to those of the untreated control cells. Based on the results of membrane depolarization and membrane disruption assay, in conjunction with the time-killing study (killing > 90% after 4 h), it can be concluded that callistemenonone A (1) acts rapidly as a bactericidal antibiotic by disturbing the bacterial membrane potential without causing membrane disruption.

Scanning electron microscopy showed that callistemenonone A (1) induced no cell lysis and membrane disruption. (A) MRSA (JCSC 2172) incubated with callistemenonone A (1) (160 μg/mL, 8 × MIC) after 4 h; (B) Untreated MRSA (JCSC 2172).
Melittin, the positive control used in several experiments, is a lytic and highly toxic peptide. It could be interesting to see if melittin and 1 in combination can lead to synergize the potency of 1 without undue toxicity from melittin. The synergism of 1 and melittin was investigated according to the Bliss independence model. Synergy value (0.025) was calculated using the formula as mentioned in Materials and Methods, this synergy value (0.025) demonstrated a very little synergistic relationship between 1 and melittin against MRSA.
In summary, these experiments have described herein the isolation, structural elucidation, and biological evaluation of a novel acylphloroglucinol named callistemenonone A (1), which was isolated from the leaves of Callistemon viminalis (Myrtaceae). The metabolite represents a new type of dimeric acylphloroglucinol with an unprecedented dearomatic dibenzofuran core which is biogenetically formed through tertiary hydroxy and ketal carbons. The oxidative coupling reaction and dearomatization were proposed to be key biotransformation steps for its biogenesis. The investigation of antibacterial mechanism, in conjunction with the time-killing study, revealed that 1 acted rapidly as a bactericidal agent by disturbing the bacterial membrane potential without causing membrane disruption. This represents a very favorable bactericidal mechanism which differs from any other antibiotic agent available either in the clinic or in development, rendering it a very promising candidate for further extensive investigation through medicinal chemistry, with an aim towards new antibiotic drug discovery.
Experimental
General procedures
Optical rotation was measured on a Perkin-Elmer 341 polarimeter (Perkin-Elmer, Boston, MA, USA). UV spectra were recorded in MeOH on a Perkin-Elmer Lambda 35 UV–vis spectrophotometer (Perkin-Elmer, Boston, MA, USA). 1D and 2D NMR spectra were recorded on a Bruker Advance-500 spectrometer with TMS as internal standard (Bruker BioSpin AG, Fällanden, Switzerland). HRESIMS data were obtained on a Bruker Bio TOF IIIQ mass spectrometer (Bruker Daltonics, Billerica, MA, USA). All solvents were analytical grade (Shanghai Chemical Plant, Shanghai, China). Silica gel (200–300 mesh) was used for column chromatography, and precoated silica gel GF254 plates (Qingdao Haiyang Chemical Plant, Qingdao, China) were used for TLC analysis. C18 reversed-phase silica gel (150–200 mesh, Merck), MCI gel (CHP20P, 75–150 μm, Mitsubishi Chemical Industries Ltd.), and Sephadex LH-20 gel (Amersham Biosciences) were also used for column chromatography. TLC spots were visualized under UV light and by dipping into 5% H2SO4 in alcohol, followed by heating. In the biological and biophysical experiments, the solvent used to dissolve 1 were DMSO.
Plant material
The leaves of C. viminalis were collected from the South China Botanical Garden, Chinese Academy of Sciences in March 2015 and identified by Dr. Yun-Fei Deng of SCBG. A voucher specimen was deposited at the Laboratory of Natural Product Chemistry Biology, SCBG.
Extraction and isolation
The chopped fresh leaves of C. viminalis (15.0 kg) were extracted with 95% Etch (30 L × 3) at room temperature for 24 hours. The pooled solvents were evaporated under reduced pressure to yield a brown syrupy residue (3.0 kg), which was suspended in H2O (3 L) and extracted successively with n-hexane (3 L × 3) and EtOAc (3 L × 3) to afford n-hexane- and EtOAc-soluble fractions. Most of spots of the two parts were similarity, which were detected by Thin layer chromatiography (TLC). So n-hexane- and EtOAc-soluble fractions (2.2 kg) were combined and subjected to silica gel column chromatography eluting with n-hexane/EtOAc with increasing polarity from 20:1 → 0:1 to yield six sub-fractions (A-F). Sub-fraction C (32.0 g) was chromatographed on a silica gel column with a gradient of CHCl3/MeOH (100:1 to 20:1) to yield three fractions (C1-C3). Sub-fraction C2 (3 g) was subjected to RP-C18 column chromatography (diameter: 60 cm, length: 254 mm; particle size: 150–200 mesh) and eluted with MeOH/H2O (90:10, 95:5, 100:0) to obtain three further sub-fractions (C21 to C23). Sub-fraction C21 (400 mg) was subjected to CC on Sephadex LH-20 (CHCl3/MeOH, 1:1, v/v) and further purified by repeated silica gel column chromatography (n-hexane/EtOAc, 20:1 → 1:1) to yield 1 (25 mg).
Callistemenonone A (1): Yellowish oil; [α]20 D = 0 (c 0.20, MeOH). UV (MeOH) λ max/nm (log ε) 238.7 (0.85), 281.0 (0.98); 1H (500 MHz) and 13C (125 MHz) NMR data, see Table 1; negative ESIMS m/z 461 [M − H]−; HRESIMS m/z 461.1819 [M − H]− (for C24H29O9, calcd. 461.1810).
Antibacterial assay
Compounds were dissolved in DMSO. The minimum inhibitory concentration (MIC) evaluation was carried out in 96-well plate according to the standard microdilution method. The minimum bactericidal concentration (MBC) was determined by sub-culturing 100 μL bacterial suspensions from the wells on the TSA plates. The plates were incubated for 24 h before the MBCs were determined. The MBC was categorized as the concentration where ≥ 99.9% reduction in bacterial cell count was observed. Test strains were S. aureus (CMCC 26003), B. cereus (CMCC 63302), and E. coli (ATCC 8739) which were purchased from Guangdong Microbiology Culture Center (Guangzhou, China). MRSA (JCSC 2172), MRSA (JCSC 4469), MRSA (JCSC 4744) were provided by T. Ito and K. Hiramatsu.
Time killing assay
An overnight culture of cells (MRSA JCSC 2172) was adjusted in 0.85% NaCl buffer to obtain a bacterial suspension with 106 to 107 CFU/mL The inoculate were treated with various concentrations (1× and 8 × MIC) of callistemenonone A (1) and vancomycin. The mixtures were incubated at 37 °C. Culture aliquots were removed at 0 h, 0.5 h, 2 h, 4 h, 8 h, and 24 h, then they were serially diluted to 100–106 times and 100 μL 10-fold serially diluted suspensions were plated on TSA plates. The TSA plates were incubated at 37 °C for 24 h, colonies were counted to calculate surviving cfu/mL. Bactericidal activity was defined as a ≥ 3-log10 CFU/mL decrease, in comparison with the baseline, after 24 h of incubation27.
Cytoplasmic membrane depolarization Assay
The effect of callistemenonone A on the membrane potential of MRSA JCSC 2172 cells was investigated using a modified version of the method described by Wu and Hancock28. Briefly, cultures of MRSA (JCSC 2172) were grown to exponential phase and then harvested. Bacteria were suspended and washed with buffer solution (5 mM HEPES at pH 7) until an optical density OD 0.1 at 600 nm (OD600) was obtained. The cell suspension was then incubated with 0.4 μM 3,3-dipropylthiacarbocyanine (DiSC3-5) for 20 min at 37 °C with shaking. Then, 100 mM KCl was added to the buffer to balance the chemical potential of K+ inside and outside the cells. The desired concentration of callistemenonone A (1) was added into a stirred cuvette, and the change in fluorescence was monitored at an excitation wavelength of 660 nm and an emission wavelength of 675 nm. DMSO and vancomycin was used as the negative controls with no effect on the depolarization. Experiments were repeated at least three times and found to be reproducible. Data from an individual experiment are presented.
SYTOX green assay
The protocol was based on the method of Rathinakumar et al.29. Briefly, MRSA (JCSC 2172) cells were suspended in 0.85% NaCl buffer until an OD600 of 0.2 was obtained. The bacterial suspension was incubated with serial concentrations of callistemenonone A (1), respectively for 4 h, and then incubated with 3 μM of SYTOX green for 5 min. The fluorescence was measured at an excitation wavelength of 504 nm and an emission wavelength of 523 nm. Melittin, a cell lytic factor, was used as the positive control. Experiments were repeated at least three times and found to be reproducible. Data from an individual experiment are presented.
Scanning electron microscopy
For the observations using scanning electron microscopy, exponential-phase bacteria were treated with the compound at 8 × MIC for 4 h at 37 °C. The cells were washed twice, suspended in PBS, and prefixed in 0.1 M phosphate buffer (pH 7.2) containing 2% glutaraldehyde and 2.5% paraformaldehyde overnight. After washing six times with 0.1 M phosphate buffer, the samples were post-fixed in 1% osmium tetroxide for 2 h. After washing another three times with 0.1 M phosphate buffer, the samples were dehydrated through a graded ethanol series and subjected to freeze-drying (JFD-310, JEOL, Tokyo, Japan). Samples were then mounted on stubs and coated with gold in a sputter coater (JFC-1600, JEOL, Tokyo, Japan), and then examined and photographed under a scanning electron microscope (JSM-6360LV, JEOL, Tokyo, Japan).
Synergistic assay
The synergy of 1 in combination with melittin was investigated in vitro against MRSA2172 using the Bliss independence model as described in previous reports30. Briefly, bacterial strains were incubated with a sub-inhibitory concentration of simvastatin and control antimicrobials for 12 h and the degree of synergy was calculated using the formula: Synergy (S) was calculated using the formula: S = (fA0/f00)(f0B/f00) − (fAB/f00).The parameter fAB refers to the optical density of the bacteria grown in the presence of 1 and melittin; parameters fA0 and f0B refer to the bacterial growth rate in the presence of 1 alone and melittin alone, respectively; the parameter f00 refers to the bacterial growth in the absence of drugs. Degree of synergy (S) values corresponds to the following cut-offs: Zero indicates neutral, values above zero (positive value) represents synergism, and values below zero (negative values) correspond to antagonism. Drug combinations with higher positive value represents high degree of synergism.
References
Anonymous. The Wealth of India – Raw Materials. Vol. 3, Council of Scientific and Industrial Research, New Delhi, India, 64–65 (1992).
Nazreen, S. et al. New flavones with antidiabetic activity from Callistemon lanceolatus DC. Fitoterapia 83, 1623–1627 (2012).
Kobayashi, K., Ishihara, T., Khono, E., Miyase, T. & Yoshizaki, F. Constituents of stem bark of Callistemon rigidus showing inhibitory effects on mouse α-amylase activity. Biol. Pharma. Bull 29, 1275–1277 (2006).
Gomber, C. & Saxena, S. Anti-staphylococcal potential of Callistemon rigidus. CEJ Med 2, 79–88 (2007).
Saxena, S. & Gomber, C. Antimicrobial potential of Callistemon rigidus. Pharm. Biol. 44, 194–201 (2008).
Chistokhodova, N. et al. Antithrombin activity of medicinal plants from central Florida. J. Ethnopharmacol. 82, 277–280 (2002).
Sangwan, N. K., Verma, B. S., Verma, K. K. & Dhindsa, K. S. Nematicidal activity of some essential oils. Pestic. Sci. 28, 331–335 (1990).
State Administration of Traditional Chinese Medicine of the People’s Republic of China. In Chinese Materia Medica; Shanghai Scientific and Technical Publishers: Shanghai; Vol. 15, p 4711, (1999).
Gohar, A. A., Maatooq, G. T., Gadara, S. R. & Aboelmaaty, W. S. One new pyrroline compound from Callistemon viminalis (Sol Ex Gaertner) G. Don Ex Loudon. Nat. Prod. Res. 27, 1179–1185 (2013).
Wu, L. et al. Isolation and biomimetic synthesis of (±)-calliviminones A and B, two novel Diels-Alder adducts, from Callistemon viminalis. Tetrahedron Lett. 56, 229–232 (2015).
Wu, L. et al. Calliviminones C-H: six new hetero- and carbon- Diels-Alder adducts with unusual skeletons from the fruits of Callistemon viminalis. RSC Adv 5, 93900–93906 (2015).
Liu, H. X., Tan, H. B. & Qiu, S. X. Antimicrobial acylphoroglucinols form the leaves of Rhodomyruts tomentosa. J. Asian Nat. Prod. Res. 18, 535–541 (2016).
Liu, H. X. et al. Isolation, synthesis, and biological activity of tomentosenol A from the leaves of Rhodomyrtus tomentosa. RSC Adv 6, 25882–25886 (2016).
Liu, H. X. et al. Isolation and biomimetic total synthesis of tomentodiones A-B, terpenoid-conjugated phloroglucinols from the leaves of Rhodomyrtus tomentosa. RSC Adv 6, 48231–48236 (2016).
Liu, H. X. et al. Rhodomentones A and B, novel meroterpenoids with unique NMR characteristics from Rhodomyrtus tomentosa. Org. Biomol. Chem. 14, 7354–7360 (2016).
Liu, H. X. et al. Acylphloroglucinols from the leaves of Callistemon viminalis. Fitoterapia 114, 40–44 (2016).
Liu, H. X. et al. Callviminols A-E, new terpenoid-conjugated phloroglucinols from the leaves of Callistemon viminalis. Fitoerapia 115, 142–147 (2016).
Cao, Q. et al. Callistrilones A and B, triketone-phloroglucinol-monoterpene hybridss with a new skeleton from Callistemon rigidus. Org. Lett. 18, 120–123 (2016).
Rattanaburi, S., Mahabusarakam, W., Phongpaichit, S. & Carroll, A. R. Acylphloroglucinols from Callistemon lanceolatus DC. Tetrahedron 69, 6070–6075 (2013).
Ghisalberti, E. L. Bioactive acylphloroglucinol derivatives from Eucalyptus species. Phytochemistry 41, 7–22 (1996).
Adam, P., Arigoni, D., Bacher, A. & Eisenreich, W. Biosynthesis of hyperforin in Hypericum perforatum. J. Med. Chem. 45, 4786–4793 (2002).
Koh, J. J. et al. Rapid bactericidal action of alpha-mangostin against MRSA as an outcome of membrane targeting. Biochim. Biophys. Acta. 1828, 834–844 (2013).
Wu, M., Maier, E., Benz, R. & Hancock, R. E. W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 38, 7235–7242 (1999).
Zhang, L. J., Dhillon, P., Yan, H., Farmer, S. & Hancock, R. E. W. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 3317–3321 (2000).
Rathinakumar, R., Walkenhorst, W. F. & Wimley, W. C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening, the importance of interfacial activity. J. Am. Chem. Soc. 131, 7609–7617 (2009).
Roth, B. L., Poot, M., Yue, S. T. & Millard, P. J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 63, 2421–2431 (1997).
Bambeke, F. Glycopeptides and glycodepsipeptides in clinical development, a comparative review of their antibacterial spectrum pharmacokinetics and clinical efficacy. Curr. Opin. Investig. Drugs 7, 740–749 (2006).
Wu, M. & Hancock, R. E. Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 274, 29–35 (1999).
Rathinakumar, R., Walkenhorst, W. F. & Wimley, W. C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening, the importance of interfacial activity. J. Am. Chem. Soc. 131, 7609–7617 (2009).
Thangamani, S., Younis, W. & Seleem, M. N. Repurposing celecoxib as a topical antimicrobial agent. Front. Microbiol 6, 560–563 (2015).
Acknowledgements
We thank the National Natural Science Foundation of China (NSFC) (No. 81502949), National Science and Technology Major Projects of China (2014ZX10005002-005), Natural Science Foundation of Guangdong Province (2015A030310482, 2016A030313149, 2016A010105015), the Stratesgic Resources Service Network Program on Plant Genetic Resources Innovation of the Chinese Academy of Sciences (No. ZSZC-005), ‘Twelfth Five-Year’ National Science and Technology Support Project of China (2015BAD15B03), and the DongGuan Innovative Research Team Program (2014607101005) for financial support. We thank Professor Emeritus Geoffrey A. Cordell for reviewing the manuscript prior to submission.
Author information
Authors and Affiliations
Contributions
X.Y.Q., L.H.X., and Z.L.Y. performed the experiments. T.H.B. and Q.S.X. conceived and supervised the experimental research. L.H.X., Z.L.Y., and X.Z.F. analyzed the results. L.H.X., Z.L.Y., T.H.B., and Q.S.X. wrote the paper. All the authors read and approved the final revision of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiang, YQ., Liu, HX., Zhao, LY. et al. Callistemenonone A, a novel dearomatic dibenzofuran-type acylphloroglucinol with antimicrobial activity from Callistemon viminalis . Sci Rep 7, 2363 (2017). https://doi.org/10.1038/s41598-017-02441-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02441-5
This article is cited by
-
Antimicrobial activity and mechanism of anti-MRSA of phloroglucinol derivatives
DARU Journal of Pharmaceutical Sciences (2024)
-
Phloroglucinols from Myrtaceae: attractive targets for structural characterization, biological properties and synthetic procedures
Phytochemistry Reviews (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
