
Abstract
The Hippo pathway is a tumor suppressor pathway that is implicated in the regulation of organ size. The pathway has three components: the upstream regulatory factors, the kinase core, and the downstream transcriptional machinery, which consists of YAP, TAZ (transcription co-activators) and TEAD (transcription factor). Formation of YAP/TAZ-TEAD complexes leads to the transcription of growth-promoting genes. Herein, we report the crystal structure of TAZ-TEAD4 complex, which reveals two binding modes. The first is similar to the published YAP-TEAD structure. The second is a unique binding mode, whereby two molecules of TAZ bind to and bridge two molecules of TEAD4. We validated the latter using cross-linking and multi-angle light scattering. Using siRNA, we showed that TAZ knockdown leads to a decrease in TEAD4 dimerization. Lastly, results from luciferase assays, using YAP/TAZ transfected or knockdown cells, give support to the non-redundancy of YAP/TAZ co-activators in regulating gene expression in the Hippo pathway.
Similar content being viewed by others


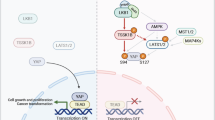
Introduction
The Hippo signaling pathway and its components were first identified and discovered through genetic screens in the fruit fly Drosophila melanogaster. It has now been established that the major function of the Hippo pathway is the control of cell number and organ size during early development and for maintaining homeostasis in adulthood1, 2. At the molecular level, the regulation of organ size is achieved through the Hippo pathway by regulating cell contact inhibition, cell proliferation and promoting apoptosis3. When this delicate control of organ size is dysregulated, cell proliferation goes unchecked and massive outgrowth of tissue occurs. This phenomenon of uncontrolled cell division is one of the hallmarks of cancer4. Hence, the Hippo pathway, when it goes awry, is linked to cancer development.
Mammalian homologues of the proteins in the Hippo pathway have been characterized and grouped into three components: the upstream regulatory factors involved in cell-to-cell signaling (FRMD6, NF2), the kinase core (MST, WW45, LATS, MOB1), and the downstream transcriptional machinery (YAP, TAZ, TEAD), which leads to the expression of genes involved in cell proliferation and anti-apoptosis. The kinases in the pathway are considered tumour suppressors and they act by phosphorylating YAP and TAZ, resulting in YAP/TAZ being sequestered in the cytoplasm and consequently promoting their turnover. Being unable to enter the nucleus upon activation of the core kinases, YAP and TAZ, both of which are transcription co-activators, are not able to bind to the transcription factor TEAD to promote transcription of genes involved in cell proliferation and anti-apoptosis5, 6.
As potent growth promoters, the overexpression of YAP and TAZ has been shown to promote cell proliferation, migration, invasion, epithelial-mesenchymal transition (EMT), tumourigenesis and is linked to poor prognosis and decreased patient survival7,8,9. In addition, they play a key role in the regulation of stem cell self-renewal and may be a critical link between stem cells and cancer cells10. Recent studies have shown that YAP and TAZ can also render cancer cells resistant to common therapeutics, such as taxol and gefitinib11,12,13. More importantly, several groups have provided evidence for the crosstalk between Hippo and other signaling pathways, such as MAPK, Wnt/β-Catenin, and TGF-β pathways; thereby implicating YAP and TAZ as central effectors of several converging pathways14,15,16,17,18.
Consequently, YAP and TAZ are candidate oncogenes, and their expression levels and/or increased nuclear accumulation are elevated in several human cancers, including human hepatocellular carcinoma and breast cancer19,20,21. Formation of the YAP/TAZ-TEAD complex within the nucleus leads to transcription of growth-promoting and pro-survival genes, thus promoting malignancy22. Conversely, disrupting the interaction between YAP/TAZ and TEAD, or the knockdown of YAP and TAZ, decreases cell proliferation and the oncogenic transforming ability of cells8, 23, 24. Small molecule inhibitors and peptides have been identified and are being developed to target the YAP/TAZ-TEAD complex25, 26. Some of the small molecule inhibitors were found to bind in a central hydrophobic pocket, where TEAD is reported to be palmitoylated. This posttranslational modification is believed to stabilize and regulate the function of TEAD27, 28.
Although YAP and TAZ share a protein sequence similarity of about 42%, are regulated by the same mechanism of the Hippo pathway and bind to similar transcription factors, such as TEAD, several reports suggest that YAP and TAZ can be independently regulated, are non-redundant and have different biological functions due to different transcriptional targets24. YAP knockout in mice results in developmental defects and are embryonically lethal, while TAZ knockout results in developmental defects of the kidney and lung29, 30. The different functional roles of YAP and TAZ might be due to their ability to promote expression of different target genes, in addition to their differential expressions. In addition, a recent study has shown that the overexpression of YAP negatively regulates TAZ, while YAP knockdown results in increased expression of TAZ. Conversely, TAZ expression levels do not modulate YAP abundance, making it a uni-directional relationship between YAP and TAZ31.
The crystal structures of both human and mouse YAP-TEAD complex have been solved previously32, 33. The structures reveal that the N-terminal of YAP adopts a helix-loop-helix structure, with helix α1 and helix α2 forming the main hydrophobic and hydrogen-bond interactions with TEAD. The loop of YAP is relatively long and is shown to form four interactions with TEAD as well. Both crystal structures, solved separately by different groups, show one molecule of YAP binding to one molecule of TEAD (1:1 complex). One of the major structural differences between YAP and TAZ is that YAP has a relatively long loop (19 residues) between helix α1 and α2, while TAZ has a shorter loop of 13 residues. Sudol et al.34 proposed that the lack of a PXXΦP motif in the TAZ linker sequence could result in a unique conformation where each TEAD molecule can interact with two TAZ molecules. In order to ascertain if TAZ and YAP binds to TEAD in the same manner and to shed light on the functional non-redundancy of the two proteins, we set out to solve the structure of TAZ-TEAD complex.
Herein, we report the crystal structure of mouse TAZ-TEAD4 complex, which reveals a distinct binding mode not observed in YAP-TEAD complex. Validation by in vitro crosslinking, multi-angle light scattering and immunoprecipitation shows that TAZ-TEAD can form a heterotetrameric complex, with two molecules of TAZ binding to two molecules of TEAD4. Coupled with results from cell-based luciferase assays, using YAP/TAZ transfected or knockdown cells, our structure of TAZ-TEAD complex provides support for the functional non-redundancy of YAP/TAZ co-activators, in regulating gene expression in the Hippo pathway.
Results and Discussion
Overall structure
We have solved the crystal structure of the mouse TAZ-TEAD4 complex to a resolution of 2.9 Å. Data collection and refinement statistics are shown in Table 1. The TAZ-TEAD complex crystallizes with four molecules of TAZ and four molecules of TEAD in the asymmetric unit. All four molecules of TEAD adopt a globular structure similar to that seen in the previously solved YAP-TEAD structures. It consists of a central β-sandwich fold and four α-helices on one side. The structures of all four molecules of TAZ reveal two short helices with a short loop of 13 residues in between. The loop lacks the PXXΦP motif that is present in YAP. Further analysis reveals two different binding modes of the TAZ-TEAD complex within the asymmetric unit. In the first binding mode, one TAZ molecule binds to one TEAD molecule to form a heterodimer in a manner similar to that observed in the structures of YAP-TEAD complex32, 33. The binding interfaces for helix α1 and helix α2 of TAZ and YAP to TEAD are virtually identical. The only difference lies in the position of the loop between helix α1 and helix α2. As the loop of TAZ is shorter than that of YAP, it adopts a downward position, while that of YAP is in an upward position (Fig. 1A).

Overall structure of TAZ-TEAD complex. (A) Comparison of mTAZ-TEAD4 and previously solved mYAP-TEAD4 (PDB: 3JUA)32 complex structures reveals different position of the loop between helix α1 and helix α2. (B) A unique binding mode is observed in the crystal structure: two TAZ molecules straddling two TEAD molecules. The TEAD molecules are related by a two-fold symmetry and two TAZ molecules criss-cross each other to bring two TEAD molecules together.
In the second binding mode, we observe two TAZ molecules straddling two TEAD molecules in a heterotetramer conformation. The two TEAD molecules are related by a two-fold symmetry and the two TAZ molecules criss-cross each other to bring the two TEAD molecules together (Fig. 1B). This conformation is achieved by the binding of TAZ helix α1 to one TEAD molecule and the binding of helix α2 of the same TAZ to a second TEAD. The loop between the two helices is stretched between the two TEAD molecules. This heterotetramer structure represents a distinct binding mode that has not been observed in the highly-related YAP-TEAD complex.
Interactions between TAZ and TEAD
Given that the TAZ-TEAD complex can adopt two different binding modes, we proceeded to look at the interaction between the TAZ and TEAD molecules to identify any differences that may give rise to the different binding modes. There are two main interaction interfaces: the first is between TAZ helix α1 and TEAD, while the second is between TAZ helix α2 and TEAD. In comparing the two binding modes, we found that the interface between TAZ helix α1 and TEAD is virtually identical for both binding modes: a hydrophobic patch of TAZ helix α1 interacts with a hydrophobic groove formed by helix α3, α4 and β6-β7 loop of TEAD. The residues involved in this hydrophobic interface are Leu28, Leu31, Phe32, Val35, Met36 of TAZ and Tyr362, Phe366, Lys369, Leu370, Leu373, Met378, Val382, Phe386 of TEAD (Fig. 2A).

Interactions between residues at the TAZ-TEAD interface. (A) The interface between TAZ helix α1 and TEAD reveals a hydrophobic patch of TAZ helix α1 (cyan) interacting with a hydrophobic groove formed by helix α3, α4 and β6-β7 loop of TEAD (purple). The side chains of residues involved in the interactions are shown in stick representation. (B) The second interface, between TAZ helix α2 and TEAD, consists of hydrophobic (grey), Van der Waals’ (red dash) and hydrogen bond (black dash) interactions. (C) In the first binding mode, where one TAZ molecule binds to one TEAD molecule, the residues in the loop region of TAZ form hydrogen bonds with TEAD. (D) In the second binding mode, where one TAZ molecule binds two TEAD molecules, the loops of two TAZ molecules (green and orange) cross each other and a unique hydrogen bond is formed between the TAZ molecules.
In the second interface between TAZ helix α2 and TEAD, we observed hydrophobic, Van der Waals’ and hydrogen bond interactions between residues. These interactions are consistent between the two binding modes. The hydrophobic interactions are formed between residues Phe52, Phe53 of TAZ and the hydrophobic groove formed by Leu288, Lys290, Trp292 and Val407 of TEAD. Van der Waals’ forces are observed between Phe52-Lys290 and Pro56-Gln418 at the TAZ-TEAD interface. Hydrogen bonds were also observed between Ser51 of TAZ and Tyr422 and Glu256 of TEAD (Fig. 2B).
Beside these two main interfaces, a few interactions were also observed between the loop region of TAZ and TEAD. In the first binding mode, where one TAZ molecule binds to one TEAD molecule, the loop region is relatively short and appears to be flexible. Hydrogen bonds are observed between the side chains of Ser41, Ser42, Trp43 of TAZ and the side chain and main chain of Glu384 of TEAD. Lys46 of TAZ also forms a hydrogen bond with Asp265 of TEAD (Fig. 2C). In the second binding mode, where one TAZ molecule straddles two TEAD molecules, only one hydrogen bond interaction is observed between the loop of TAZ and TEAD (Trp43-Glu384), as the loop region stretches between two TEAD molecules and is not in close proximity to TEAD to form many interactions. Instead, the loops of the two TAZ molecules cross each other in this conformation and a hydrogen bond is observed between Asn33 of each of the TAZ molecules (Fig. 2D).
According to Chan et al.8, the following pairs of residues of TAZ, when mutated to Alanines, resulted in reduced ability to bind with TEAD: Asp27-Leu28, Leu31-Phe32, Trp43-Arg44, Leu48-Phe49, and Phe52-Phe53. From our TAZ-TEAD crystal structure, we show that Leu28, Leu31, Phe32, Leu48, Phe52, and Phe53 of TAZ are important in forming hydrophobic interactions with TEAD. Trp43 and Phe52 of TAZ are also essential in forming Van der Waals’ interactions with TEAD (Fig. 2A–C). These residues and interactions are of great significance because abrogation of these interactions results in the loss of the transforming ability of cells. Point mutations of TEAD4 residues W292, K290 and Y422 (Fig. 2B) were also shown to cause a great reduction in YAP binding and transformation ability32. Most significantly, the Y422 residue of mouse TEAD4 is equivalent to Y421 of human TEAD1 and the Y421H missense mutation is the underlying cause of Sveinsson’s chorioretinal atrophy disease in humans35. Our TAZ-TEAD structure shows that this tyrosine residue forms a crucial hydrogen bond with Ser51 of TAZ. Hence, our structure provides additional structural basis for the cause of Sveinsson’s chorioretinal atrophy disease.
Comparison of TAZ-TEAD and YAP-TEAD structures
The comparison of our TAZ-TEAD structure with the two previously published YAP-TEAD structures reveals many similarities and some differences. Several important residues involved in the interaction of the two complexes are highly conserved between TAZ and YAP. Thus, not surprisingly, the hydrophobic interactions at the interface between TAZ/YAP helix α1 and TEAD are virtually identical. The myriad of hydrophobic, Van der Waals’ and hydrogen bond interactions at the interface between TAZ/YAP helix α2 and TEAD are also highly similar. For the first binding mode, where one TAZ molecule binds to one TEAD molecule, the total buried accessible surface area of the three major contact areas (helix α1, loop, helix α2) averages to approximately 1326 Å2, 36. This is similar to that observed for the YAP-TEAD structure (~1300 Å)32.
The only difference observed between the TAZ-TEAD and YAP-TEAD complexes lies in the loop region. The loop of YAP displays the following interactions with TEAD: Val65-Phe330, Pro66-Phe330, Thr68-Asn385 and Pro70-Glu384 (Fig. 3A). These residues found in the PXXΦP motif of YAP are missing in the shorter loop of TAZ. Hence, in the TAZ-TEAD structure, we observed a different set of interactions: between Ser41, Ser42, Trp43, Lys46 of TAZ and Asp265, Glu384 of TEAD (Fig. 3B). Moreover, in the second binding mode of the TAZ-TEAD structure, we observed fewer interactions than that of YAP-TEAD, as the loop of TAZ extends across two TEAD molecules but is not in close contact to TEAD to form interactions.

Comparison of YAP-TEAD and TAZ-TEAD structures. (A) Interactions between residues on the loop of YAP (pink) and TEAD (red), differ from that of the (B) residues on the shorter loop of TAZ (cyan) and TEAD (purple).
Mutagenesis studies done by Chen et al.32 revealed that the interactions between the PXXΦP motif in the loop of YAP and TEAD is essential for the transforming activity of cells, as a mutated loop (PXXΦP mutated to AXXAA) or deleted loop (loss of PXXΦP motif) resulted in significantly reduced or completely loss of ability of cells to grow in soft agar. However, TAZ, which has a shorter loop that does not consist of a PXXΦP motif, still possess transforming ability. From our TAZ-TEAD structure, we show that the lack of a PXXΦP motif does not affect TAZ binding to TEAD, as other residues (Ser41, Ser42, Trp42, Lys46) of the TAZ loop are able to maintain interactions with TEAD. Hence, a possible explanation for this discrepancy would be that the maintenance of the helix-loop-helix conformation of YAP/TAZ – not the PXXΦP motif – is essential for TEAD binding and consequently the transforming ability of cells37.
TAZ-TEAD forms a heterotetramer in vitro
In order to validate the distinct binding mode of TAZ-TEAD, we did specific crosslinking of the protein complex and determined the molecular weight of the complex using multi-angle light scattering. Since Asn33 of TAZ is the only residue that is in close proximity (~7.4 Å between Cα) to each other in the heterotetramer conformation, we mutated the residue to cysteine. We also mutated a surface-exposed cysteine (Cys360) of TEAD to serine and proceeded to do specific disulfide crosslinking of the Asn33Cys residue using copper phenanthroline. From the results, we observed that upon specific crosslinking, the proteins form a complex of about 65.0 kD (±1.6%), which is approximately double the size of the protein complex without crosslinking (27.7 kD ± 6.9%). This means that upon specific crosslinking, the proteins likely adopt a heterotetramer conformation of two TAZ molecules binding to two TEAD molecules. Reversing the crosslinking, using the reducing agent DTT, reverts the protein complex to a heterodimer conformation where one TAZ binds to one TEAD, as shown by the analytical gel filtration elution profile (Fig. 4). Thus, the TAZ-TEAD complex probably exists in a dynamic equilibrium between the heterodimer and heterotetramer states in solution.

Specific Crosslinking and multi-angle light scattering analysis of TAZ-TEAD complex. (A) Analytical gel filtration shows that crosslinking (red) causes the TAZ-TEAD complex to elute earlier than control (without crosslinking, black), indicating an increase in molecular weight. Reversal of crosslinking by addition of DTT (blue) reverts the size of the protein complex to that of the control. (B) Differential refractive index of the control (top) and crosslinked (bottom) protein complexes plotted as a function of time. The weight average molecular weight of the protein complex, calculated from light-scattering measurements, is shown as a red continuous line.
The TAZ-TEAD heterotetramer conformation observed in our crystal structure is further supported by cell-based data published by Muakami et al.38. Using co-immunoprecipitation, TAZ, but not YAP, was shown to adopt a homodimer conformation in cells, with the coiled-coil domain of TAZ being important for homodimer formation. Although the longer construct of TAZ, including the coiled-coil domain, did not co-crystallize with TEAD in our hands, the published cell-based data suggests that the coiled-coil domain can precipitate TAZ homodimer formation, which can lead to TAZ-TEAD adopting a heterotetramer conformation. Published results from Muakami et al.38 might also explain why the two YAP-TEAD crystal structures, solved separately by two groups, did not reveal the possibility of YAP forming a homodimer to bring two TEAD molecules together.
Most recently, a group published the crystal structure of mouse Vgll4-TEAD4 protein complex, which revealed a unique conformation whereby one Vgll4 molecule interacts with two TEAD4 molecules39. Although the primary sequence of the TEAD-interacting domain of Vgll differs greatly from that of YAP/TAZ, the first published structure of mouse Vgll1-TEAD4 complex revealed similar binding mode as that of YAP-TEAD: one molecule of Vgll1 binds to one molecule of TEAD via the same interface – helix α1 and hydrophobic groove – of the YAP-TEAD structure40. Since the Vgll1-TEAD structure was so similar to that of the YAP-TEAD structure, it came as a surprise that the Vgll4-TEAD structure adopts a different conformation. The major difference between the two homologues is that human Vgll1 has a highly conserved TEAD-interacting motif at its N-terminal (residues 27–51), while human Vgll4 has two partially-conserved TEAD-interacting motifs at its C-terminal (residues 208–219 and 236–247). Hence, in the Vgll4-TEAD structure, one TEAD-interacting motif of Vgll4 binds to one TEAD molecule and the second motif binds to a second TEAD molecule. The linker between the two motifs stretches between two TEAD molecules and brings them close together, but not close enough for any interactions to form between the TEAD molecules. Therefore, the ability of Vgll4 to adopt this unique conformation lends support to our TAZ-TEAD heterotetramer structure.
Functional non-redundancy of YAP and TAZ
Having established that TAZ-TEAD can adopt a different conformation from YAP-TEAD complex, we then carried out cell-based luciferase assays to give support to the functional non-redundancy of YAP and TAZ. By knocking down YAP or TAZ expression in H1299 cells, we are able to probe the importance of the individual co-activator TAZ or YAP in promoting transcription by TEAD. It is important to note that when one of the co-activators is knocked down, the expression level of the other co-activator does not increase to compensate for the knock down, but remains the same as that of control cells (Fig. 5A). Nonetheless, the results revealed that cells with TAZ alone not only showed higher reporter activity than cells with YAP alone, but also when compared to control cells (Fig. 5A). This suggests that TAZ possesses stronger co-activator ability, than YAP, to activate TEAD transcription in the presence of the 8xGTIIC promoter41. Alternatively, the knock down of YAP could have relieved the repression on TAZ, resulting in significantly higher reporter activity in YAP knock down cells as compared to control cells. Therefore, YAP and TAZ are not only non-redundant in cells, but a knockdown of individual co-activator results in a different outcome, with respect to the promotion of transcription by TEAD.

Functional non-redundancy of YAP and TAZ revealed by cell-based luciferase assay. (A) YAP knockdown cells show higher reporter activity than TAZ knockdown cells, in the presence of a synthetic 8xGTIIC TEAD luciferase promoter. YAP or TAZ knock down efficiency was checked with antibodies and GAPDH was used as loading control. (B) In the presence of a single TEAD binding site at the enhancer, control, YAP and TAZ transfected cells showed similar reporter activity. In the presence of tandem TEAD binding sites, YAP transfected cells showed double the reporter activity (additive), while TAZ transfected cells showed more than double the reporter activity. Each bar represents the mean and standard deviation of three independent experiments. (C) Proposed model of how the formation of a TAZ-TEAD heterotetramer complex may result in a stronger additive effect in the presence of tandem TEAD binding sites (blue block arrows).
In order to explain the structure-function relationship of our TAZ-TEAD structure, we performed luciferase assay using YAP or TAZ transfected cells, in the presence of an enhancer containing single or tandem TEAD binding sites. The results showed that in the presence of single TEAD binding site at the enhancer, both YAP and TAZ transfected cells gave rise to similar reporter activity (Fig. 5B). When the enhancer contained a tandem TEAD binding site, YAP transfected cells showed double the reporter activity as compared to single TEAD binding site. This indicates that the binding of two YAP-TEAD heterodimer complexes on the enhancer results in an additive effect. Interestingly, TAZ transfected cells showed more than double the reporter activity in the presence of tandem as compared to single TEAD binding site. The higher reporter activity might be due to the formation of a TAZ-TEAD heterotetramer complex on the tandem TEAD binding site.
Based on previously published results, our TAZ-TEAD crystal structure and our functional cell-based data, we propose that in the presence of single TEAD binding site on DNA, YAP-TEAD and TAZ-TEAD adopt a similar heterodimer conformation. In the presence of tandem TEAD binding sites on DNA, two YAP-TEAD heterodimers can bind to produce an additive effect. On the contrary, TAZ can adopt a criss-cross homodimer conformation to bring two TEAD molecules together to produce an even stronger additive effect (Fig. 5C). Such a conformation could be important for the non-redundancy of YAP and TAZ in their functional roles of promoting differential expression of target genes. An example of how a transcription co-activator can modify the DNA-binding specificity of a transcription factor was revealed by Halder et al.42 in their study of the Vestigial-Scalloped protein complex (Vg-Sd are Drosophila homologues of mammalian Vgll-TEAD). Specifically, the Vg-Sd complex appears to form a heterotetramer complex on tandem binding sites on DNA. Thus, Vg (co-activator) switches the DNA target preference of Sd (transcription factor) from a single to a tandem binding site. The authors also speculate that a single molecule of Vg could bridge two Sd molecules to increase the binding affinity and stability of the complex to DNA. Their speculation has now been confirmed by the Vgll4-TEAD structure39 that shows exactly such a unique conformation that brings two transcription factors together.
Concluding remarks
Our TAZ-TEAD crystal structure reveals two binding modes (1:1 and 2:2 complexes) of the co-activator with its transcription factor. The structure not only reveals the similar mode of action between YAP and TAZ, but also provides additional support for the non-redundancy of YAP and TAZ transcription co-activators. In particular, our cell-based data shows that the formation of a TAZ-TEAD heterotetramer complex may produce a stronger additive effect for the expression of certain target genes, especially those possessing two adjacent binding sites for the transcription factor. Further experiments will be necessary to ascertain if the YAP-TEAD protein complex can also adopt a different confirmation for the expression of different target genes. Most recently, Lee et al. showed that the DNA-binding domain of TEAD adopts a homodimer conformation in the crystal structure43. The results allude to TEAD switching between monomer and dimer to regulate DNA target selectivity. Crystal structures of YAP/TAZ-TEAD in complex with their DNA targets are thus needed to provide evidence for their binding conformations in the presence of different DNA targets. More work in this area will eventually allow us to understand how the structures of the YAP-TEAD and TAZ-TEAD complexes can give rise to functional non-redundancy and regulation of YAP/TAZ-dependent gene expression.
Material and Methods
Protein expression and purification
mTAZ (residues 25–57) and mTEAD4 (residues 210–427) were cloned into a modified pETDuet-1 vector (Novagen), containing a 3C protease-cleavable His6-tag at the N-terminal of mTAZ. The proteins were co-expressed in E. coli BL21 (DE3) strain by induction with 0.5 mM isopropyl β-D-thiogalactopyranoside at 18 °C overnight. Harvested cells were resuspended in buffer A (20 mM Tris pH 8 and 150 mM sodium chloride) supplemented with 1 mM phenylmethylsulfonyl fluoride, 25 ug/ml DNaseI, and 2 mM magnesium chloride. The TAZ-TEAD complex was purified on TALON Co2+ resin equilibrated in buffer A and eluted on a stepwise gradient of 250 mM to 500 mM imidazole. Desalting was carried out to remove imidazole before addition of 3 C protease to cleave off the His6-tag at 4 °C overnight. Uncleaved protein and protease were removed by a second affinity chromatography step. The cleaved protein was then subjected to size exclusion chromatography on a Superdex75 gel filtration column using a buffer containing 20 mM Tris pH 8, 150 mM sodium chloride, and 5 mM β-mercaptoethanol. The protein complex was concentrated to 20 mg/ml, frozen in liquid nitrogen and stored at −80 °C. For the crosslinking experiment, TAZ Asn33 and TEAD surface-exposed Cys360 were replaced with cysteine and serine respectively, in the abovementioned TAZ-TEAD construct. The protein complex was expressed and purified as described above.
Crystallization and structure determination
Large crystals of TAZ-TEAD complex appeared after 3–5 days in hanging drops by mixing 4 μl of protein with 4 μl of reservoir solution containing 6% Polyethylene glycol 10000, 0.05 M magnesium acetate, and 0.1 M tri-sodium citrate pH 5.2 at 20 °C. Micro streak seeding was carried out to produce single crystals. Dehydrating solution (12% Polyethylene glycol 10000, 0.09 M magnesium acetate, 0.12 M tri-sodium citrate pH 5.4A, 10% glycerol) was added slowly to the crystal drop until the total volume of the drop was twice the original. The drop was then equilibrated against air for 30 minutes at room temperature. A block-shaped crystal with dimensions of approximately 0.5 × 0.2 × 0.1 mm was then flash frozen in liquid nitrogen.
The diffraction data for the TAZ-TEAD complex were measured at the National Synchrotron Radiation Research Center (NSRRC), Taiwan on beamline BL13C1. The data was processed using iMosflm44 and scaled using Scala45 from the CCP446 suite of programs, to a resolution of 2.9 Å. The calculation of Rfree used 5% of data. The structure of the complex was solved by molecular replacement with Phaser47 from the CCP4 suite of programs using the mTEAD4 molecule in the mYAP-mTEAD4 structure as a search model (PDB code: 3JUA)32. Initial refinement was carried out with Refmac548 using rigid body refinement followed by restrained refinement. Electron density and difference density maps were inspected and the model was improved using Coot49. Further refinement was carried out using Phenix50. The position and conformation of TAZ in the second binding mode was confirmed by the generation of a Fo-Fc omit map (Figure S1). Protein domain interfaces were analyzed using PDBePISA36. Figures were prepared using PyMOL51.
Specific crosslinking and multi-angle light scattering
Prior to crosslinking, desalting was carried out to remove β-mercaptoethanol from the protein buffer. Specific disulfide crosslinking of the protein (100 μg) was initiated by the addition of 1.5 mM CuCl2 and 5 mM phenanthroline for 1 hour at 25 °C. The reaction was stopped by the addition of 50 mM EDTA for 15 minutes at 25 °C. Reversal of crosslinking was carried out by adding 25 mM DTT for 1 hour at 25 °C. The elution profiles of the products were monitored by analytical gel filtration using the Superdex 75 10/300 GL column (GE Healthcare). Analytical size exclusion chromatography and multi-angle light scattering (MALS) was performed using a Superdex 75 5/150 GL column (GE Healthcare), connected upstream of the UV/Vis detector, Wyatt DAWN HELEOS II light scattering detector and Wyatt Optilab refractive index monitor. The system was equilibration with 20 mM Tris pH 8 and 150 mM sodium chloride, before injecting 25 ul of the protein sample (6 mg/ml) into the column. Data analysis was carried out using ASTRA software (Wyatt). Graphs were prepared using Prism 6 (GraphPad).
Luciferase assay
H1299 cells were stably transduced with control shRNA, YAP-shRNA and TAZ-shRNA21. The YAP or TAZ protein knock down efficiency was checked with antibodies (Cell Signaling Technology). Cells were grown in RPMI media supplemented with 10% fetal bovine serum and puromycin for selection. Cells in 24-well plates were transfected with 0.5 ug of synthetic 8xGTIIC TEAD luciferase promoter41 and 0.1 ug of Renilla luciferase control reporter, using Lipofectamine2000 (Thermo Fisher Scientific). Luciferase assay was performed 24 hours after transfection using the Dual-Luciferase Reporter Assay system (Promega), according to manufacturer’s guidelines.
Single (TGTGGAATGTGT) and tandem (TGTGGAATGTGTGGAATGTGT) TEAD binding sites were cloned upstream of the SV40 promoter of the pGL3-promoter vector. Non-transduced H1299 cells were transfected with 0.1 ug co-activator (pCI control vector, YAP or TAZ), 2.5 ug of pGL3-promotor (control, single or tandem binding sites), and 0.5 ug of Renilla luciferase reporter, using Lipofectamine2000. Luciferase assay was performed 24 hours after transfection. Graphs were made using Prism 6 and all results shown are of triplicates.
References
Pan, D. Hippo signaling in organ size control. Genes & development 21, 886–897, doi:10.1101/gad.1536007 (2007).
Pan, D. The hippo signaling pathway in development and cancer. Developmental cell 19, 491–505, doi:10.1016/j.devcel.2010.09.011 (2010).
Wu, S., Huang, J., Dong, J. & Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456, doi:10.1016/S0092-8674(03)00549-X (2003).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70, doi:10.1016/S0092-8674(00)81683-9 (2000).
Huang, J., Wu, S., Barrera, J., Matthews, K. & Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122, 421–434, doi:10.1016/j.cell.2005.06.007 (2005).
Lei, Q. Y. et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 28, 2426–2436, doi:10.1128/MCB.01874-07 (2008).
Richardson, A. L. et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 9, 121–132, doi:10.1016/j.ccr.2006.01.013 (2006).
Chan, S. W. et al. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J Biol Chem 284, 14347–14358, doi:10.1074/jbc.M901568200 (2009).
Knight, J. F. et al. TEAD1 and c-Cbl are novel prostate basal cell markers that correlate with poor clinical outcome in prostate cancer. Br J Cancer 99, 1849–1858, doi:10.1038/sj.bjc.6604774 (2008).
Lian, I. et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev 24, 1106–1118, doi:10.1101/gad.1903310 (2010).
Lai, D., Ho, K. C., Hao, Y. & Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer research 71, 2728–2738, doi:10.1158/0008-5472.CAN-10-2711 (2011).
Xu, W. et al. Up-regulation of the Hippo pathway effector TAZ renders lung adenocarcinoma cells harboring EGFR-T790M mutation resistant to gefitinib. Cell & bioscience 5, 7, doi:10.1186/2045-3701-5-7 (2015).
Zhao, Y. et al. YAP-induced resistance of cancer cells to antitubulin drugs is modulated by a Hippo-independent pathway. Cancer research 74, 4493–4503, doi:10.1158/0008-5472.CAN-13-2712 (2014).
Grannas, K. et al. Crosstalk between Hippo and TGFbeta: Subcellular Localization of YAP/TAZ/Smad Complexes. Journal of molecular biology 427, 3407–3415, doi:10.1016/j.jmb.2015.04.015 (2015).
Heallen, T. et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461, doi:10.1126/science.1199010 (2011).
Hong, X. et al. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. The EMBO journal 33, 2447–2457, doi:10.15252/embj.201489385 (2014).
Rosenbluh, J. et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473, doi:10.1016/j.cell.2012.11.026 (2012).
Reddy, B. V. & Irvine, K. D. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Developmental cell 24, 459–471, doi:10.1016/j.devcel.2013.01.020 (2013).
Overholtzer, M. et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proceedings of the National Academy of Sciences of the United States of America 103, 12405–12410, doi:10.1073/pnas.0605579103 (2006).
Zender, L. et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267, doi:10.1016/j.cell.2006.05.030 (2006).
Chan, S. W. et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer research 68, 2592–2598, doi:10.1158/0008-5472.CAN-07-2696 (2008).
Vassilev, A., Kaneko, K. J., Shu, H., Zhao, Y. & DePamphilis, M. L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes & development 15, 1229–1241, doi:10.1101/gad.888601 (2001).
Zhao, B. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes & development 22, 1962–1971, doi:10.1101/gad.1664408 (2008).
Zhang, H. et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. The Journal of biological chemistry 284, 13355–13362, doi:10.1074/jbc.M900843200 (2009).
Pobbati, A. V. et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 23, 2076–2086, doi:10.1016/j.str.2015.09.009 (2015).
Zhou, Z. et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 29, 724–732, doi:10.1096/fj.14-262980 (2015).
Noland, C. L. et al. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 24, 179–186, doi:10.1016/j.str.2015.11.005 (2016).
Chan, P. et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nature chemical biology 12, 282–289, doi:10.1038/nchembio.2036 (2016).
Morin-Kensicki, E. M. et al. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol 26, 77–87, doi:10.1128/MCB.26.1.77-87.2006 (2006).
Hossain, Z. et al. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci USA 104, 1631–1636, doi:10.1073/pnas.0605266104 (2007).
Finch-Edmondson, M. L. et al. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. The Journal of biological chemistry 290, 27928–27938, doi:10.1074/jbc.M115.692285 (2015).
Chen, L. et al. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes & development 24, 290–300, doi:10.1101/gad.1865310 (2010).
Li, Z. et al. Structural insights into the YAP and TEAD complex. Genes & development 24, 235–240, doi:10.1101/gad.1865810 (2010).
Sudol, M., Shields, D. C. & Farooq, A. Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin Cell Dev Biol 23, 827–833, doi:10.1016/j.semcdb.2012.05.002 (2012).
Fossdal, R. et al. A novel TEAD1 mutation is the causative allele in Sveinsson’s chorioretinal atrophy (helicoid peripapillary chorioretinal degeneration). Hum Mol Genet 13, 975–981, doi:10.1093/hmg/ddh106 (2004).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. Journal of molecular biology 372, 774–797, doi:10.1016/j.jmb.2007.05.022 (2007).
Hau, J. C. et al. The TEAD4-YAP/TAZ protein-protein interaction: expected similarities and unexpected differences. Chembiochem 14, 1218–1225, doi:10.1002/cbic.201300163 (2013).
Murakami, M., Nakagawa, M., Olson, E. N. & Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proceedings of the National Academy of Sciences of the United States of America 102, 18034–18039, doi:10.1073/pnas.0509109102 (2005).
Jiao, S. et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer Cell 25, 166–180, doi:10.1016/j.ccr.2014.01.010 (2014).
Pobbati, A. V., Chan, S. W., Lee, I., Song, H. & Hong, W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 20, 1135–1140, doi:10.1016/j.str.2012.04.004 (2012).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183, doi:10.1038/nature10137 (2011).
Halder, G. & Carroll, S. B. Binding of the Vestigial co-factor switches the DNA-target selectivity of the Scalloped selector protein. Development 128, 3295–3305 (2001).
Lee, D. S., Vonrhein, C., Albarado, D., Raman, C. S., Veeraraghavan, S. A potential structural switch for regulating DNA-binding by TEAD transcription factors. Journal of molecular biology (2016).
Powell, H. R. The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr D Biol Crystallogr 55(Pt 10), 1690–1695, doi:10.1107/S0907444999009506 (1999).
Evans, P. R. Scaling and assessment of data quality. Acta Cryst D62, 72–82 (2005).
Collaborative Computational Project, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst, 760-763 (1994).
McCoy, A. J. et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674, doi:10.1107/S0021889807021206 (2007).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255, doi:10.1107/S0907444996012255 (1997).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, doi:10.1107/S0907444904019158 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D-Biological Crystallography 66, 213–221 (2010).
Schrodinger, L. L. C. The PyMOL Molecular Graphics System, Version 1.3r1 (2010).
Acknowledgements
We thank Jean, Yuch-Cheng and other beamline scientists for their support at the NSRRC beamline BL13C1. We also thank Kit Yeung (Pär Nordlund’s lab) for her help in using and analyzing data for MALS. This work is financially supported by the Agency for Science, Technology, and Research (A*STAR), and NMRC Open Fund Individual Research (OF-IRG) Grant (No. NMRC/OFIRG/0024/2016) in Singapore.
Author information
Authors and Affiliations
Contributions
H.Y.K.K. and S.W.C. designed and performed the experiments, wrote the paper and prepared the figures. S.K.J.T, F.G., and C.J.L. provided technical assistance and performed the experiments. W.H. and H.S. conceived and coordinated the study, and revised the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaan, H.Y.K., Chan, S.W., Tan, S.K.J. et al. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci Rep 7, 2035 (2017). https://doi.org/10.1038/s41598-017-02219-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02219-9
This article is cited by
-
Direct and selective pharmacological disruption of the YAP–TEAD interface by IAG933 inhibits Hippo-dependent and RAS–MAPK-altered cancers
Nature Cancer (2024)
-
Complex roles of Hippo-YAP/TAZ signaling in hepatocellular carcinoma
Journal of Cancer Research and Clinical Oncology (2023)
-
TEADs serve as potential prognostic biomarkers and targets for human gastric cancer
BMC Gastroenterology (2022)
-
Context-dependent roles of YAP/TAZ in stem cell fates and cancer
Cellular and Molecular Life Sciences (2021)
-
The essential role of TAZ in normal tissue homeostasis
Archives of Pharmacal Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
