
Abstract
This report describes the morphological characterization and genome analysis of an induced prophage (PLg-TB25) from a dairy strain of Lactococcus garvieae. The phage belongs to the Siphoviridae family and its morphology is typical of other lactococcal phages. A general analysis of its genome did not reveal similarities with other lactococcal phage genomes, confirming its novelty. However, similarities were found between genes of its morphogenesis cluster and genes of Gram-positive bacteria, suggesting that this phage genome resulted from recombination events that took place in a heterogeneous microbial environment. An in silico search for other prophages in 16 L. garvieae genomes available in public databases, uncovered eight seemingly complete prophages in strains isolated from dairy and fish niches. Genome analyses of these prophages revealed three novel L. garvieae phages. The remaining prophages had homology to phages of Lactococcus lactis (P335 group) suggesting a close relationship between these lactococcal species. The similarity in GC content of L. garvieae prophages to the genomes of L. lactis phages further supports the hypothesis that these phages likely originated from the same ancestor.
Similar content being viewed by others

Introduction
Bacterial viruses (phages) are considered the most abundant and diverse biological entities in our biosphere. Yet, most phages can be classified into just two main categories. Virulent phages can only replicate through a lytic cycle, which leads to cell lysis and the release of new virions ready to infect other sensitive hosts. On the other hand, temperate phages also have the ability to complete a lysogenic cycle in which they integrate their genome into the bacterial host chromosome and, thereby, replicate with the genome of the cell. Once the genome of a temperate phage is injected into the cytoplasm of its bacterial host, depending on the metabolic state of the cell, the phage genes involved in the lysogenic cycle may be expressed to favour this lifestyle. The lysogeny state will be maintained until stress conditions cause activation of the prophage through the transcription of lytic genes and the beginning of the lytic cycle1, 2.
The number of studies on phage-bacteria interactions has increased in the past decade. Many of these studies are related to a revival in the potential use of lytic phages as alternatives to antimicrobials for a myriad of applications, including inactivating antibiotic-resistant bacterial pathogens3. There has also been an increased interest in further understanding the defense mechanisms used by bacteria to combat phages4, 5.
Studies on temperate phages have mostly focused on their ability to contribute to bacterial evolution and adaptation to different environments rather than on their antimicrobial activities. Generally, prophages are responsible for changing the host’s behaviour, including granting immunity against infection by the same or closely related phages, disrupting bacterial gene(s) during genome integration, as well as modulating host gene expression through phage promoters. In some cases, temperate phages carry genes coding for toxins, regulatory and effector proteins, adhesins, exonucleases and superantigens. These sequences are often flanked by a specific transcriptional promoter and terminator, allowing gene expression during the lysogenic cycle6. The presence of a new prophage in a bacterial strain may lead to the so-called “lysogenic conversion”, where a non-pathogenic strain is converted into a pathogen by the integration of a temperate phage genome carrying genes coding for toxin or virulence factors. One well-characterized example is Escherichia coli O157:H7, where new clones have emerged following the acquisition of two Shiga toxin-encoding prophages (Sp5 and Sp15)7. Strains of Vibrio cholera have also acquired the cholera toxin through a filamentous phage8.
Lactococcus garvieae is one of the most important pathogens in the aquaculture sector9. This bacterial species is also found in different food matrices10. In addition, clinical cases associated with L. garvieae infection, albeit rare, have been reported in humans11. Little information is currently available about the pathogenic potential of L. garvieae. Much is known about its evolutionary history and ability to colonize diverse environments, and L. garvieae and the industrially-relevant dairy species Lactococcus lactis have a phylogenetic relationship, with more than 900 genes in common12. Analysis of its Mobile Genetic Elements (MGEs) showed a high degree of variability that can be linked to the lifestyle of this species. In particular, the distribution of insertion sequences (IS) has been used to characterize different ecotypes13. Finally, not much is known about phages infecting L. garvieae. To our knowledge, only two virulent phages of L. garvieae have been described and studied at the genomic level14, 15, and more recently, a temperate L. garvieae phage induced from a strain isolated from a marine fish in Japan was characterized16.
Here, we describe the isolation and characterization of a new temperate phage from a dairy strain of L. garvieae. We also searched for prophages in 16 L. garvieae genomes available in public databases, leading to the identification of three novel prophages.
Results
General features of the temperate phage PLg-TB25
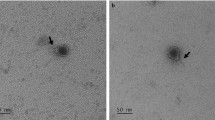
L. garvieae TB25 was previously isolated from an Italian cheese. A mitomycin C induction assay led to the isolation of an inducible prophage we named PLg-TB25. As shown in Fig. 1, phage PLg-TB25 is characterized by a 60 ± 6 nm icosahedral capsid and a non-contractile tail of 222 ± 6 nm long, 13 ± 3 nm wide, indicating it belongs to the Siphoviridae family. It has a double-stranded DNA genome of 38,122 bp and its GC content was calculated to be 34.5%, slightly lower than the GC content (38.1%) of its L. garvieae TB25 host17. The PLg-TB25 sequence shared no homology with the limited number of known L. garvieae phages. However, we identified homology with only very short DNA fragments of other L. lactis phage genomes. Thus, the L. garvieae temperate phage PLg-TB25 is a new member of the Siphoviridae family.

Electron micrograph of the phage PLg-TB25 induced from strain TB25.
Genome analysis
The search for orfs using ORF Finder and RAST Server was limited to those encoding proteins of more than 30 amino acids and flanked by an upstream potential Shine-Dalgarno sequence. The functions of the ORFs were presumed by comparing (BLASTp) deduced protein sequences with the GenBank database as well as by identifying conserved domains. Gene order was started by identifying the gene coding the integrase as the first orf (orf1), as done previously for other lactococcal phages18. Therefore, the PLg-TB25 genome starts with the divergently oriented lysogenic module (orf1 to orf5), followed by the replication/transcription module (orf6 to orf26), the morphogenesis genes (orf27 to orf48) and finally the lysis module (orf49 and orf50). Similar gene organization has been reported for other lactococcal phages18. Of note, while the draft genome of the host L. garvieae TB25 strain is available17, fragments of the inducible phage genome were found on different contigs of TB25. Yet, the phage gene order was the same on the various bacterial contigs (data not shown) as the gene order obtained in the single assembled contig from the induced phage.
In total, we identified 58 orfs covering 91% of the phage genome (Table 1, Fig. 2). The most common starting codon was AUG (87%), followed by UUG (10%) and GUG (3%). A typical RBS (AGGAGA) preceded only eight orfs (orf5, orf10, orf11, orf21, orf30, orf34, orf38, and orf50). We did not identify any tRNA or recognizable virulence factors in the genome of phage PLg-TB25. Predicted functions were attributed to 31 ORFs (53%). The proteins encoded by the 27 remaining ORFs had no homology with other phage proteins, confirming that phages are sources for novel genes and that the inducible phage PLg-TB25 is new.

Map of the phage PLg-TB25 genome. Each arrow and number identifies an open reading frame. Black arrows identify the lysogeny module. For specific functions see Table 1.
Within the lysogeny and replication modules, the majority of the ORFs best matched proteins found in strains of L. garvieae and L. lactis. Conversely, the deduced ORFs involved in the phage’s morphological structure are similar to proteins found in other Gram positive bacteria, such as Staphylococcus spp., Enterococcus spp., and Fructobacillus spp., although with low amino acid identity (31–54%). A 6-kb region containing 8 genes with low GC content (31.5%) was located downstream of the lysis module. One of the genes seemed to code for a cold shock protein19. While the function of cold-shock proteins is not fully understood, they often bind nucleic acids and may provide a mechanism for coping with stress and adapting to changing environmental conditions. This additional region at the end of the genome was likely acquired through recombination events or imprecise excision of the prophage.
A comparison between the genomes of phage PLg-TB25 and the PLgT-1 temperate phage from a L. garvieae marine fish isolate, revealed similar length (38 kb for PLg-TB25 and 40 kb for PLgT-1) and GC content (35.4% for the marine isolate and 34.5% for the dairy isolate). The 66 orfs found in PLgT-1 are organized in modules similar to PLg-TB25 but the gene/protein content is completely different.
Search for temperate phages in L. garvieae genomes
The search for prophages was extended to 16 L. garvieae genomes available in GenBank (Supplementary Table S1). As reported in Table 2, eight seemingly complete prophages were found in the genomes of seven L. garvieae strains isolated from dairy and fish environments. The genome sizes ranged from 30 to 40 kb and, when possible, the integration site (att core) was also determined. Six prophages had lower GC content (34.1–35.9%) compared to the rest of the bacterial genome (37–38%).
To verify whether L. garvieae strains colonizing a similar ecological niche carried similar prophages, we compared the genome of inducible prophage PLg-TB25 with prophages found in the genomes of the two L. garvieae strains of dairy origin, IPLA 31405 and NBRC 100934. Very low sequence identity was found between these prophages. Moreover, the prophage from NBRC 100934 (PLg-100934) shared low nucleotide identity with other phage genomes available in GenBank. In fact, the closest (with 11% identity) phage genome was the L. lactis temperate phage BK5-T (P335 group, Fig. 3)20.

Similarity matrix of 32 lactococcal phages and prophages based on the presence/absence of genes. The heatmap is generated based on the number of proteins shared by phages. Deeper shade of blue indicates a closer relationship.
The genome of PLg-100934 was 36,265 bp in length with a GC content of 37.5%, a value close to its host (38.5%) (Supplementary Table S2). A total of 54 orfs were detected, covering 90% of the genome. The majority of the ORFs use AUG as the starting codon (85%), followed by UUG (11%) and GUG (4%). A RBS (AGGAGA) was found upstream of 11 orfs (orf4, orf11, orf18, orf19, orf21, orf28, orf32, orf37, orf45, orf48, and orf52). Genome analysis identified one tRNA (Lys) and no recognizable virulence factors. The genome of PLg-100934 was also divided into four modules: lysogeny (orf1 to orf6), replication/transcription (orf7 to orf31), morphogenesis (orf32 to orf50), and lysis (orf51 and orf52). Predicted functions were attributed to 23 of the 54 orfs (42%), including orf31, which was predicted to be related to a L. lactis homing endonuclease thought to be involved in horizontal gene transfer21, 22. As reported for phage PLg-TB25, the PLg-100934 genome carries two extra genes with low GC content (31.8%) downstream of the lysis module. The function of the deduced proteins is unknown.
Two prophages were found in the genome of dairy strain L. garvieae IPLA 3140523, having homology to L. lactis phages. The genome of PLg-IPLA31405a was 34,986 bp in length with a GC content of 36.4%. A total of 53 orfs were detected, covering 90% of the genome. The genome of the second prophage, PLg-IPLA31405b, was 30,579 bp in length with a GC content of 35.1% and 46 orfs covering 85% of the genome. One of the prophages, PLg-IPLA31405a, had >90% nucleotide homology with the virulent L. lactis phage ul3618 and its mutant ul36.k1 (Fig. 3), the latter being resistant to the AbiK abortive infection mechanism24. Half of the deduced ORFs (26/53) had between 32 and 97% amino acid identity to proteins from these L. lactis phages (Fig. 4). The morphogenesis module was particularly conserved, suggesting the same morphological features. Both L. lactis phages (ul36 and ul36.k1) are virulent members of the P335 group, which contains both temperate and lytic phages4. The gene coding for a dUTPase proposed to be used to detect P335 phages was not found in the PLg-IPLA31405a genome18.

Genomic comparison between L. garvieae phage IPLA31405b and L. lactis phage ul36.k1. Color shading was used to discriminate between ≥70% amino acid identity (dark color) and ≤69% amino acid identity (light color). The absence of shading indicates no significant similarity. The percent of amino acid identity inside the shading is representative of the aligned region only. Black arrows identify the lysogeny module.
Interestingly, the deduced protein of a gene found after an orf coding for a putative XRE regulator in the PLg-IPLA3145a genome had 72% amino acid identity with the Sak protein of L. lactis phages ul36.k124 and ul36.125. Sak is involved in sensitivity/insensitivity to the lactococcal AbiK abortive infection system (Fig. 4). Surprisingly, a gene (orf53) coding for a protein sharing a conserved domain with the lactococcal abortive infection system, AbiF (COG4823)26, was detected downstream of the lysis module27. A phylogenetic analysis was performed using the amino acid sequences of ORF53 (AbiF conserved domain), phage PLg-IPLA 3405a and 20 Abi systems from L. lactis 27,28,29. The proteomic phylogenetic tree constructed using MEGA5 software and the neighbour-joining method revealed that the L. garvieae Abi-like protein was grouped with other lactococcal Abi systems tested, but diverged in a separate branch (Supplementary Fig. S1).
The other L. garvieae IPLA 31405 prophage, PLg-IPLA 31405b, was related to the temperate phage r1t from L. lactis 30 (Figs 3 and 5). Phage r1t also belongs to the P335 group (subgroup III)31. The highest amino acid identity was found with proteins involved in the morphogenesis module (75%). While a gene coding for a dUTPase was not found, an additional gene, located 700 bp downstream from the lysis module, appeared to code for a protein with a conserved cold-shock DNA-binding domain (pfam00313).

Genomic comparison between L. garvieae phage IPLA31405b and L. lactis phage r1t. Color shading was used to discriminate between ≥70% amino acid identity (dark color) and ≤69% amino acid identity (light color). The absence of shading means no significant similarity. The percent of amino acid identity inside the shading is representative of the aligned region only. Black arrows identify the lysogeny module.
Similar comparative genome analyses were performed with prophages harboured by L. garvieae strains isolated from fishes, such as ATCC 49156, Lg2 and UNIUD 074 (Table 2). The prophages from L. garvieae ATCC 49156 and Lg2 are closely related (99% nucleotide identity) and have significant nucleotide identity (95% over 41% of the genome) with the prophage found in L. garvieae strain UNIUD 074 (Fig. 3). Interestingly, they are all related to the temperate phage ɸTP712 found in the widely used plasmid-free laboratory strain L. lactis MG1363 and derived from the dairy L. lactis strain NCDO 71232, 33. Phage ɸTP712 is also related to the sequenced temperate genome, PLgT-1, isolated from a marine environment (Fig. 3). These genomes have a similar size and genome organization. The morphogenesis module is the most conserved region and while we cannot confirm at this time that they are inducible and functional, it is tempting to speculate that at some point they had the same morphological features.
Finally, L. garvieae strains 8831 and PAQ102015-99, both isolated from rainbow trout, may have an identical prophage. Due to the genome status (contigs) of strain 8831 we were unable to find the complete phage genome sequence delimited by the att sites of PLg-PAQ102015-99 (Table 2). Still, both prophages do not have any significant identity with other known phage genomes but their organization was similar to those discussed above (Supplementary Table S3). Most ORFs seemingly involved in replication and transcription have various levels of similarity with the host proteins of L. garvieae. However, the morphogenesis cluster presents the highest nucleotide variability. Seven deduced orfs (orf23, orf24, orf26 to orf29, orf38) matched (with an amino acid identity ranging from 53 to 82%) proteins found in three species of the genus Weissella (hellenica, oryzae and koreensis)34. Moreover, seven orfs (orf 25, orf30 to orf34, orf37) displayed similarities with deduced proteins from strains of Enterococcus gilvus and E. faecalis 35, 36. As noted above, a putative homing endonuclease (orf11) and a 4.2 kb DNA fragment with lower GC content (31%) were located downstream of the lysis module. Comparison of L. garvieae phages with members of the currently recognized 10 L. lactis phage groups4 revealed that while GE1 is more closely related with phage Q54 (Q54 species) and c2 (c2 species), L. garvieae prophages are more related to L. lactis phages of the P335 group.
Overall, our comparison of prophages from L. garvieae strains isolated from dairy and fish samples indicated low nucleotide identity, highlighting the diversity of lactococcal phages, particularly L. garvieae prophages.
Discussion
The recent isolation of a lytic phage infecting a strain of L. garvieae with significant similarities to dairy L. lactis phages belonging to the c2 and Q54 groups14, raised the question of whether the same was true for temperate L. garvieae phages and prophages. Moreover, since little data is available on MGEs that contribute to the evolution and adaptability of the L. garvieae species, we characterized an inducible temperate phage and analysed several prophages found within the genomes of L. garvieae strains available in public databases. Phage genome sequencing has revealed the presence of several novel genes with unknown functions. While these genes provide limited information on the biology of these phages, their analysis can shed light on their origin and provide underlying information on phage-bacteria interactions.
L. garvieae strain TB25 was previously isolated from an Italian cheese sample and was found to possess an inducible prophage belonging to the Siphoviridae family. Comparative analyses of the genome of phage PLg-TB25 with the genome of the recently described temperate phage PLgT-1 from a fish L. garvieae isolate indicated low nucleotide identity. However, the genome of PLg-TB25 had similar features (genome size, gene organization and GC content) to those observed in other L. lactis temperate phages18. Yet, the overall low nucleotide identity of phage PLg-TB25 with other phage genomes available in public databases confirmed that it represents a newly functional lactococcal phage. Of note, the inducible phage PLg-TB25 did not infect a panel of 56 strains of L. lactis (data not shown).
The analysis of 16 sequenced L. garvieae genomes revealed at least three other novel prophage groups. Within the different genomic modules, several genes encode for putative proteins with similarities to deduced proteins from phylogenetically distant genera, such as Lactobacillus, Weissella, and Enterococcus. In all likelihood, these novel phages are the result of genetic recombination events that have taken place in an environment containing multiple bacterial genera and species, and that have led to subsequent adaptation to a L. garvieae host.
The other prophages found in the genome of L. garvieae strains show similarity with temperate phages of L. lactis, belonging to the P335 group. L. lactis phages are currently classified into 10 groups based on genome analysis and phage morphology4 but only one group (P335) appears to contain virulent and temperate phages. Some authors have proposed to divide the diverse P335 phage group into subgroups37. These observations suggest an evolutionary history in an environment where these two lactococcal bacterial species can thrive, perhaps the dairy ecosystem. Since the GC content of these L. garvieae (pro)phages is lower as compared to the GC of their hosts and, in fact, much closer to the GC content of L. lactis strains and phages, it is tempting to speculate that they originated from L. lactis, while on-going adaptation to a L. garvieae host. The analysis of four phage genomes harboured by dairy L. garvieae strains also revealed the presence of seemingly additional genes after the lysis module. These genes encode for putative proteins involved in responses to environmental stresses or host strains (cold-shock proteins and defense mechanisms).
Since the L. garvieae temperate phage PLgT-1 was previously described to be capable of transduction, thereby possibly playing a role in the genetic evolution and diversification of L. garvieae marine strains16, it is conceivable to suggest the involvement of the prophages characterized in this study in spreading genes which might contribute to the adaptation of L. garvieae to the dairy environment. Mobile Genetic Elements found in strain IPLA 31405 have already been proposed to play an important role in adaptation in milk, through dissemination of the gene for lactose utilization38.
Perhaps of interest, no known virulence factors were found in the prophages characterized in this study, even if some of the strains were isolated from infected fishes. While it remains unclear if these L. garvieae strains were directly responsible for the reported illnesses, it suggests that the virulence factors are either elsewhere in the bacterial genomes or that new molecules contributing to the pathogenicity of this organism have yet to be discovered.
In conclusion, this study highlights the diversity of L. garvieae phages and, in particular, its prophages. While most of our current knowledge about lactococcal phages is derived from the characterization of phages infecting L. lactis strains in the cheese and fermented milk industries39,40,41, it appears that the Lactococcus phage population is more diverse than previously estimated. In fact, it is plausible that some L. garvieae phages might have originated from L. lactis while others are the results of recombination events with phages infecting other bacterial genera.
Methods
Induction assay and morphology studies
L. garvieae strain TB25, previously isolated from an Italian cheese17, was grown statically at 30 °C in M17 broth (Pronadisa) containing 1% glucose (GM17) to an optical density at 600 nm (OD600) of 0.3. Mitomycin C (Sigma) was added to a concentration of 5 µg/ml and the OD600 was measured (in quadruplicate) every 30 min for over 5 hours using a BioTek PowerWave XS2 spectrophotometer (BioTek). Typical induction curves observed with the mitomycin C-containing cultures were characterized by an initial increase in OD600 followed by a sharp reduction, compared to the control without mitomycin C.
The presence of induced phages was confirmed by transmission electronic microscopy (TEM). Briefly, the phage lysate was filtered through a 0.45 µm syringe filter and 1 ml was centrifuged at 24,000 × g for 1 h at 4 °C (Beckman). The supernatant (approximately 800 µl) was gently discarded and the remaining lysate (approximately 200 µl) was washed twice with 800 µl of ammonium acetate (0.1 M, pH 7.5) then centrifuged (1 h at 24,000 × g at 4 °C) and discarded. Next, 10 μl of the remaining phage solution (200 μl) was mixed with 10 μl of 2% uranyl-acetate and deposited on a nickel, Formvar-carbon-coated grid (Pelco International). The liquid was removed after 1 min by touching the edge of the grid with blotting paper. Phage morphology was observed at 80 kV using a JEOL1230 transmission electron microscope (Platforme d’Imagerie Moléculaire et Microscopie of the Université Laval). Capsid size, tail length and tail width were determined by measuring at least 10 phage specimens31. The phage was named PLg-TB25.
Phage DNA extraction
DNA of phage PLg-TB25 was isolated as described previously42, with the modifications described here. After DNase treatment to remove free DNA in the phage lysate, the DNAse was inactivated at 65 °C for 30 min. To facilitate the release of phage DNA from the capsid, 200 µl of SDS (20% stock solution) was added, along with 20 µl of proteinase K (stock solution: 20 mg/ml), and samples were incubated at 37 °C for 15 min and then at 60 °C for 30 min.
To sequence the genome of phage PLg-TB25, 90 mL of induced lysate was filtered, and polyethylene glycol (8000, 10% final concentration) and NaCl (final concentration of 0.6 M) were added to the lysate. This mixture was centrifuged at 24,000 × g (Beckman) for 1 h at 4 °C. The phage pellet was resuspended in 1 ml of phage buffer (10 mM Tris-HCl pH 7.4, 100 mM NaCl, 10 mM MgSO4) and treated with SDS/proteinase K as described above. The DNA was purified using an UltraCleanTM Microbial DNA Isolation Kit (MO BIO Laboratories, Inc.).
Phage DNA sequencing and analysis
A PLg-TB25 sequencing library was first prepared with the Nextera XT DNA Sample Prep Kit (Illumina) according to the manufacturer’s instructions. The library was sequenced using a MiSeq system (2 × 250 nt paired-end). De novo assembly was performed with the ABySS v1.5.2 assembler and CLC v7. Open reading frame (ORF) prediction was carried out using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and RAST Server43. An ORF was considered valid only if the start codon was AUG, UUG or GUG and coded for at least 30 amino acids (aa). The presence of a ribosomal binding site (RBS) similar to the standard Shine-Dalgarno sequence (AGGAGA) was also determined. Functions and domains were attributed by comparison of the translated products with the database using BLASTp44. PSI-BLAST and InterProScan at EMBL-EBI (http://www.ebi.ac.uk/) were used to search for more distant homologous proteins and conserved domains, respectively. The ProtParam tool (http://web.expasy.org/protparam/) was used to determine theoretical molecular masses (MM) and isoelectric points (pI) of the deduced phage proteins. Transfer RNA (tRNA) were predicted using the tRNAscan-SE server45 and confirmed using the ARAGORN program46. Virulence Factor Databases47, together with DBETH48, were used to search for virulence factors. Online bioinformatics tools were used with the default settings. Prophage and phage genome maps were generated with BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html) and manually edited in Adobe Illustrator.
In silico search for prophages in the genomes of L. garvieae strains
The nucleotide sequences of 16 L. garvieae genomes in the NCBI database (Supplementary Table S3) were searched for prophages using PHAST with the default parameters49. Sequences of at least 30 kb with genes involved in integration, DNA replication and morphogenesis, were suggestive of complete prophages. Homology searches were performed using BLASTn and BLASTp with default parameters44, 50.
Similarity matrix
The similarity matrix was genereated as previously described51. Briefly, all proteins of L. garvieae and L. lactis (pro)phage were grouped into cluster of orthologous genes using COGsoft52 requiring an e-value lower than 1e-3 and a protein alignment covering at least 75% of the length of the longest protein. COGsoft output was parsed to generate a presence/absence binary matrix that was used to calculate de distance between each phages according to the Jaccard index (dist function in R). The order of the rows and columns was manually adjusted when needed.
Nucleotide sequence accession number
The complete annotated genomic sequence of temperate PLg-TB25 phage from L. garvieae strain TB25 was deposited in GenBank under accession number KX833905.
References
Davies, E. V., Winstanley, C., Fothergill, J. L. & James, C. E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 363(5), fnw015, doi:10.1093/femsle/fnw015 (2016).
Shabbir, M. A. B. et al. Bacteria vs. bacteriophages: parallel evolution of immune arsenals. Front. Microbiol. 7, 1292, doi:10.3389/fmicb.2016.01292 (2016).
Bakhshinejad, B. & Sadeghizadeh, M. Bacteriophages and their applications in the diagnosis and treatment of hepatitis B virus infection. World J. Gastroenterol. 20, 11671–11683, doi:10.3748/wjg.v20.i33.11671 (2014).
Deveau, H., Labrie, S. J., Chopin, M.-C. & Moineau, S. Biodiversity and classification of lactococcal phages. Appl. Environ. Microbiol. 72, 4338–4346, doi:10.1128/AEM.02517-05 (2006).
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712, doi:10.1126/science.1138140 (2007).
Fortier, L. C. & Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365, doi:10.4161/viru.24498 (2013).
Hayashi, T. et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 96–107 (2001).
Waldor, M. K. & Mekalanos, J. J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914, doi:10.1126/science.272.5270.1910 (1996).
Vendrell, D. et al. Lactococcus garvieae in fish: A review. Comp. Immunol. Microbiol. Infect. Dis. 29, 177–198, doi:10.1016/j.cimid.2006.06.003 (2006).
Ferrario, C., Ricci, G., Borgo, F., Rollando, A. & Fortina, M. G. Genetic investigation within Lactococcus garvieae revealed two genomic lineages. FEMS Microbiol. Lett. 332, 153–161, doi:10.1111/j.1574-6968.2012.02591.x (2012).
Reguera-Brito, M. et al. Genetic analysis of human clinical isolates of Lactococcus garvieae: relatedness with isolates from foods. Infect. Genet. Evol. 37, 185–191, doi:10.1016/j.meegid.2015.11.017 (2016).
Ferrario, C. et al. Lactococcus garvieae: where is it from? A first approach to explore the evolutionary history of this emerging pathogen. PLoS One 8(12), e84796, doi:10.1371/journal.pone.0084796 (2013).
Eraclio, G., Ricci, G. & Fortina, M. G. Insertion sequence elements in Lactococcus garvieae. Gene 555, 291–296, doi:10.1016/j.gene.2014.11.019 (2015).
Eraclio, G. et al. A new virulent phage infecting Lactococcus garvieae, with homology to Lactococcus lactis phages. Appl. Environ. Microbiol. 81, 8358–8365, doi:10.1128/AEM.02603-15 (2015).
Ghasemi, S. M., Bouzari, M., Yoon, B. H. & Chang, H.-I. Comparative genomic analysis of Lactococcus garvieae phage WP-2, a new member of Picovirinae subfamily of Podoviridae. Gene 551, 222–229, doi:10.1016/j.gene.2014.08.060 (2014).
Hoai, T. D., Nishiki, I. & Yoshida, T. Properties and genomic analysis of Lactococcus garvieae lysogenic bacteriophage PLgT-1, a new member of Siphoviridae, with homology to Lactococcus lactis phages. Virus Res. 222, 13–23, doi:10.1016/j.virusres.2016.05.021 (2016).
Ricci, G., Ferrario, C., Borgo, F., Rollando, A. & Fortina, M. G. Genome sequences of Lactococcus garvieae TB25, isolated from Italian cheese, and Lactococcus garvieae LG9, isolated from Italian rainbow trout. J. Bacteriol. 194, 1249–1250, doi:10.1128/JB.06655-11 (2012).
Labrie, S. & Moineau, S. Complete genomic sequence of bacteriophage ul36: demonstration of phage heterogeneity within the P335 quasi-species of lactococcal phages. Virology 296, 308–320, doi:10.1006/viro.2002.1401 (2002).
Keto-Timonen, R. et al. Cold shock proteins: a minireview with special emphasis on Csp-family of enteropathogenic Yersinia. Front. Microbiol 7, 1151, doi:10.3389/fmicb.2016.01151 (2016).
Lakshmidevi, G., Davidson, B. E. & Hillier, A. J. Molecular characterization of promoters of the Lactococcus lactis subsp. cremoris temperate bacteriophage BK5-T and identification of a phage gene implicated in the regulation of promoter activity. Appl. Environ. Microbiol. 56, 934–942 (1990).
Weisberg, R. A., Gottesman, M. E., Hendrix, R. M. & Little, J. W. Family values in the age of genomics: Comparative analyses of temperate bacteriophage HK022. Annu. Rev. Genet. 33, 565–602, doi:10.1146/annurev.genet.33.1.565 (1999).
Belfort, M. & Bonocora, R. P. Homing endonucleases: from genetic anomalies to programmable genomic clippers. Methods Mol. Biol. 1123, 1–26, doi:10.1007/978-1-62703-968-0_1 (2014).
Flórez, A. B. et al. Genome sequence of Lactococcus garvieae IPLA 31405, a bacteriocin-producing, tetracycline-resistant strain isolated from a raw-milk cheese. J. Bacteriol. 194, 5118–5119, doi:10.1128/JB.00975-12 (2012).
Labrie, S. J. & Moineau, S. Abortive infection mechanisms and prophage sequences significantly influence the genetic makeup of emerging lytic lactococcal phages. J. Bacteriol. 189(4), 1482–1487, doi:10.1128/JB.01111-06 (2007).
Bouchard, J. D. & Moineau, S. Homologous recombination between a lactococcal bacteriophage and the chromosome of its host strain. Virology 270, 65–75, doi:10.1006/viro.2000.0226 (2000).
Garvey, P., Fitzgerald, G. F. & Hill, C. Cloning and DNA sequence analysis of two abortive infection phage resistance determinants from the lactococcal plasmid pNP40. Appl. Environ. Microbiol. 61, 4321–4328 (1995).
Chopin, M. C., Chopin, A. & Bidnenko, E. Phage abortive infection in lactococci: variations on a theme. Curr. Opin. Microbiol. 8, 473–479, doi:10.1016/j.mib.2005.06.006 (2005).
Durmaz, E. & Klaenhammer, T. R. Abortive phage resistance mechanism AbiZ speeds the lysis clock to cause premature lysis of phage-infected Lactococcus lactis. J. Bacteriol. 189, 1417–1425, doi:10.1128/JB.00904-06 (2007).
Haaber, J., Moineau, S., Fortier, L. C. & Hammer, K. AbiV, a novel antiphage abortive infection mechanism on the chromosome of Lactococcus lactis subsp. cremoris MG1363. Appl. Environ. Microbiol. 74, 6528–6537, doi:10.1128/AEM.00780-08 (2008).
Van Sinderen, D. et al. Sequence analysis and molecular characterization of the temperate lactococcal bacteriophage r1t. Mol. Microbiol. 19, 1343–1355, doi:10.1111/mmi.1996.19.issue-6 (1996).
Samson, J. E. & Moineau, S. Characterization of Lactococcus lactis phage 949 and comparison with other lactococcal phages. Appl. Environ. Microbiol. 76, 6843–6852, doi:10.1128/AEM.00796-10 (2010).
Wegmann, U., Overweg, K., Jeanson, S., Gasson, M. & Shearman, C. Molecular characterization and structural instability of the industrially important composite metabolic plasmid pLP712. Microbiology 158, 2936–2945, doi:10.1099/mic.0.062554-0 (2012).
Roces, C. et al. Lack of the host membrane protease FtsH hinders release of the Lactococcus lactis bacteriophage TP712. J. Gen. Virol. 94, 2814–2818, doi:10.1099/vir.0.057182-0 (2013).
Fusco, V. et al. The genus Weissella: taxonomy, ecology and biotechnological potential. Front. Microbiol 6, 155, doi:10.3389/fmicb.2015.00155 (2015).
Tyrrell, G. J. et al. Enterococcus gilvus sp. nov. and Enterococcus pallens sp. nov. isolate from human clinical specimens. J. Clin. Microbiol. 40, 1140–1145, doi:10.1128/JCM.40.4.1140-1145.2002 (2002).
Zhang, C., Du, J. & Pen, Z. Correlation between Enterococcus faecalis and persistent intraradicular infection compared with primary intraradicular infection: a systematic review. J. Endod 41, 1207–1213, doi:10.1016/j.joen.2015.04.008 (2015).
Mahony, J. et al. Identification of a new P335 subgroup through molecular analysis of lactococcal phages Q33 and BM13. Appl. Environ. Microbiol. 79, 4401–4409, doi:10.1128/AEM.00832-13 (2013).
Flórez, A. B. & Mayo, B. The plasmid complement of the cheese isolate Lactococcus garvieae IPLA 31405 revealed adaptation to the dairy environment. PLoS One 10(5), e0126101, doi:10.1371/journal.pone.0126101 (2015).
Garneau, J. E. & Moineau, S. Bacteriophages of lactic acid bacteria and their impact on milk fermentations. Microb. Cell Fact. 10(Suppl 1), S20, doi:10.1186/1475-2859-10-S1-S20 (2011).
Samson, J. E. & Moineau, S. Bacteriophages in food fermentations: new frontiers in a continuous arms race. Annu. Rev. Food Sci. Technol. 4, 347–368, doi:10.1146/annurev-food-030212-182541 (2013).
Mahony, J., McDonnell, B., Casey, E. & van Sinderen, D. Phage-host interactions of cheese-making lactic acid bacteria. Annu. Rev. Food Sci. Technol. 7, 267–285, doi:10.1146/annurev-food-041715-033322 (2016).
Moineau, S., Pandian, S. & Klaenhammer, T. R. Evolution of a lytic bacteriophage via DNA acquisition from the Lactococcus lactis chromosome. Appl. Environ. Microbiol. 6, 1832–1841 (1994).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75, doi:10.1186/1471-2164-9-75 (2008).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402, doi:10.1093/nar/25.17.3389 (1997).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964, doi:10.1093/nar/25.5.0955 (1997).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32, 11–16, doi:10.1093/nar/gkh152 (2004).
Chen, L., Xiong, Z., Sun, L., Yang, J. & Jin, Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–645, doi:10.1093/nar/gkr989 (2012).
Chakraborty, A., Ghosh, S., Chowdhary, G., Maulik, U. & Chakrabarti, S. DBETH: a database of bacterial exotoxins for human. Nucleic Acids Res. 40, D615–620, doi:10.1093/nar/gkr942 (2012).
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J. & Wishart, D. S. PHAST: a Fast Phage Search Tool. Nucleic Acids Res 39, W347–W352, doi:10.1093/nar/gkr485 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Hamdi, S. et al. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci Rep 7, 40349, doi:10.1038/srep40349 (2017).
Kristensen, D. M. et al. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487, doi:10.1093/bioinformatics/btq229 (2010).
Acknowledgements
We are grateful to Giovanni Ricci, Stefania Arioli and Jessy Bélanger for technical help as well as Giacomo Eraclio for suggestions. We would like to thank Barbara-Ann Conway (Medical Writer & Editor) for editorial assistance. S.M. acknowledges funding from the Natural Sciences and Engineering Research Council of Canada. S.M. holds a Tier 1 Canada Research Chair in Bacteriophages.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: G.E., M.G.F. Supervised the study: S.M. Performed the experiments: G.E., D.M.T. Analyzed the data: G.E., M.G.F., S.J.L., S.M. Wrote the paper: G.E., M.G.F., S.J.L., S.M.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eraclio, G., Fortina, M., Labrie, S.J. et al. Characterization of prophages of Lactococcus garvieae . Sci Rep 7, 1856 (2017). https://doi.org/10.1038/s41598-017-02038-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02038-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
