
Abstract
Doubly uniparental inheritance (DUI) describes a mode of mtDNA transmission widespread in gonochoric freshwater mussels (Bivalvia: Palaeoheterodonta: Unionida). In this system, both female- and male-transmitted mtDNAs, named F and M respectively, coexist in the same species. In unionids, DUI is strictly correlated to gonochorism and to the presence of the atypical open reading frames (ORFans) F-orf and M-orf, respectively inside F and M mtDNAs, which are hypothesized to participate in sex determination. However, DUI is not found in all three Unionida superfamilies (confirmed in Hyrioidea and Unionoidea but not in Etherioidea), raising the question of its origin in these bivalves. To reconstruct the co-evolution of DUI and of ORFans, we sequenced the mtDNAs of four unionids (two gonochoric with DUI, one gonochoric and one hermaphroditic without DUI) and of the related gonochoric species Neotrigonia margaritacea (Palaeoheterodonta: Trigoniida). Our analyses suggest that rearranged mtDNAs appeared early during unionid radiation, and that a duplicated and diverged atp8 gene evolved into the M-orf associated with the paternal transmission route in Hyrioidea and Unionoidea, but not in Etherioidea. We propose that novel mtDNA-encoded genes can deeply influence bivalve sex determining systems and the evolution of the mitogenomes in which they occur.
Similar content being viewed by others


Introduction
Bivalves are the only known group of animals in which both sexes are capable of stably transmitting their mitochondrial (mt) genome to their progeny through doubly uniparental inheritance (DUI)1. In DUI species, some embryos retain sperm mitochondria that carry a male-transmitted or M-type mtDNA, actively segregate them in their germ line during early development and grow as males, producing M-homoplasmic sperm as adults. By contrast, other embryos eliminate the M mtDNA, or highly reduce its copy number during early development, segregate only the female-transmitted mitochondria with the F-type mtDNA in their germ line and develop as females, in a developmental path comparable to that of other animal species1. The F and M mtDNAs can be extremely divergent in nucleotide (nt) sequence (up to ~40%2), and can also have different gene order and content. In particular, they contain additional lineage-specific open reading frames (ORFs) without recognizable homologies to known genes (hereafter called ORFans3). These F- and M-specific ORFans have been respectively called F-orf and M-orf 4,5,6,7,8,9. Another particular feature often found in DUI species are modifications to the cox2 gene: for example, all DUI species of freshwater mussels (Palaeoheterodonta: Unionida) possess a 3′-elongated cox2 gene in their M mtDNA2, 10.
DUI and the ORFans carried by the two mtDNAs have been hypothesized to be part of a sex determination system involving mitochondria, in part because bivalves lack heteromorphic sex chromosomes in their nuclear genomes and also because freshwater mussels with DUI that switched from the ancestral gonochoric reproductive mode to a derived hermaphroditism lost the M mt genome in the process6. The mtDNA retained in these hermaphrodites, named H, is derived from an ancestral F mitogenome and its F-orf is highly modified into a so-called H-orf 6. The predicted functions of the proteins encoded by the F-orf and M-orf ORFans (we will use ‘F-ORF’ and ‘M-ORF’ to refer to these proteins) support their direct involvement in the DUI mechanism: for example, F-ORFs are suggested to interact with nucleic acids, adhere to membranes, and have roles in signalling, and M-ORFs are suggested to interact with the cytoskeleton and take part in ubiquitination and apoptosis processes8, 11, 12. However, the precise nature of the link between DUI (and M and F ORFans) and the maintenance of gonochorism remains unknown.
To date, DUI has been detected in >100 gonochoric bivalve species, with a somewhat scattered phylogenetic distribution13. The complexity of this system led to the hypothesis that it evolved once in a basal, ancestral lineage of bivalves, followed by subsequent losses in the common ancestor to some modern taxa1, 14, 15. With a single origin of DUI and with no recombination between the two mitogenomes, all sequences of the F and M lineages should be reciprocally monophyletic, in a pattern named ‘gender-joining’15. Up to now however, the gender-joining clustering has been consistently observed only in species of the order Unionida (e.g., ref. 13), suggesting that DUI was present in their common ancestor. In other orders of bivalves, the F and M mtDNAs of a single species are frequently distinct from one another yet they cluster together in a ‘taxon-joining’ pattern15. This phylogenetic pattern may be due to ‘masculinization’ or ‘role reversal events’, a phenomenon directly demonstrated only in Mytilus. Masculinization involves an F mtDNA that invades the paternal route of inheritance and unseats the old M, thus becoming a new M (reviewed in ref. 16). An alternative hypothesis could be independent origins of DUI. For example, studies on F-ORF and M-ORF proteins suggested that ORFans might have various origins (viral or mitochondrial), and that their fixation might have triggered multiple rises of DUI in bivalves8, 11, 12, 17.
The Palaeoheterodonta is one of the most ancient lineages of Bivalves (over 470 million years; ref. 18 and references therein). It is currently represented by two orders, Trigoniida and Unionida (taxonomy from ref. 19). The order Trigoniida is constituted by only a few living species of marine clams, all belonging to the genus Neotrigonia, whereas the Unionida is composed of six families of freshwater mussels19: Iridinidae, Etheriidae, and Mycetopodidae (superfamily Etherioidea); Hyriidae (superfamily Hyrioidea20); and Margaritiferidae and Unionidae (superfamily Unionoidea). No general agreement about the phylogenetic relationships among these six families has been reached yet20. However, the occurrence of DUI in Palaeoheterodonta has been explored by ref. 14. Their results indicated that DUI is absent from the superfamily Etherioidea, and this observation led them to ask (1) whether DUI is the basal condition of Unionida that was lost in the Etherioidea lineage, or (2) if DUI was gained only in the ancestor(s) to the Unionoidea and Hyrioidea lineages14. The authors also tested for the presence of the M type mtDNA in the trigoniid Neotrigonia margaritacea, but the negative results left uncertain the presence of DUI in the order Trigoniida14. An update on this question was provided by ref. 21, who reported the presence of two sex-associated mitotypes with extremely low divergence from one another in N. margaritacea. The sequences obtained, however, are not publicly available at the time of writing this article, and the low divergence found calls for more in-depth studies to confirm the presence of DUI in N. margaritacea.
To reconstruct the evolution of DUI in the ancient bivalve clade Palaeoheterodonta and to better understand the link between sex determination and ORFans in F and M mtDNAs, we sequenced seven new mt genomes from five palaeoheterodont species, with or without DUI and with different reproductive strategies. For Trigoniida we obtained the mt genome, presumably the F, of N. margaritacea (gonochoric, DUI status uncertain), while for Unionida we sequenced the mtDNAs of the following species: Mutela dubia (Iridinidae) (gonochoric without evidence of DUI14, 22), Anodontites trapesialis (Mycetopodidae) (hermaphroditic23 without evidence of DUI; Hoeh W.R. personal communication), and the M and F genomes of Hyridella menziesii (Hyriidae) and Cumberlandia monodonta (Margaritiferidae) (both gonochoric with DUI14). These new sequences allowed us to reconstruct a robust phylogeny of Palaeoheterodonta and infer the origin and evolution of DUI and the ORFans in Unionida.
Results
Overview of mitochondrial genomes
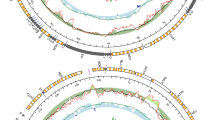
Complete mtDNA sequences have been obtained for N. margaritacea (16,739 bp; putatively the F, if this species has DUI), H. menziesii (F: 16,031 bp; M: 18,140 bp), C. monodonta (F: 16,099 bp; M: 17,575 bp), and M. dubia (non-DUI mtDNA: 16,168 bp). A nearly complete mtDNA sequence was retrieved for A. trapesialis (non-DUI mtDNA: 15,117 bp), as we were not able to sequence two small segments between cox2 and nad2 and within the 16S rDNA. All sequences have been deposited in GenBank (accession numbers KU873118-KU873124; see Table 1). The structures of the genomes are shown in Fig. 1. All mtDNAs possess the standard set of 37 genes. H. menziesii and C. monodonta possess the additional unionid F-orf and M-orf in their F and M genomes, respectively, and the 3′-elongated cox2 gene in their M mtDNAs (1,380 bp in H. menziesii and 1,269 bp in C. monodonta). The cox2 genes of N. margaritacea, M. dubia and A. trapesialis do not have a 3′ elongated region. tRNA-Glu is duplicated in C. monodonta F mtDNA (see Supplementary Fig. S1 in Supplementary Information 1). The gene order of the seven mt genomes is largely similar, with the main difference being an inversion of cox2 and nad3 between N. margaritacea and unionids (Fig. 1 and Supplementary Fig. S1). Supplementary Information 1 contains a comparison of nucleotide content among the seven new mtDNAs and those of the freshwater mussel species listed in Table 1 (Supplementary Table S1 and Supplementary Fig. S2), as well as codon usage descriptions (Supplementary Tables S2 and S3, Supplementary Fig. S3). Repeated sequences were found in all genomes, except for N. margaritacea and H. menziesii F mtDNAs (Fig. 1). The two most notable features are (1) three large tandem repeats in A. trapesialis (331–310 bp, between nad4L and the end of nad6) that comprise pseudogenized fragments of tRNA-Asp, atp8, and nad6, and (2) a complex, ~380 bp-long repeat region in the 3′ half of H. menziesii M-orf. Alignments of these repeats and several other minor features are shown in Supplementary Information 2. For H. menziesii and C. monodonta mtDNAs, the overall intraspecific p-distance values between F- and M-encoded protein coding genes (PCGs) (~40%) and their proteins (48–49%) and the overall d N/d S values between F and M genes (H. menziesii: ~0.33; C. monodonta: ~0.40), are comparable with previous observations on other DUI freshwater mussels, and equivalent values are found also for the interspecific comparisons (Supplementary Tables S4 and S5 in Supplementary Information 3) (see ref. 2 for a comparison).

Maps of the seven mt genomes sequenced in this study. The corresponding GenBank accession numbers are given inside each genome map. Genomes are not in scale among each other, see main text for their length. Outer ring comprises all standard and putative coding sequences, identified with the following colour code: yellow, genes encoding electron transport chain and ATP-synthase subunits; dark blue, tRNA genes (see Supplementary Fig. S1 in Supplementary Information 1 for their names); pale green, rRNA genes; red, F-orf; bright blue, M-orf; bright green, additional ORFs cited in the text. Sequences are located on the outer or inner side of this circle according to their coding direction, respectively forward (clockwise) and reverse (anti-clockwise). In A. trapesialis mtDNA, the position of the two sequencing gaps is indicated on this ring. Middle ring comprises the repetitive regions, indicated in grey (not found in N. margaritacea and H. menziesii F): in A. trapesialis are specified the names R1–3 for the large tandem repeats found between nad4L and atp6; in H. menziesii M, two regions of high similarity found in different locations are marked with an asterisk (*), and a large palindrome sequence with “p”. Alignments of all repetitive regions can be found in Supplementary Information 2. Innermost ring displays the position of the unassigned regions as black segments.
Phylogenetic analyses
Phylogenies of Palaeoheterodonta were reconstructed using the nt and amino acid (aa) sequences of 12 PCGs (except atp8) of the mt genomes in Table 1. Maximum likelihood (ML) and Bayesian inference (BI) were utilized. The resulting ML trees are shown in Fig. 2: minor differences at subfamily or species level between them and BI trees are described in Fig. 2 legend. In the nt-based phylogeny (Fig. 2a), N. margaritacea mtDNA is sister to all Unionida mtDNAs, which are separated into two major clades: the first comprises only M mt genomes of superfamilies Hyrioidea and Unionoidea, while the second the non-DUI mtDNAs of Etherioidea as a sister group to F and H mtDNAs of Hyrioidea and Unionoidea. In both of these main branches, H. menziesii mt genomes are always sister to Unionoidea ones. The three superfamilies of freshwater mussels are thus monophyletic in both main Unionida clades, as well as the families for which more than one species is available (i.e., Margaritiferidae and Unionidae; Table 1). A polytomy is found at the base of Unionidae M mtDNAs in the ML tree that is resolved in the BI one (not shown), where these genomes have the same relationships as the F mtDNAs of the respective species. In the aa-based phylogenies (Fig. 2b), Palaeoheterodonta mt genomes are split in two major branches: one in which N. margaritacea is sister to a clade comprising Etherioidea plus the F and H mt genomes of all other freshwater mussels, and another containing all Unionida M mtDNAs (Hyrioidea and Unionoidea again have the same relationships in both branches). Unionida mt genomes are therefore split on two different branches because of N. margaritacea nested position. The topology of the Hyrioidea + Unionoidea F/H clade in both trees is identical to that of the BI nt-based tree described above and in the figure legend. In the ML tree shown in Fig. 2b, the same polytomy found in the nt-based tree in the M clade is present, which is again resolved in the BI tree (not shown) as described above.

Phylogeny of Palaeoheterodonta. Trees were constructed using (a) nucleotide sequence of 12 mtDNA-encoded genes and (b) the respective inferred protein sequences of species in Table 1. Only ML trees are shown: topology of BI trees was largely congruent; the few small differences are only described in the main text and below. Support values at a node are shown only if (1) they were not 100% bootstrap support on the ML tree and 1.0 for the posterior probability values on the BI tree, or (2) when a node was only present in the ML tree. Support values, when shown, are presented next to the node as ‘ML bootstrap value/BI posterior probability’. Species names in the middle column are coded with the following colours, according to taxonomy and/or mtDNA type: black, non-Palaeoheterodonta outgroups; brown, Trigoniida; red, Etherioidea; pink, F mtDNA of DUI species; blue, M mtDNA of a DUI species; violet, H mtDNA of a secondarily hermaphroditic unionid. Branches inside Palaeoheterodonta are coloured according to taxa: brown, Trigoniida; red, Etherioidea; bright green, Hyriidae (Hyrioidea); dark green, Margaritiferidae (Unionoidea); aqua blue, Unionidae (Unionoidea). (a): A dissimilarity in the ‘Unionoidea F’ clade between BI and ML trees resides in the different position of the branch comprising five species of the subfamily Gonideinae (i.e., H. cumingii, H. schlegelii, I. japanensis, S. carinatus, and S. oleivora; Table 1) relative to all other Unionidae F/H mtDNAs: in the ML tree here shown, this clade is sister to the cluster containing Ambleminae and Lampsilinae (see Table 1 for those species), while in the BI (not shown) it is sister to the clade containing the other ten F and H Unionidae mt genomes. Because of the positions in both ML and BI trees of L. tortuosa, A. woodiana and U. pictorum, subfamilies Gonideinae and Unioninae (Table 1) are not supported as being monophyletic. (b): Both ML and BI protein-based trees differ from the nucleotide-based one in the relative position of L. tortuosa, which branches differently inside the same cluster (compare the two trees in figure). Again, Gonideinae and Unioninae are not monophyletic (Table 1).
Search for additional ORFs and functional characterization
Excluding the F-orf and M-orf in H. menziesii and C. monodonta mtDNAs, we found a total of 375 new possible ORFs coding for >10 aas in the unassigned regions (URs) of the new mt genomes. Specifically, we found 79 ORFs in N. margaritacea, 58 in A. trapesialis, 50 in M. dubia, 34 in H. menziesii F mtDNA and 60 in its M, and 41 in C. monodonta F mtDNA and 53 in its M (Supplementary Information 4). None of the 375 ORFs translated protein sequences contain statistically supported conserved domains. Among F- and M-ORFs, only H. menziesii M-ORF exhibited conserved domains (Supplementary Table S6 in Supplementary Information 4). To search for homologs, we compared the putative new ORF products, plus the H. menziesii and C. monodonta F- and M-ORFs, to the standard 13 mtDNA-encoded proteins, to proteins in databases, to lineage-specific ORFans of known DUI species, and to themselves (Supplementary Information 4). No significant hits were found in the comparison with standard mtDNA-encoded proteins. Searches on the Swiss-Prot database gave no results, while TrEMBL gave a total of 266 hits for 30 of the new ORFs. Most of the hits are to putative uncharacterized proteins without a functional description; however, for certain ORFs, some hits were to characterized proteins (Supplementary Information 4). The new ORFs proteins and the F- and M-ORF from H. menziesii and C. monodonta have no similarities to either known F- or M-ORFs of the DUI taxa Mytilus spp., Ruditapes philippinarum, or Musculista senhousia. For eight ORFs in H. menziesii M mtDNA located in two distant URs (UR3 and UR17), their similarity appears to be due to the presence of a sequence block shared between those URs (Fig. 1 and Supplementary Information 2). C. monodonta M-ORF has a hit with CmonM_UR_24_11 (phmmer24 results: E-value = from 3.0E-05 to 6.2E-05, score = from 19.0 to 19.6; Supplementary Information 4), a new ORF just upstream of it between tRNA-Asp and atp8 (Fig. 1). A structural alignment of these two proteins made with T-Coffee Expresso25,26,27 showed high similarity, with many identical aas (score = 91, cons = 9; alignment shown in Supplementary Information 5, Supplementary Fig. S4). Finally, Hidden Markov model (HMM) profiles of unionid F-ORFs and M-ORFs produced by ref. 12 were also used to search for similarities with the translated proteins of the new ORFs (Supplementary Information 4). Apart from F- and M-ORFs, no other hits were found using the F-ORF profiles, but one hit was found with M-ORF profiles for Atra_UR_22_18 protein (hmmsearch24 results: E-value = 0.048, score = 6.5), an ORF located between tRNA-Asp and atp8 (like CmonM_UR_24_11 in C. monodonta M mtDNA described above) inside the second large repeat in A. trapesialis mtDNA (Fig. 1), in correspondence of atp8 pseudogenized fragments (Supplementary Information 2).
Structural characterization of proteins
We performed a structural characterization of H. menziesii and C. monodonta MCOX2, since they are the first examples of such elongated proteins for Hyriidae and Margaritiferidae, and of the F- and M-ORFs of the same species to compare them with the putative proteins CmonM_UR_24_11, Atra_UR_21_9, and Atra_UR_22_18 given their overall similarity described above. Supplementary Information 5 contains all structural alignments cited below (shown in Supplementary Fig. S4) and details of the characterization, which is summarized in Fig. 3. H. menziesii and C. monodonta MCOX2 extensions (Fig. 3a,b) have a different number of predicted transmembrane sections (four versus three), more supported in H. menziesii than in C. monodonta, but the portion containing the first two helices in both proteins aligns with good quality. H. menziesii and C. monodonta F-ORFs (Fig. 3c,d) share a largely identical structure but they align well only in the second half of the sequence, and the first helix is predicted as having transmembrane properties only in H. menziesii. M-ORFs (Fig. 3e,f) are on the contrary quite variable, both in length and structure. The first half of H. menziesii M-ORF broadly resembles the whole protein of C. monodonta (and this part has a better overall alignment); a signal peptide in this region is recognized only in H. menziesii. The second half of H. menziesii M-ORF encoded by the repeat region described above (from position ~140) may form some helices and beta-sheets, but it is also characterized as largely disordered. C. monodonta CmonM_UR_24_11 protein structure (Fig. 3g) is largely overlapping with that of the same species M-ORF (Fig. 3f; see above), but contrary to it, a signal peptide is found at CmonM_UR_24_11 N-terminus. Atra_UR_21_9 putative protein structure (Fig. 3h) is comparable to that of C. monodonta M-ORF and CmonM_UR_24_11, and to the first half of H. menziesii M-ORF. A signal peptide is predicted at its N-terminus, as well as a small disordered C-terminal segment. Despite the presence of atp8 fragments in Atra_UR_21_9, it does not align well with A. trapesialis ATP8 protein. Overall, the proteins scoring the best alignments with Atra_UR_21_9 are C. monodonta F-ORF and M-ORF, followed by H. menziesii M-ORF and F-ORF. H. menziesii M-ORF has the highest number of identical aas aligned compared to all other proteins considered (24 aas). The short length of the Atra_UR_22_18 protein (22 aas; Supplementary Information 4) prevented a structural characterization. Its best alignments are with Atra_UR_21_9 protein and CmonM_UR_24_11. As for Atra_UR_21_9, the sequence of Atra_UR_22_18 aligns with A. trapesialis ATP8 with low scores. These results indicate that structural similarities are maintained in the elongated MCOX2, as well as in the lineage-specific F- and M-ORFs proteins, of hyriids and margaritiferids. Also, they evidence the strict relationships of the putative proteins encoded by CmonM_UR_24_11, Atra_UR_21_9, and Atra_UR_22_18 with M-ORFs.

Structural characterization of proteins encoded by lineage-specific genes and ORFs. Visual summary of the characterization made with Quick2D50 on MCOX2, F-ORF, M-ORF, and the putative proteins encoded by the ORFs CmonM_UR_24_11 and Atra_UR_21_9 from Hyridella menziesii, Cumberlandia monodonta, and Anodontites trapesialis mtDNAs. Complete Quick2D outputs are displayed in Supplementary Information 5. In each panel, on the X axis are the amino acid positions of the protein, while on the Y is the support for each feature (described below) shown in terms of how many methods indicated a certain characteristic at a given position. Colour code of features: full green areas, transmembrane regions; striped red areas, regions forming helices; striped blue areas, regions forming beta-sheets; purple lines, signal peptide signature; orange lines, disordered regions. The two grey squares on the background of panels (a) and (b) represent, from left to right, the boundaries of the conserved transmembrane and periplasmic domains of COX2.
Discussion
The sequencing of mt genomes representing Palaeoheterodonta taxa for which no complete sequences were available until now sheds new light on the evolution of mtDNA and on mitochondrial inheritance systems in this ancient bivalve taxon. First, mt genome organization in Palaeoheterodonta seems to be rather stable compared to other bivalve taxa, in which important rearrangements are frequently observed among relatively close taxa within the same family or genus28. Indeed, the major difference between N. margaritacea and Unionida is only an inversion between cox2 and nad3, and mt genomes of Etherioidea (i.e., M. dubia and A. trapesialis) practically have the same organization as the F ones of Hyrioidea and Unionoidea, apart from the absence of F-orf (Fig. 1). A. trapesialis mtDNA, however, shows a highly rearranged area between nad4L and atp6 composed of three tandem repeats, resulting in an organization and gene order similar to those of M mt genomes in DUI unionids (Fig. 1). Specifically, our results suggest that the position of tRNA-Asp and atp8, identical to that of M mtDNAs4, is most probably the outcome of a tandem duplication of the segment containing the original functional copies followed by pseudogenization of supernumerary ones that are still recognizable in the repeated sequences (i.e., a tandem duplication-random loss event29). For example, ORFs Atra_UR_21_9 and Atra_UR_22_18, located in the repeats, contain recognizable parts of atp8. Because of these rearrangements, Atra_UR_21_9 is located in exactly the same position as the M-orf in DUI freshwater mussels (between nad4L and tRNA-Asp4; Fig. 1), and its putative protein also has structural analogies to the M-ORF of DUI unionids (Fig. 3). Again in support of the tandem duplication hypothesis, the protein translated from Atra_UR_22_18 (between tRNA-Asp and atp8; Fig. 1) has similarities to both the ATP8 produced by the same genome and to the putative product of Atra_UR_21_9. The similarity between the two A. trapesialis ORFs and their location mirror what is observed in the M mtDNA of the DUI margaritiferid C. monodonta (Fig. 1), whose M-orf and additional ORF CmonM_UR_24_11 (Fig. 1) are located in the same positions and encode for proteins comparable in structure (Fig. 3). H. menziesii M mtDNA shows the longest cox2 and M-orf genes found in the M mt genome of any DUI unionid. The M-orf of this species has a complex repeat region in its 3′ half downstream from the region whose protein product is more similar to other M-ORFs (see Fig. 3). Until now, tandem repeats in DUI-related ORFans have been observed only in the H-orf of hermaphroditic freshwater mussels that lost DUI secondarily6, 12, but to our understanding, this feature of the M-orf does not seem to have affected the DUI system of H. menziesii.
By removing the sperm transmitted mt genomes from the trees in Fig. 2, we can see that the relationships among families indicated by female-transmitted mtDNAs are exactly the same in both phylogenies. The evolutionary tree of Palaeoheterodonta based on egg-transmitted mtDNAs obtained in this way (shown in Fig. 4) confirms the long-established sister group relationship of orders Trigoniida and Unionida. Inside the monophyletic order Unionida, the superfamily Etherioidea appears to be a monophyletic sister group to a clade comprising the monophyletic superfamilies Hyrioidea and Unionoidea as sister taxa. The relationship of Hyriidae with the other five families of Unionida has been highly debated20, but all our reconstructions have strong support (Fig. 2). As observed in previous studies (e.g., ref. 13), our phylogenies gave a gender-joining15 topology for palaeoheterodont mt genomes (Fig. 2). Because protein sequences are more conserved at this taxonomic level and less prone to saturation18, we consider this latter reconstruction to be the more supported and we thus infer that DUI was present prior to the divergence of the orders Trigoniida and Unionida. However, this interpretation must be tempered by the uncertain presence of DUI in N. margaritacea 21, the sister taxon to all Unionida in our analysis (Fig. 4). If DUI can be clearly demonstrated in this species, then the hypothesis that it was present in the Trigoniida-Unionida common ancestor can be supported, thus dating the presence of DUI in Palaeoheterodonta at >200 MYA (based on the estimates by ref. 30). If not currently present in the Trigoniida, DUI could have been lost independently in this order or, alternatively, DUI may not have been present in the last common ancestor of Trigoniida-Unionida and gained independently in Unionida (a conclusion different to what the aa-based tree indicates, but in accord with the nt-based tree; Fig. 2). Following a strict interpretation of the phylogeny in Fig. 2b, the two etherioids M. dubia (gonochoric) and A. trapesialis (hermaphroditic) must have lost DUI at some point during their evolution, maintaining only a female-transmitted mt genome. It is unknown, however, if this inferred loss might have been ancestral to all Etherioidea or if there were two independent events, and if such events affected their sex determination mechanism. Given the high plasticity of sex determining systems in bivalves (Breton S. et al. in preparation), and the extremely long evolutionary time that might separate present-day M. dubia and A. trapesialis from the putative loss event(s), the gonochorism and hermaphroditism of these two species22, 23 might not be directly related to the putative loss of DUI. Without additional information, it is presently more parsimonious to infer that DUI was lost once, in the common ancestor to the Etherioidea.

Model for the evolution of mt genomes and DUI in Palaeoheterodonta. The backbone tree represents the phylogeny obtainable by removing the M mtDNAs from the trees in Fig. 2. DUI presence/absence is specified for all major clades. At the tip of each final branch, the names of the species for which we obtained the mtDNA sequences in this study are indicated together with the respective family (no species are enlisted for Unionidae); superfamily affiliations are specified with bars on the right side of the figure. Circles beside a final branch represent a schematic mt genome structure with the following colour code: black, mtDNA of a non-DUI species; red, F mtDNA in a DUI species/family; blue, M mtDNA in a DUI species/family. Purple circles in parentheses represent the typical H mtDNA of secondarily hermaphroditic species in a given family. For N. margaritacea mt genome, the interrogation point highlights the uncertain presence of DUI in this species, while the red colour the fact that the genome we obtained, if DUI is present, would be its F. Bars on the mtDNAs represent atp8-derived ORFs (green), M-orfs (blue), F-orfs (red), or H-orfs (purple), while blue triangles represent the elongated cox2 genes in M mtDNAs. The position of these latter features on the schematic mtDNAs reflects their actual location in the mt genomes (compare with Fig. 1). Bars inside the Unionida branches of the tree indicate evolutionary events and/or character states of lifestyle-related features as enlisted by ref. 14 (grey bars) or evolutionary events of mt genomes (black bars), described inside the figure.
The hypothetical origin of freshwater mussels M-orf genes from a duplication of atp8 recently proposed by ref. 12 is supported by our model of DUI evolution (Fig. 4), in which the M-orfs of H. menziesii and C. monodonta (and of all DUI unionid species in general) are suggested to share the same ancestry with A. trapesialis Atra_UR_21_9 and Atra_UR_22_18. These latter ORFs contain clearly recognizable pseudogenized fragments of atp8 derived from its duplication, and the protein encoded by Atra_UR_22_18 was recognized by the M-ORF HMM profiles. The architecture of A. trapesialis mtDNA and of M mt genomes of distantly related DUI species (Fig. 1) must have arisen in the first phases of Unionida radiation. Given its close phylogenetic relationship to the F mtDNA of DUI unionids (Fig. 2), the rearranged mt genome of A. trapesialis most probably originated from a female-transmitted genome in the common ancestor of the two main lineages Etherioidea and Hyrioidea + Unionoidea (Fig. 4, step A). We hypothesize this ancestor to have been gonochoric, since our phylogeny suggests that it had DUI (Figs 2 and 4), and DUI has never been observed in hermaphroditic species6, 14. If true, a rearranged mt genome structure (1) was fixed in the maternal route of inheritance in the ancestor of etherioids, which contemporarily lost DUI, and Iridinidae lost this M-like architecture during their evolution (Fig. 4, steps C and D), and (2) invaded the paternal route of transmission in the Hyrioidea + Unionoidea lineage substituting a pre-existing M (i.e., a role reversal event16). Alternatively, if we hypothesize that the last common ancestor of the two main unionid lineages did not have DUI, then DUI appeared only in the Hyrioidea + Unionoidea lineage in which the rearranged mt genome became male-transmitted. This option, which would be in line with the hypothesis of multiple origins of DUI in bivalves1, 8, would move the origin of DUI in freshwater mussels at the earliest to the split between Hyrioidea and Unionoidea. However, this hypothesis is currently not supported by our phylogenetic analyses (Fig. 2).
The switch of the rearranged mt genome to the paternal route of inheritance in the Hyrioidea + Unionoidea line (Fig. 4, step F) might have been caused by the two new atp8-derived ORFs. The original ability of the ATP8 protein to be incorporated into the ATPase complex might have been maintained in the duplicated gene products, giving them the capacity to alter membrane potential by modulating ATPase activity12, 31 or, at least, to be inserted into mitochondrial membranes. Our results (Supplementary Table S6) and previous studies8, 12 corroborate a proposed involvement of M-ORFs in membrane association, microtubule binding and RNA interactions, activities that can be directly related to the peculiar behaviour of sperm mitochondria in DUI species during early male development (see references in ref. 32) and to the proposed sex determining capabilities of M mtDNA. The atp8-derived ORFs might have granted selfish characteristics to their host mtDNA or developed the ability to distort sex determination to maleness17, transforming them into M-orfs (we will refer to them as M-orf1, between tRNA-Asp and nad4L, and M-orf2, between atp8 and tRNA-Asp, for simplicity). This M lineage subsequently evolved in its own particular ways in the different families. In Hyriidae (H. menziesii) and Unionidae, M-orf2 was lost (Fig. 4, step H), and M-orf1 acquired repetitive sequences and became unusually long in the first but not in the latter of these families (Figs 1 and 4e). Only Margaritiferidae (C. monodonta) retained the ancestral state in their M mtDNA by maintaining both M-orf1 and M-orf2 (Figs 1 and 4, step G): their translated proteins are very similar in structure and sequence, which can be due to either their common ancient origin from ATP8 (see above) and/or to convergent functions and convergent evolution. Since it is, however, the first observation of an M-orf2 gene, more complete M mt genomes from DUI margaritiferids will be required to clarify its evolution. Moreover, the inferred independent loss of M-orf2 in Hyriidae and Unionidae (see Fig. 4) questions (1) the role of this ORFan for the DUI system, if any, in Margaritiferidae, and (2) if there was a complete transfer to the nucleus in Hyriidae and Unionidae or if the second M-orf was simply lost from their M mtDNA.
At the same time, in the female-transmitted line, the atp8-derived ORFs are not maintained, the F-orf appears upstream nad2 and is fixed (Fig. 4, step G) (whether this ORFan was the result of another gene duplication, specifically of nad2, is discussed in ref. 12). Previous studies suggested that mitochondrial ORFans in bivalves could act as sex ratio distorters and restorers, i.e., they would be key elements of a sex determination system involving F and M mt (and nuclear) genomes6, 12, 17. The role of the F-orf is still unknown, but the macromutations it accumulates when a unionoid becomes hermaphroditic and loses DUI (i.e., when a F-orf becomes an H-orf; Fig. 4, step I) support a concerted action between F-orf and M-orf in maintaining gonochorism6, 12. Concurrently, an elongated cox2 was also fixed in the M line (Fig. 4, step F): although no molecular evidence has been gathered yet to support this hypothesis, it was suggested that the presence of the Mcox2 elongation in DUI freshwater mussels could prevent role reversals10, a phenomenon observed in Mytilus species who have DUI and lack such elongated gene1. The prevention of role reversals and recombination might have been positively selected to avoid the rise of new mt genomes possessing both F- and M-orf and keep the gonochoristic sex determination system safe. The connection among ORFans, DUI, and gonochorism in freshwater mussels is thus forged: this bond is surely strong, as F and M lineages have been evolving separately for several hundred million years2, 33, and when a modification in one of these three elements rarely occurs, it unequivocally forces the other two to degenerate6 (Fig. 4, step I).
Conclusion
In this study we propose a model for the evolution of the unique mitochondrial inheritance system that is DUI in bivalves, in particular in the order Unionida. We suggest that present-day sperm-transmitted mtDNAs evolved from a deviant mt genome, with possibly selfish and/or sex distorting qualities6, 17, in a supposedly DUI gonochoric ancestor of all freshwater mussels. Specifically, two atp8-derived ORFs are suggested to have triggered a change in inheritance route of the mt genome carrying them. One of them became the M-orf, an ORFan gene thought to be involved in the maintenance of gonochorism in freshwater mussels with DUI6. How these ORFs might have established a link between mtDNA and sex determination is however still obscure. Deviant mt genomes with sex distorting qualities have been observed also outside bivalves34, but the general influence of mitochondria and their genome on sex determination in animals remains largely unknown. In bivalves, who lack heteromorphic sex chromosomes and are mostly gonochoric but show a myriad of variations on hermaphroditism (Breton S. et al. in preparation), divergent selfish mtDNAs carrying novel ORFans would have an easy way into moulding their flexible sex determination system to achieve constant transmission through the generations17. Our results indeed demonstrate how plastic the architecture of the small mt genome can potentially be, and how it could deeply influence the biology of its ‘host’ on large evolutionary scales beyond the mere production of energy for the cell35.
Methods
Animal sampling and genome sequencing
Animals, all of which (1) are members of the bivalve clade Palaeoheterodonta (see Table 1 for a detailed taxonomy of the species), (2) have different reproductive strategies, and (3) do or do not show DUI evidence, were collected from the following locations: Neotrigonia margaritacea Lamarck 1804 (Trigoniida: Trigoniidae; gonochoric, DUI status uncertain) from Gulf St. Vincent (South Australia); Mutela dubia Gmelin 1791 (Unionida: Iridinidae; gonochoric without DUI) from the Nile River (Al Jizah, Egypt); Anodontites trapesialis Lamarck 1819 (Unionida: Mycetopodidae; hermaphroditic without DUI) from the Rio Madre de Dios (Madre de Dios, Peru); Hyridella menziesii Gray 1843 (Unionida: Hyriidae; gonochoric with DUI) from the Taieri River (South Island, New Zealand); and Cumberlandia monodonta Say 1829 (Unionida: Margaritiferidae; gonochoric with DUI) from the Clinch river (Claiborne County, TN, USA). Specimens were sexed through microscopical examination of the gonads to check for the type of gametes produced. Total DNA was extracted from gonadal tissue of one hermaphroditic, or one female and one male individual for each species (except for N. margaritacea for which only one female was available) with a QIAGEN DNeasy animal kit (QIAGEN Inc., Valencia, CA, USA) using the animal tissue protocol. Sequencing of the seven mt genomes (i.e., both F and M for H. menziesii and C. monodonta, one from M. dubia and A. trapesialis, and the putative F from N. margaritacea) was performed by Genome Sequencer FLX sequencing service (McGill University, Montréal, Quebec, Canada).
Annotation and characterization of mtDNAs
PCGs and rRNA coding genes were annotated with MacVector Sequence Analysis Software 10.0 (Accelrys Inc, San Diego, California, USA). ARWEN36 and tRNAscan37 were used to annotate tRNA genes. When ARWEN and tRNAscan failed to recognize a tRNA, the program MiTFi38 (implemented in MITOS39) was used, because it implements an alternate structure-based covariance model to detect tRNA folding. Gene order comparisons among the newly sequenced mt genomes were performed manually. Tandem repeat sequences and other repeats in the mtDNAs were defined using Tandem repeats finder40 and BLASTn41, using for the latter a single whole genome sequence as both query and subject with default parameters. Diagrams of mtDNA structures were generated using the program GenomeVx42. Nucleotide content of N. margaritacea and all freshwater mussel mt genomes in Table 1, as well as the codon usage (expressed as relative synonymous codon usage, RSCU) in the 13 standard PCGs of the seven newly sequenced mtDNAs, were calculated using MEGA 5.2.243. Pairwise-distances (p-distances) for standard PCGs (i.e., those that code for subunits of the electron transport chain or ATPsynthase) and rRNA genes, PCGs translated proteins, as well as d N and d S statistics for the 13 PCGs, were calculated with MEGA 5.2.2. Alignments for these analyses were produced by MEGA 5.2.2 with MUSCLE44, using codon alignment for PCGs and DNA for rRNA genes. Intraspecific p-distance comparisons between F and M mtDNAs of H. menziesii and C. monodonta for PCGs were calculated, as well as the interspecific ones (i.e., a genome of one species versus the genome of another). For rRNA genes, p-distances were only calculated for the 12S and 16S genes for H. menziesii and C. monodonta. All the obtained statistics were processed with R version 3.1.045 on RStudio 0.9846, which was also used for drawing graphs.
Characterization of ORFs
URs of all newly sequenced mt genomes were extracted and given a sequential identification number, choosing as number 1 the UR upstream from cox1 (see Fig. 1) and then numbering all others in a clockwise direction. We then searched the URs for additional ORFs with the EMBOSS getorf program47, considering only ORFs at least 33 nts in length (i.e., a minimum of 10 codons encoding aas plus a stop codon) using the invertebrate mitochondrial genetic code. These ORFs were translated into the corresponding protein sequences in two ways (again with getorf): (1) using the corresponding aa in the presence of alternative start codons, and (2) always using methionine as the first aa regardless of the predicted first codon in the ORF. Both sets of translations, plus H. menziesii and C. monodonta lineage-specific F-orf and M-orf translated protein sequences, were used for the subsequent analyses. The set of ORF proteins was searched for conserved domains against the CDD database48 (last accessed June 2015) with Batch Web CD-search tool49, using the ‘live search’ mode with default settings and a cutoff E-value of 0.001. We locally ran phmmer (present in the HMMER24 suite of programs) to compare the set of protein sequences against: (1) the 13 standard proteins encoded by the newly sequenced mtDNAs, to find unannotated, or degenerated but still recognizable, duplicated genes; (2) the proteins present in the Swiss-Prot and TrEMBL databases (downloaded October 2015), to find homologs; (3) all known F- and M-ORF proteins of non-unionid DUI species (i.e., Mytilus edulis, M. galloprovincialis, M. trossulus, M. californianus, M. senhousia, and R. philippinarum 5, 8) to find similarities in evolutionarily distant taxa; (4) F-ORF, M-ORF, and H-ORF proteins of freshwater mussel species used in the phylogenetic analyses (see below), to find similarities among unionids; and (5) the protein set itself, to find similar proteins encoded in the same or in different mtDNAs. Default parameters were used for all the phmmer searches (i.e., an E-value cutoff of 0.001). We also ran hmmsearch24 locally to search the new ORF proteins against unionoids F-ORF and M-ORF HMM profiles produced from the respective protein alignments; these profiles were built for an extensive in silico characterization of freshwater mussel lineage-specific ORFans by ref. 12. An E-value cutoff of 1 was used in this case to allow the profiles to detect homologs other than F- and M-ORFs themselves. Quick2D50 was used to characterize the structure of MCOX2, M-ORF, and F-ORF of H. menziesii and C. monodonta, plus other putative proteins that were found to be comparable to lineage-specific ORFan products. Structural alignments for ORF proteins of particular interest were produced by T-Coffee Expresso25,26,27 using default parameters. The two COX2 conserved domains inside the H. menziesii and C. monodonta MCOX2 proteins were identified with BLASTp41.
Phylogenetic analyses
We performed a phylogenetic analysis on Palaeoheterodonta bivalves using all standard PCG sequences, except atp8 because of its high variability and lack of confidence in alignment, from the seven newly sequenced mtDNAs and 26 mt genomes of other freshwater mussels belonging to 20 species (15 F, 5 H, and 6 M mtDNAs; see Table 1 for species list, mtDNAs, and references). To root the trees in the subsequent analyses, we chose to use PCG sequences from the mtDNAs of the Protobranch bivalves Solemya velum (Solemyida) (GenBank accession number: JQ72844751) and Nucula nucleus (Nuculida) (GenBank accession number: EF211991; Dreyer H. and Steiner G. personal communication), plus those from the aplacophoran mollusc Chaetoderma nitidulum (GenBank accession number: EF211990; Dreyer H. and Steiner G. personal communication). Thus, a total of 36 taxa was used to reconstruct the phylogenies. The T-Coffee algorithm25 was used for single alignments. For aas, we carried out structural alignments through PSI-BLAST52 in the succession PSI-Coffee > Expresso > accurate. For nts we used M-Coffee, starting from MAFFT53 and MUSCLE44 libraries. Alignments were masked to discard phylogenetic noise using Aliscore 2.054, BMGE 1.155 (using BLOSUM95 for aas), Gblocks 0.91b56, and Noisy57, with default settings. Only sites kept by at least three out of four of these programs were retained for further analyses. We selected the best-fitting partitioning scheme and molecular evolution models using PartitionFinderProtein and PartitionFinder 1.1.058 under the Bayesian Information Criterion (BIC) and a greedy approach. The phylogenetic tree was estimated with both ML and BI approaches. The software RAxML 8.2.059 was chosen for the ML inference under the CAT model and using 500 bootstrap replicates. BI was conducted with MrBayes 3.2.160 as in ref. 18. The evolutionary models chosen with the BIC for the nt alignments of the PCGs were the following: GTR + I + G for atp6, cox1, cox2, cox3, cytb, nad1, nad2, nad4, and nad5; HKY + I + G for nad3 and nad6; and HKY + G for nad4L. Trees were graphically edited using PhyloWidget61 and Dendroscope 3.3.262.
References
Zouros, E. Biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 40, 1–31, doi:10.1007/s11692-012-9195-2 (2013).
Doucet-Beaupré, H. et al. Mitochondrial phylogenomics of the Bivalvia (Mollusca): searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol. Biol. 10, 50, doi:10.1186/1471-2148-10-50 (2010).
Fischer, D. & Eisenberg, D. Finding families for genomic ORFans. Bioinformatics 15, 759–762, doi:10.1093/bioinformatics/15.9.759 (1999).
Breton, S. et al. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics 183, 1575–1589, doi:10.1534/genetics.109.110700 (2009).
Breton, S. et al. Evidence for a fourteenth mtDNA-encoded protein in the female-transmitted mtDNA of marine Mussels (Bivalvia: Mytilidae). PLoS One 6, e19365, doi:10.1371/journal.pone.0019365 (2011).
Breton, S. et al. Novel protein genes in animal mtDNA: a new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol. Biol. Evol 28, 1645–1659, doi:10.1093/molbev/msq345 (2011).
Passamonti, M., Ricci, A., Milani, L. & Ghiselli, F. Mitochondrial genomes and Doubly Uniparental Inheritance: new insights from Musculista senhousia sex-linked mitochondrial DNAs (Bivalvia Mytilidae). BMC Genomics 12, 442, doi:10.1186/1471-2164-12-442 (2011).
Milani, L., Ghiselli, F., Guerra, D., Breton, S. & Passamonti, M. A comparative analysis of mitochondrial ORFans: new clues on their origin and role in species with doubly uniparental inheritance of mitochondria. Genome Biol. Evol 5, 1408–1434, doi:10.1093/gbe/evt101 (2013).
Ghiselli, F. et al. Structure, transcription, and variability of metazoan mitochondrial genome: perspectives from an unusual mitochondrial inheritance system. Genome Biol. Evol 5, 1535–1554, doi:10.1093/gbe/evt112 (2013).
Curole, J. P. & Kocher, T. D. Ancient sex-specific extension of the cytochrome c oxidase II gene in bivalves and the fidelity of doubly-uniparental inheritance. Mol. Biol. Evol. 19, 1323–1328, doi:10.1093/oxfordjournals.molbev.a004193 (2002).
Milani, L., Ghiselli, F., Maurizii, M. G., Nuzhdin, S. V. & Passamonti, M. Paternally transmitted mitochondria express a new gene of potential viral origin. Genome Biol. Evol 6, 391–405, doi:10.1093/gbe/evu021 (2014).
Mitchell, A., Guerra, D., Stewart, D. & Breton, S. In silico analyses of mitochondrial ORFans in freshwater mussels (Bivalvia: Unionoida) provide a framework for future studies of their origin and function. BMC Genomics 17, 597, doi:10.1186/s12864-016-2986-6 (2016).
Gusman, A., Lecomte, S., Stewart, D. T., Passamonti, M. & Breton, S. Pursuing the quest for better understanding the taxonomic distribution of the system of doubly uniparental inheritance of mtDNA. PeerJ 4, e2760, doi:10.7717/peerj.2760 (2016).
Walker, J. M. et al. Taxonomic distribution and phylogenetic utility of gender-associated mitochondrial genomes in the Unionoida (Bivalvia). Malacologia 48, 265–282 (2006).
Theologidis, I., Fodelianakis, S., Gaspar, M. B. & Zouros, E. Doubly uniparental inheritance (DUI) of mitochondrial DNA in Donax trunculus (Bivalvia: Donacidae) and the problem of its sporadic detection in Bivalvia. Evolution 62, 959–970, doi:10.1111/j.1558-5646.2008.00329.x (2008).
Stewart, D. T., Breton, S., Blier, P. U. & Hoeh, W. R. Masculinization Events and Doubly Uniparental Inheritance of Mitochondrial DNA: A Model for Understanding the Evolutionary Dynamics of Gender-Associated mtDNA in Mussels In Evolutionary Biology (ed. Pontarotti, P.) 163–173, doi:10.1007/978-3-642-00952-5_9 (Springer, 2009).
Milani, L., Ghiselli, F. & Passamonti, M. Mitochondrial selfish elements and the evolution of biological novelties. Curr. Zool. doi:10.1093/cz/zow044 (2016).
Plazzi, F., Puccio, G. & Passamonti, M. Comparative Large-Scale Mitogenomics Evidences Clade-Specific Evolutionary Trends in Mitochondrial DNAs of Bivalvia. Genome Biol. Evol 8, 2544–2564, doi:10.1093/gbe/evw187 (2016).
Bieler, R., Carter, J. G. & Coan, E. V. Classification of Bivalve Families. Malacologia 52, 113–133 (2010).
Graf, D. L., Jones, H., Geneva, A. J., Pfeiffer, J. M. 3rd & Klunzinger, M. W. Molecular phylogenetic analysis supports a Gondwanan origin of the Hyriidae (Mollusca: Bivalvia: Unionida) and the paraphyly of Australasian taxa. Mol. Phylogenet. Evol. 85, 1–9, doi:10.1016/j.ympev.2015.01.012 (2015).
Glavinic, A. Doubly Uniparental Inheritance (DUI) of mitochondrial DNA in Neotrigonia margaritacea (Bivalvia: Palaeoheterodonta) and its evolution in Bivalvia in Systematics, Phylogeny, Phylogeography and Reproduction of Neotrigonia (Bivalvia: Palaeoheterodonta) 97–133 (Flinders University, School of Biology, Faculty of Science and Engineering, 2010).
Walker, J. M., Bogan, A. E., Garo, K., Soliman, G. N. & Hoeh, W. R. Hermaphroditism in the Iridinidae (Bivalvia: Etherioidea). J. Mollus. Stud. 72, 216–217, doi:10.1093/mollus/eyi072 (2006).
Callil, C. T. & Mansur, M. C. D. Gametogênese e dinâmica da reprodução de Anodontites trapesialis (Lamarck) (Unionoida, Mycetopodidae) no lago Baía do Poço, planície de inundação do rio Cuiabá, Mato Grosso, Brasil. Rev. Bras. Zool. 24, 825–840, doi:10.1590/S0101-81752007000300033 (2007).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–W37, doi:10.1093/nar/gkr367 (2011).
Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217, doi:10.1006/jmbi.2000.4042 (2000).
Armougom, F. et al. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res 34, W604–W608, doi:10.1093/nar/gkl092 (2006).
Di Tommaso, P. et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39, W13–W17, doi:10.1093/nar/gkr245 (2011).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101, 301–320, doi:10.1038/hdy.2008.62 (2008).
Boore, J. L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals in Comparative Genomics, 1 st edition, vol. 1 (ed. Sankoff, D. & Nadeau, J. H.) 133–147, doi:10.1007/978-94-011-4309-7_13 (Kluwer Academic, 2000).
Hoeh, W. R., Stewart, D. T., Saavedra, C., Sutherland, B. W. & Zouros, E. Phylogenetic evidence for role-reversals of gender-associated mitochondrial DNA in Mytilus (Bivalvia: Mytilidae). Mol. Biol. Evol. 14, 959–967, doi:10.1093/oxfordjournals.molbev.a025839 (1997).
Brown, S. V., Hosking, P., Li, J. & Williams, N. ATP synthase is responsible for maintaining mitochondrial membrane potential in bloodstream form Trypanosoma brucei. Eukaryot. Cell 5, 45–53, doi:10.1128/EC.5.1.45-53.2006 (2006).
Guerra, D., Ghiselli, F., Milani, L., Breton, S. & Passamonti, M. Early replication dynamics of sex-linked mitochondrial DNAs in the doubly uniparental inheritance species Ruditapes philippinarum (Bivalvia Veneridae). Heredity 116, 324–332, doi:10.1038/hdy.2015.105 (2016).
Breton, S., Doucet Beaupré, H., Stewart, D. T., Hoeh, W. R. & Blier, P. U. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23, 465–474, doi:10.1016/j.tig.2007.05.011 (2007).
Perlman, S. J., Hodson, C. N., Hamilton, P. T., Opit, G. P. & Gowen, B. E. Maternal transmission, sex ratio distortion, and mitochondria. P. Natl. Acad. Sci. USA 112, 10162–10168, doi:10.1073/pnas.1421391112 (2015).
Breton, S. et al. A resourceful genome: updating the functional repertoire and evolutionary role of animal mitochondrial DNAs. Trends Genet. 30, 555–564, doi:10.1016/j.tig.2014.09.002 (2014).
Laslett, D. & Canbäck, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175, doi:10.1093/bioinformatics/btm573 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res 25, 955–964 (1997).
Jühling, F. et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res 40, 2833–2845, doi:10.1093/nar/gkr1131 (2012).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319, doi:10.1016/j.ympev.2012.08.023 (2013).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580, doi:10.1093/nar/27.2.573 (1999).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410, doi:10.1016/S0022-2836(05)80360-2 (1990).
Conant, G. C. & Wolfe, K. H. GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862, doi:10.1093/bioinformatics/btm598 (2008).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739, doi:10.1093/molbev/msr121 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797, doi:10.1093/nar/gkh340 (2004).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria http://www.R-project.org/ (2014).
RStudio Team. RStudio: Integrated Development Environment for R. RStudio, Inc., Boston, MA http://www.rstudio.com/ (2015).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277, doi:10.1016/S0168-9525(00)02024-2 (2000).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res 43, D222–D226, doi:10.1093/nar/gku1221 (2015).
Marchler-Bauer, A. & Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic Acids Res 32, W327–W331, doi:10.1093/nar/gkh454 (2004).
Alva, V., Nam, S. Z., Söding, J. & Lupas, A. N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res 44, W410–W415, doi:10.1093/nar/gkw348 (2016).
Plazzi, F., Ribani, A. & Passamonti, M. The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics 14, 409, doi:10.1186/1471-2164-14-409 (2013).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402, doi:10.1093/nar/25.17.3389 (1997).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780, doi:10.1093/molbev/mst010 (2013).
Misof, B. & Misof, K. A Monte Carlo Approach Successfully Identifies Randomness in Multiple Sequence Alignments: A More Objective Means of Data Exclusion. Syst. Biol. 58, 21–34, doi:10.1093/sysbio/syp006 (2009).
Criscuolo, A. & Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 10, 210, doi:10.1186/1471-2148-10-210 (2010).
Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 17, 540–552, doi:10.1093/oxfordjournals.molbev.a026334 (2000).
Dress, A. W. et al. Noisy: Identification of problematic columns in multiple sequence alignments. Algorithm. Mol. Biol. 3, 7, doi:10.1186/1748-7188-3-7 (2008).
Lanfear, R., Calcott, B., Ho, S. Y. W. & Guindon, S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 29, 1695–1701, doi:10.1093/molbev/mss020 (2012).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313, doi:10.1093/bioinformatics/btu033 (2014).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 61, 539–542, doi:10.1093/sysbio/sys029 (2012).
Jordan, G. E. & Piel, W. H. PhyloWidget: web-based visualizations for the tree of life. Bioinformatics 24, 1641–1642, doi:10.1093/bioinformatics/btn235 (2008).
Huson, D. H. & Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. 61, 1061–1067, doi:10.1093/sysbio/sys062 (2012).
Soroka, M. & Burzynski, A. Complete female mitochondrial genome of Anodonta anatina (Mollusca: Unionidae): confirmation of a novel protein-coding gene (F ORF). Mitochondr. DNA 26, 267–269, doi:10.3109/19401736.2013.823176 (2015).
Wang, G., Cao, X. & Li, J. Complete F-type mitochondrial genome of Chinese freshwater mussel Lamprotula tortuosa. Mitochondr. DNA 24, 513–515, doi:10.3109/19401736.2013.770508 (2013).
Huang, X.-C. et al. The Complete Maternally and Paternally Inherited Mitochondrial Genomes of the Endangered Freshwater Mussel Solenaia carinatus (Bivalvia: Unionidae) and Implications for Unionidae Taxonomy. PLoS ONE 8, e84352, doi:10.1371/journal.pone.0084352 (2013).
Huang, X.-C., Zhou, C.-H., Ouyang, S. & Wu, X.-P. The complete F-type mitochondrial genome of threatened Chinese freshwater mussel Solenaia oleivora (Bivalvia: Unionidae: Gonideinae). Mitochondr. DNA 26, 263–264, doi:10.3109/19401736.2013.823190 (2015).
Serb, J. M. & Lydeard, C. Complete mtDNA Sequence of the North American Freshwater Mussel, Lampsilis ornata (Unionidae): An Examination of the Evolution and Phylogenetic Utility of Mitochondrial Genome Organization in Bivalvia (Mollusca). Mol. Biol. Evol. 20, 1854–1866, doi:10.1093/molbev/msg218 (2003).
Soroka, M. & Burzynski, A. Complete sequences of maternally inherited mitochondrial genomes in mussels Unio pictorum (Bivalvia, Unionidae). J. Appl. Genet 51, 469–476, doi:10.1007/BF03208876 (2010).
Acknowledgements
We would like to thank Ana Glavinic and Kirsten Benkendorff for providing us Neotrigonia margaritacea samples. This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (grant no. RGPIN/435656–2013 to S.B. and grant no. RGPIN/217175–2013 to D.T.S.).
Author information
Authors and Affiliations
Contributions
D.G. and S.B. conceived the study. S.B. and D.G. annotated the mt genomes. D.G. and F.P. performed all the analyses. All authors participated in writing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession codes: The annotated mtDNA sequences obtained in this study have been deposited in GenBank under the accession numbers KU873118-24.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guerra, D., Plazzi, F., Stewart, D.T. et al. Evolution of sex-dependent mtDNA transmission in freshwater mussels (Bivalvia: Unionida). Sci Rep 7, 1551 (2017). https://doi.org/10.1038/s41598-017-01708-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01708-1
This article is cited by
-
Mito-nuclear coevolution and phylogenetic artifacts: the case of bivalve mollusks
Scientific Reports (2022)
-
No evidence of DUI in the Mediterranean alien species Brachidontes pharaonis (P. Fisher, 1870) despite mitochondrial heteroplasmy
Scientific Reports (2022)
-
Boundaries and hybridization in a secondary contact zone between freshwater mussel species (Family:Unionidae)
Heredity (2021)
-
Unorthodox features in two venerid bivalves with doubly uniparental inheritance of mitochondria
Scientific Reports (2020)
-
Variability of mitochondrial ORFans hints at possible differences in the system of doubly uniparental inheritance of mitochondria among families of freshwater mussels (Bivalvia: Unionida)
BMC Evolutionary Biology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
