
Abstract
The uptake of formaldehyde (HCHO) on mineral dust affects its budget as well as particle properties, yet the process has not yet been fully investigate. Here, TiO2 and nitrate-doped TiO2 aerosols were used as proxies for mineral dust, and the uptake of HCHO was explored in a chamber under both dark and illuminated conditions. The uptake loss of HCHO on UV-illuminated aerosols is 2–9 times faster than its gaseous photolysis in our experimental system. The uptake coefficient in the range of 0.43–1.68 × 10−7 is 1–2 orders of magnitude higher than previous reports on model mineral dust particles. The reaction rate exhibits a Langmuir-Hinshelwood-type dependence on nitrate content and relative humidity, suggesting the competitive role of nitrate salts, water vapor and HCHO on the TiO2 surface. The reaction produces carbon dioxide as the main product and gaseous formic acid as an important intermediate. The hydroxyl radical produced on illuminated TiO2 primarily drives the fast oxidation of HCHO. The nitrate radical arising from the TiO2-catalyzed photoreaction of nitrate synergistically promotes the oxidation process. This study suggests a novel oxidation route for HCHO in the atmosphere, taking into account high abundance of both mineral dust and anthropogenic TiO2 aerosols.
Similar content being viewed by others


Introduction
Formaldehyde (HCHO) is the most abundant carbonyl compound in the atmosphere; its presence affects both the radical budget and secondary aerosol formation1, 2. For instance, photolysis of HCHO is an important source of HOx (=OH radical +HO2 radical), which plays a critical role in the photochemical reactions in the troposphere3. Meanwhile, oxidation of HCHO produces formic acid, which increases the acidity of aerosol particles via gas-particle partitioning, promoting the formation of secondary organic aerosols (SOAs)4, 5. The oxidation of long-chain organic compounds, such as isoprene, is the major chemical source of HCHO in the low troposphere, while the oxidation of methane is the major chemical source in remote areas6,7,8. Although the chemical source dominates, direct emissions from combustion and vegetation are not negligible9. In addition, emission of HCHO from processes in the snow pack greatly affects the emission budget in snow-covered areas10. The sink of HCHO includes deposition, oxidative decay by hydroxyl radicals (·OH) and most importantly its photolysis in the gas phase6,7,8. The atmospheric lifetime of HCHO against its chemical decay is approximately one day, while its photolysis lifetime around noon is as short as 1–2 hours. Despite the detailed knowledge on the HCHO budget, a substantial discrepancy between field observations and model outputs has been found, both for box and multi-dimensional models6,7,8. The discrepancy suggests, in fact, an incomplete understanding of the budget of HCHO, such as the heterogeneous sink of HCHO on aerosols in regions of heavy aerosol loading8, 11,12,13,14.
The uptake and following oxidation or oligomerization of HCHO on aerosols has been explored in the laboratory as a potential sink of HCHO11,12,13. The uptake coefficient of HCHO on various aerosol components, including sulfuric acid, organic matters, and mineral dust, has been measured11, 12, 15,16,17. Generally, previous evaluations based on the laboratory measurements suggest that the heterogeneous sink of HCHO can be neglected11, 12, 16, 18. However, we argue here that previous studies have not fully explored the photo-enhancement effect of mineral dust (e.g., TiO2), nor possible synergistic effects of other aerosol constituents, such as nitrate, under environmentally relevant conditions11, 12, 16, 18, 19.
TiO2 is a minor but still an important component of mineral dust with a mass fraction in the range from 0.1–10 wt.%20. The injection of dust into the atmosphere is ~1,000–3,000 Tg per year21, 22. In addition, TiO2 is now increasingly used as an environmental self-cleaning and depolluting coating on the outer layer of buildings, glasses, airport roofs, etc. due to its photocatalytic properties23,24,25. Bang and Murr identified a TiO2 particle with a size of about 50 nm in atmospheric particulate matter in El Paso, Texas, with its source suspected to be anthropogenic26. Many laboratory studies have been conducted to explore the photocatalytic features of TiO2 in heterogeneous reactions. The results show that UV-illuminated pure TiO2 or mineral dust greatly enhanced the uptake of O3, NOx (NO + NO2), SO2, volatile organic compounds (VOCs), compared to that under dark conditions27,28,29,30,31,32,33,34,35,36. Under a typical flux of 103–104 photons·cm−2 · nm−1 between 300–390 nm in the solar radiation37, excited TiO2 is theoretically able to generate photoactive species that drive rapid redox reactions of HCHO on mineral dust aerosols and photocatalytic anthropogenic surfaces.
Here, the reactions of HCHO on TiO2 and nitrate-doped TiO2 aerosols were investigated in a 400 L environmental aerosol chamber under 8–80% RH. The uptake of HCHO and formation of products were monitored as the chamber was illuminated under environment-relevant UV radiation in the region of 300–420 nm.
Results
Below we demonstrate that the oxidation of HCHO is significantly photo-enhanced, with a synergistic effect from the photoreaction of nitrate on the aerosol. The uptake loss of HCHO in our experimental system is even faster compared to its gas-phase photolysis, inferring a novel sink of HCHO of potential atmospheric significance.
Fast Photochemical Oxidation of HCHO
Firstly, experiments without aerosol introduction were conducted. HCHO was introduced into the chamber under dark conditions without the presence of aerosol particles. After a ~1 hr equilibrium period, the concentration of HCHO was mostly stable and no clear trend was observed (see Supplementary Fig. S1a); when the UV lamps were turned on, a slow decrease of HCHO concentration was observed (see Supplementary Fig. S1b). The time series of HCHO concentration in experiments with TiO2 or KNO3-TiO2 aerosols under both dark and illuminated conditions are shown in Fig. 1a. No decay of HCHO concentration was observed in dark conditions, even when TiO2 or KNO3-TiO2 aerosol was present. A linear decay of HCHO concentration was observed only under illuminated conditions (R2 = 0.99). The fitted zero-order reaction rate constant of HCHO on TiO2 and KNO3-TiO2 aerosols was 7.8 × 1017 molecule m−3 min−1 and 10.8 × 1017 molecule m−3 min−1, respectively, after the gas-phase photolysis of HCHO was subtracted. This observation suggests that no significant uptake of HCHO on TiO2 or KNO3-TiO2 aerosols exists under dark conditions, which agrees with previous reports of the minor importance of heterogeneous uptake of HCHO on aerosols in dark conditions11, 12, 16, 18. In contrast, this observation also suggests a significant but variable photo-enhanced uptake on both TiO2 and KNO3-TiO2 aerosols, which is even faster than HCHO photolysis loss in the gas phase (1.2 × 1017 molecule m−3 min−1, as can be deduced from Supplementary Fig. S1b). The enhanced uptake rate is ~6.5 and 9.0 times higher on TiO2 and KNO3-TiO2 aerosols, respectively, relative to the gaseous photolysis of HCHO. The photocatalytic effect of TiO2 and the synergistic effect of co-existed nitrate is expected and will be discussed below.

Kinetic curves of reactant and products. (a) Concentrations of formaldehyde. (b) Concentrations of CO2. (c) Concentrations of formic acid. (d) Concentrations of total carbon. “TiO2 dark” is TiO2 aerosol in dark condition, “TiO2 Illumination” is TiO2 aerosol in illuminated condition, “4 wt.% KNO3-TiO2 illumination” is 4 wt.% KNO3 coated TiO2 aerosol in illuminated condition. The RH inside the Chamber is 8%.
Similar experiments with other aerosols, including SiO2, KNO3-SiO2, (NH4)2SO4-TiO2 and K2SO4-TiO2 particles, were also carried out to examine the photocatalytic effect of TiO2 and the synergistic effect of the co-existed nitrate. Table 1 shows the rate constants on these particles with photolysis rate constant being subtracted. SiO2 aerosols presented slight uptake of HCHO, due to some reactive sites existing on the surface of SiO2. The comparison between SiO2 and TiO2 aerosols highlighted the photocatalytic effect of TiO2. KNO3-SiO2 particle showed nearly no uptake of HCHO, maybe because of the blocking effect of KNO3. This implies that the synergistic effect of nitrate on HCHO photo-degradation could only occur when TiO2 and nitrate anions are present simultaneously. The photodegradation of HCHO on 4 wt.% K2SO4-TiO2 aerosols was observed to be smaller than on pure TiO2 particles, suggesting that K2SO4 might block reactive sites on TiO2 aerosols, and hinder the uptake of HCHO.
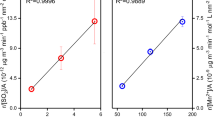
(NH4)2SO4-TiO2 particles have a much stronger inhibiting effect compared to K2SO4-TiO2 on the photodegradation of HCHO. The significant inhibiting effect of (NH4)2SO4 might be ascribed to NH4 +. Previous studies found that the photogenerated holes, generated by the excitation of TiO2, can react with adsorbed H2O to produce hydroxyl radicals38, which would oxidize negative trivalent N to higher valence nitrogen species, such as NO2 − or NO3 − 39, 40. In order to verify this photocatalytic oxidation of NH4 +, XPS N1s spectra of 20 wt.% (NH4)2SO4-TiO2 aerosol sample before and after photoreaction with HCHO were measured (Fig. 2). The XPS N1s spectrum of the fresh (NH4)2SO4-TiO2 sample presented a clear peak around 401.69 eV, which could be defined as NH4 + 41. After the (NH4)2SO4-TiO2 particles reacted with HCHO under ultraviolet irradiation, the NH4 + peak was reduced and there were two new peaks emerging at 399.75 eV defined as N− 42, 43 and 406.97 eV defined as NO3 − 44. The results directly illustrate that the ammonium ion could be oxidized to NO3 − by the photogenerated holes so as to compete with HCHO photooxidation, thereby having an inhibiting effect on the photodegradation of HCHO. This can also be confirmed by the lower decay rate of HCHO on NH4NO3-TiO2 composite aerosols compared to that on KNO3-TiO2 aerosols (Table 1).

XPS N1s spectra of 20 wt.% (NH4)2SO4-TiO2. (a) Before photoreaction with formaldehyde. (b) After the photoreaction with formaldehyde.
Figure 1b–d shows the time series of CO2, gaseous formic acid, and the total carbon in the illumination experiments along with the photo-enhanced decay of HCHO. The total carbon involving HCHO, CO2 and formic acid was near stable throughout the experiments, implying that the mass balance of carbon was almost closed, and that CO2 and formic acid were the major products of HCHO photochemical oxidation on aerosols in our experiments (Fig. 1d). CO2 appeared to be the main product, as the formation of CO2 was much faster than that of formic acid and increased with the photo-enhanced decay of HCHO throughout the illumination experiments (Fig. 1b). The formation rate of CO2 in experiments with KNO3-TiO2 or TiO2 aerosols is 1.1 × 1018 molecule · m−3 · min−1 and 7.8 × 1017 molecule · m−3·min−1, respectively. These rates were nearly the same as the HCHO decay rates in these experiments. Gaseous formic acid appeared to be an intermediate, as the formation of formic acid on TiO2 aerosols reached its maximum value at about 40 min illumination time and then decreased afterwards, while the formation of formic acid on KNO3-TiO2 aerosols showed a more complex trend (Fig. 1c). The production of formic acid in the ‘KNO3-TiO2’ experiment was lower than that in the ‘TiO2’ experiment, in contrast to the production of CO2. This infers a fast formic acid to CO2 conversion in the presence of nitrate in TiO2 aerosols, which may be attributed to the synergistic effect of co-existed nitrate.
Overall, both the TiO2 particle and the co-existed nitrate promoted the photochemical oxidation of HCHO at a rate dramatically faster than just the HCHO gas phase photolysis in our experiments, with CO2 as the main product and formic acid as an intermediate. TiO2 is the necessary substance for enhanced HCHO oxidation on the surface, and the synergistic effect relies on the co-existence of nitrate and TiO2.
Rate Dependency on Nitrate Content and RH
As TiO2 was coated by secondary nitrate at various RH in ambient conditions, the dependency of the heterogeneous oxidation rate of HCHO on nitrate content and relative humidity was key to extrapolating any laboratory results to a real environment. KNO3-TiO2 and NH4NO3-TiO2 aerosols with different nitrate mass fractions were used in the illumination experiments; the dependence of the oxidation rate constant of HCHO on nitrate content is shown in Fig. 3. The reaction rate constants first increased with nitrate mass fraction at low nitrate loadings, and then decreased with nitrate fraction if nitrate loadings were higher than 12 wt.% for KNO3-TiO2 or 4 wt.% for NH4NO3-TiO2. As the nitrate content is above 50 wt.% for KNO3-TiO2 and 8 wt.% for NH4NO3-TiO2, the nitrate-TiO2 aerosols demonstrate no stronger reactivity than pure TiO2 aerosol. Possible reasons will be discussed below. Similarly, a saddle-shaped dependence of oxidation rate constants of HCHO on RH was observed in the experiments with KNO3-TiO2 and TiO2 aerosols (Fig. 4). The reaction rate constant first increased with RH and then decreased with an optimal value at 30% and 50% for KNO3-TiO2 and TiO2 aerosols, respectively. In the low RH region, KNO3-TiO2 aerosols presented higher reactivity towards HCHO than pure TiO2 aerosols, while at a high RH beyond 50%, the reaction rate constants on the two type of aerosols were similar. On NH4NO3-TiO2 aerosols, the oxidation rate constant of HCHO deceases with RH in the range of 8–80%, and is lower than that on pure TiO2 aerosols at RH beyond 20%.

Variations of photoreaction rate constants with nitrate content. Formaldehyde photoreaction rate constants on KNO3-TiO2 or NH4NO3-TiO2 aerosol as a function of composited nitrate content under 8% RH.

Variations of photoreaction rate constants with relative humidity. Formaldehyde photoreaction rate constants on TiO2, 4 wt.% KNO3-TiO2 or 4 wt.% NH4NO3-TiO2 aerosol as a function of relative humidity.
The dependence of the oxidation rate constants of HCHO on nitrate contents and RH shed some light on the underlying mechanism of HCHO oxidation on the aerosols. Previous studies have found that the photogenerated holes on excited TiO2 would react with surface H2O to produce hydroxyl radicals38,39,40, which is the oxidant for HCHO oxidation. Therefore, H2O adsorbed on TiO2 surface is essential for this HCHO oxidation reaction. Similarly, NO3 radicals are produced from complexed nitrate on illuminated TiO2 aerosol, and the NO3 radical also behaves as an oxidant, providing an additional driving force for HCHO oxidation on the aerosol (see below). Therefore, the competitive adsorptions of H2O with HCHO, nitrate with HCHO ought to be important in determining the oxidation rate of HCHO in this study. On the one hand, the competitive adsorption of nitrate with HCHO on photogenerated holes of TiO2 particles might explain the nitrate content dependence (Fig. 3). As the nitrate is accumulatively coated on TiO2 aerosols, it blocks the reactive sites and reduces the reaction probability of HCHO with oxidants of both origins. The different dependent patterns of KNO3-TiO2 aerosols and NH4NO3-TiO2 aerosols in Fig. 3 might be due to another pair of competitive reactions, i.e., reactions of oxidants with HCHO and NH4 +, which is also reflected by previous analyses of Fig. 2. On the other hand, the competitive adsorption of H2O with HCHO might explain the RH dependence for the case of KNO3-TiO2 and TiO2 aerosols (Fig. 4). An optimal amount of H2O on the aerosols facilitates the decomposition of H2O by photogenerated holes on excited TiO2, thus the formation of ·OH oxidants. Conversely, excessive adsorbed water, like excessive nitrate, affects the reaction by inhibiting the contact between HCHO and the oxidants45, or changing the oxide coordinated nitrate to water solvated nitrate, thus reducing the reaction rate46. The different hydroscopic properties of nitrate salts make the dependence more complex. For example, the KNO3 in KNO3-TiO2 particles helps to adsorb moisture, leading to a greater number of OH radicals being generated at low surface water content, but more active sites being blocked by the nitrate solution layer on aerosols under high surface water content, relative to pure TiO2 aerosols. The Langmuir-Hinshelwood type of dependence of the reaction rate on RH is not observed in the ‘NH4NO3-TiO2’ experiment. NH4NO3 salt has low deliquescence point and adsorbed water may make NH4 + easily dissolved. This would make the reaction between NH4 + and OH radical (as discussed before) easier than the reaction between HCHO and OH radicals. So the adsorbed water showed negative effect on HCHO photodegradation over NH4NO3-TiO2. Another possible reason for no peak of RH is the inability to control RH below 8% in our environmental chamber.
Discussion
Based on the data above and some references, a mechanism for the oxidation of HCHO on TiO2 particles under illuminated conditions can be proposed. As shown in Supplementary Eqs S1–S13 11, 18, 47,48,49,50. When TiO2 was irradiated with light energy higher than its bandgap (that is, with wavelengths lower than 387 nm), electron-hole pairs generated on the surface could react with H2O and O2, producing reactive oxygen species (ROS) (such as ·OH, HO2· and H2O2) (see Supplementary Eqs S1–S5), as suggested elsewhere18, 49. The ROS, especially OH radicals, played important roles in oxidizing HCHO. Xu et al. detected H2COO as the transitional product (shown in Supplementary Eqs S6–S9)11. HCHO can also directly react with HO2· to generate HCOOH48 (see Supplementary Eq. S10). The formic acid/formate could further be oxidized to CO2 by OH (see Supplementary Eqs S11–S13). In addition, the photogenerated holes could also react directly with adsorbed HCHO and produce CO2 47. Competition reactions between HCHO and other molecules have not been shown.
As illustrated in Fig. 1, a substantial enhancement in HCHO uptake on aerosol was observed in the presence of nitrate salts. This might be attributed to the electron-trapping effect of nitrate anion, which limited charge-carrier recombination at the TiO2 surface and thus increased the availability of holes for oxidative process of adsorbed HCHO51. NO3 − could also react directly with photogenerated holes to generated NO3 radicals (Eqs 1 and 2), according to previous investigations of the photochemistry of illuminated nitrate-TiO2 particles52. NO3 radicals generated on the surface could effectively oxidize HCHO, with CHO· and HNO3 as the products (Eq. 3)53. CHO· further participated in a series of reactions to finally generate CO2 (Eqs 6 and 7). Meanwhile, NO3 radicals underwent a rapid photolysis under visible light irradiation to generate NO and NO2 through Eqs (4) and (5), with Eq. (4) as the prominent pathway54, 55. This gave us an opportunity to verify the existence and effect of NO3 radicals in our experiment system. Figure 5 shows the photoreaction rate constants of HCHO on TiO2 and KNO3-TiO2 aerosols under “365 nm lamp” illumination and under both “365 nm lamp” and yellow fluorescence lamp (450–750 nm) illumination. The oxidation rate constants of HCHO on TiO2 aerosol were comparable under these two illumination conditions. The probable reason for this is that TiO2 is not sensitive to visible light. However, the rate constant on KNO3-TiO2 aerosol under illumination of both lamps was lower than that under only the “365 nm lamp”, indicating a reduced oxidation rate due to NO3 radical photolysis by visible light. The observation of the rate decrease of HCHO due to NO3 photolysis provides experimental evidence for the existence of NO3 for the first time.

Photoreaction rate constants with light illumination. Formaldehyde photoreaction rate constants on TiO2 or 4 wt.% KNO3-TiO2 aerosol in the condition of 8% RH under light illumination of “365 nm” or “365 nm + yellow fluorescence”, respectively.
Taking into account that both OH and NO3 radicals are coming from photogenerated holes reacted with adsorbed H2O and nitrate, respectively, there would exist a competition of the two processes. The mass effect of TiO2 on the reaction rate of HCHO is depicted in Supplementary Fig. S2. The reaction rate kept increasing with added mass of TiO2 (without any nitrate doping) below 60 mg (see Supplementary Fig. S2(a)). After that, the reaction rate levelled off and reached a plateau. Below this threshold of 60 mg, the almost linear trend is due to the increase of photogenerated holes and therefore of OH radicals (through the following reaction h+ + H2O → HO· + H+), responsible for the oxidation of HCHO. At higher TiO2 loadings, the plateau is due to radical self-reactions and various recombination processes. These observations nicely fit standard trends in TiO2 based photocatalysis, even at the lower mass investigated here, clearly indicating that it controls the degradation of HCHO. While photocatalysis dominates the degradation of HCHO, the exact pathway in presence of nitrate depends on its actual mass, as shown in Supplementary Fig. S2(b). At low TiO2 masses, the nitrate anions react with available holes (NO3 − + h+ vb → NO3·) and reduce the amount of generated OH radicals, overall this leads to a reduction of the observed reaction rate (nitrate radicals being less reactive than OH radicals). At different loadings, however the situation is quite different as a clear enhancement can be observed when significant amount of NO3 can be produced. Thus nitrate presents positive effect on the photodegradation of HCHO for KNO3-75 mgTiO2 sample. Therefore, there is a competition of photogenerated holes to produce OH and NO3 radicals, which are both responsible for the oxidation of HCHO, with OH radical as the major one. This is another evidence that NO3 radicals are produced in the system and can present its synergistic effect with appropriate amount of TiO2 loading.
Enhanced oxidative decays of HCHO on both TiO2 aerosols and nitrate-TiO2 aerosols were observed under illuminated conditions. The photocatalytic effect of TiO2 plays a key role in the enhanced oxidation of HCHO. The synergistic effect of nitrate is ascribed to the generation of NO3 radicals on excited TiO2 aerosols. Currently, it is out of reach to measure in-situ the surface concentration of nitrate radicals with state-of-the-art analytical tools. However, we have experimentally proved the presence of NO3 radicals, as discussed above. NH4 + reacts competitively with photogenerated holes on the TiO2 aerosol, and produces high valence nitrogen species. The Langmuir-Hinshelwood type of dependence of the oxidation rate of HCHO on nitrate and water content highlights the competitive adsorption and reaction of the reactants, i.e. HCHO with nitrate, HCHO with H2O, on the reactive sites of TiO2 aerosol.
The oxidation rate of HCHO on both TiO2 aerosol and nitrate-TiO2 aerosol is around one order of magnitude faster than the photolysis of HCHO in the gas phase. The uptake coefficient is calculated to be in the range of 0.43–1.68 × 10−7 under our experimental conditions (see Supplementary: Calculation of uptake coefficient and Table S1), which is ~1–2 orders of magnitude higher than previous reports11, 12, 16, implying a novel HCHO sink of potential atmospheric significance. The photocatalytic oxidation rate of HCHO might linearly depend on TiO2 mass fraction in dust aerosol in ambient conditions, which is much lower than the TiO2 mass fraction used in our experiments. Assuming a TiO2 mass fraction of 5% in mineral dust and a comparable surface density of mineral dust aerosol to our experimental value (2 × 104 μm2 cm−3), the heterogeneous oxidation decay of HCHO on dust aerosol is at least comparable to its photolysis in the gas phase in typical dust events20. Furthermore, the enhanced uptake of HCHO on low NO3 − loading TiO2 aerosol, as indicated in our study, suggests the importance of this kind of reaction in dust storm episodes when the mass percentage of NO3 − is less than 40%56. On non-dust days, the heterogeneous oxidation decay of HCHO is less important, but cannot be neglected in HCHO budget analysis. Finally, our results demonstrate the need for more experiments employing ambient dust aerosols of various atmospheric aging processes under various environmental conditions, to parameterize the oxidative decay of HCHO on mineral dust better.
Methods
Environmental Chamber
An environmental chamber was built for studying the heterogeneous reaction of HCHO with TiO2 and nitrate-doped TiO2 aerosols (see Supplementary Fig. S3). The main body of the chamber is a 400 L FEP bag (Mitsubishi, Japan, designed by Safelab company, Beijing) having a pillow shape after inflation with a size of 1.15 m (length) × 1.40 m (height) × 0.55 m (width). It is vertically hung on an aluminum frame and is covered with a light-shading black cloth.
The chamber is capable of temperature, light intensity and relative humidity control, and is equipped with contamination-free carrier gas supply and a particle introduction system. Temperature was controlled to be around 295 K in the laboratory by air conditions. One light source with a solar-mimicking UV spectrum of 320–400 nm was used, with the main wavelength of 365 nm, being called the 365 nm lamp hereafter. In some experiments, one yellow fluorescence lamp in the wavelength range from 450–750 nm was also used for comparison, being called the yellow fluorescence lamp hereafter. The output spectra of both light sources are presented in Supplementary Fig. S4. Zero air was obtained from compressed air after purification with activated carbon, filtering aerosol (Jiechi purification equipment Limited, Shanghai) and drying with porous silicon. Saturated water vapor was obtained via a water bubbler, and introduced into the chamber together with dry air at certain flow rates to control the desired relative humidity. Aerosol was introduced through the particle introduction system. This consists of a high-pressure pipe and particle holder tube. Sample powder was first placed inside the holder tube and then sprayed into the chamber by a transient high-pressure high pure nitrogen gas (99.9999%, Huayuan Gas Company, Beijing).
The chamber is also equipped with various inlets, outlets and detection instruments. The particle size distribution was measured by a Scanning Nano Particle Spectrometer (SNPS-n20, HCT, Korea, Sheath air flow: Sample air flow = 10:1). Other outflow from the chamber was first filtered prior to any further analysis to protect the instruments from contamination with particles. Reactant HCHO was generated by thermolysis of paraformaldehyde at 70 °C and was brought into the chamber by the carrier gas at a flow rate of 100 mL/min and detected via acetyl acetone spectrophotometric method using a UV-Vis spectrophotometer (T6, PERSEE, Beijing). CO2 was detected by gas chromatography (SP3429, FID detector, Molecular sieve column, Column temperature, 100 °C, Injection temperature, 100 °C) equipped with an oxidation furnace. Gaseous formic acid was first scrubbed in a porous-glass-plate absorption tube by deionized water and then detected by ion chromatography (US Dionex, ICS-900, AS-14 anion column with AG-14 protection column, ASRS-4 mm anion suppressor, ECD-1 detector) with 1.25 mM sodium borate solution as eluent.
Particulate Sample Preparation and Chemicals
In our experiments, two nitrate salts, potassium nitrate or ammonium nitrate, were complexed with TiO2 aerosols to evaluate the nitrate effect on HCHO uptake and possible influences of the cations. First, a series of nitrate solutions of different concentrations was prepared. Then, 300 mg of TiO2 particles were mixed in 1 mL nitrate solution to obtain a mash. The mash was dried in an oven and then ground carefully to ensure a uniform composite of particles. Since ammonium nitrate decomposes at 110 °C, the drying temperature was set to 90 °C. The nitrate content in the composite aerosol samples ranged from 0.2–80 wt.%. For the purpose of comparison, 4 wt.% (NH4)2SO4-TiO2, 20 wt.% (NH4)2SO4-TiO2, 4 wt.% K2SO4-TiO2 and 4 wt.% KNO3-SiO2 samples were also prepared. The blank TiO2 or SiO2 samples were solved in pure water with the same procedure as mentioned above.
The chemicals used are commercial products. Titanium dioxide is Degussa P25 from Germany (50 nm, 54 m2/g), SiO2, KNO3, NH4NO3, (NH4)2SO4 and K2SO4 are all analytical reagents (>96%).
Experiments of Formaldehyde with Particles
Before each experiment, the chamber was cleaned by pumping and inflating with zero air for two cycles. The deflated chamber was inflated by 200 L zero air, following by the introduction of HCHO, and then the chamber was filled up with zero air to about 400 L. The concentrations of HCHO, CO and CO2 were detected as a function of time. It took about 60 min for the HCHO concentration to be stable. After the HCHO concentration became stable, particulate samples were introduced, and the particle size distribution of aerosol inside the chamber was also monitored as a function of time. The concentration of HCHO decreased once particles were introduced, and it took another 30 min to reach stable plateau again. Meanwhile, large aerosol particles tended to settle down on the inner wall of the chamber within 30 min of the introduction; thereafter a relative stable particle number density of about 4000 cm−3 in a mono-modal distribution with a peak particle size around 100 nm was observed (see Supplementary Fig. S5). Compared to an ambient dust aerosol observation in Beijing, our laboratory-generated TiO2 aerosol contains a comparable number of particles beyond 40 nm, but fewer particles smaller than 40 nm57. The surface density of aerosols during the whole experiment stage was stable at around 2 × 104 μm2 cm−3, which was comparable to that observed in dust events in Beijing, 1–2 orders of magnitude higher than observations in the urban area, and up to three orders of magnitude higher than observations in the background area8, 57. After the concentrations of both HCHO and aerosol became stable, the lamps were turned on and the concentrations of HCHO, CO, CO2 and gaseous formic acid were monitored.
The blank experiment without aerosol (Supplementary Fig. S1) shows that the photolysis frequency of HCHO in the gas phase fitted to be around 2 × 10−5 s−1. The photolysis frequency was roughly comparable to the “standard” photolysis frequency of HCHO during typical tropical summer conditions on the ground (solar elevation angle θ = 0°)58, suggesting that the light intensity in our experiment was environmentally relevant. In addition, the solar light-mimicking spectrum of our light source in the 300–365 nm region overlaps with the HCHO spectrum in the UV region. Overall, the characteristics of the light source enabling the photochemical reaction are environmentally relevant.
References
Sumner, A. L. et al. A study of formaldehyde chemistry above a forest canopy. J. Geophys. Res. Atmos 106, 24387–24405 (2001).
Possanzini, M., Palo, V., Petricca, M., Fratarcangeli, R. & Brocco, D. Measurements of lower carbonyls in Rome ambient air. Atmos. Environ. 30, 3757–3764 (1996).
Mahajan, A. S. et al. DOAS observations of formaldehyde and its impact on the HOx balance in the tropical Atlantic marine boundary layer. J. Atmos. Chem. 66, 167–178 (2010).
Jang, M. & Kamens, R. M. A predictive model for adsorptive gas partitioning of SOCs on fine atmospheric inorganic dust particles. Environ. Sci. Technol. 33, 1825–1831 (1999).
Jang, M. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science 298, 814–817 (2002).
Vigouroux, C. et al. Ground-based FTIR and MAX-DOAS observations of formaldehyde at Reunion Island and comparisons with satellite and model data. Atmos. Chem. Phys. 9, 9523–9544 (2009).
Luecken, D. J., Hutzell, W. T., Strum, M. L. & Pouliot, G. A. Regional sources of atmospheric formaldehyde and acetaldehyde, and implications for atmospheric modeling. Atmos. Environ. 47, 447–490 (2012).
Li, X. et al. Modeling of HCHO and CHOCHO at a semi-rural site in southern China during the PRIDE-PRD2006 campaign. Atmos. Chem. Phys. 14, 12291–12305 (2014).
Li, Y., Shao, M., Lu, S., Chang, C. & Dasgupta, P. Variations and sources of ambient formaldehyde for the 2008 Beijing Olympic games. Atmos. Environ. 44, 2632–2639 (2010).
Preunkert, S. et al. The atmospheric HCHO budget at Dumont d’Urville (East Antarctica): Contribution of photochemical gas-phase production versus snow emissions. J. Geophys. Res. Atmos 118, 13319–13337 (2013).
Xu, B. Y., Zhu, T., Tang, X. Y. & Shang, J. Heterogeneous reaction of formaldehyde on the surface of TiO2 particles. Sci. China. Chem 53, 2644–2651 (2010).
Xu, B. Y., Shang, J., Zhu, T. & Tang, X. Y. Heterogeneous reaction of formaldehyde on the surface of γ-Al2O3 particles. Sci. China Chem 45, 3569–3575 (2011).
Toda, K. et al. Formaldehyde content of atmospheric aerosol. Environ. Sci. Technol. 48, 6636–6643 (2014).
Ji, Y. M., Wang, H. H., Li, G. Y. & An, T. Ch. Theoretical investigation on the role on mineral dust aerosol in atmospheric reaction: A case of the heterogeneous reaction of formaldehyde with NO2 onto SiO2 dust surface. Atmos. Environ. 103, 207–214 (2015).
Jayne, J. T., Worsnop, D. R., Kolb, C. E., Swartz, E. & Davidovits, P. Uptake of gas-phase formaldehyde by aqueous acid surfaces. J. Phys. Chem. 100, 8015–8022 (1996).
Sassine, M., Burel, L., D’Anna, B. & George, C. Kinetics of the tropospheric formaldehyde loss onto mineral dust and urban surfaces. Atmos. Environ. 44, 5468–5475 (2010).
Li, Z., Schwier, A. N., Sareen, N. & McNeill, V. F. Reactive processing of formaldehyde and acetaldehyde in aqueous aerosol mimics: surface tension depression and secondary organic products. Atmos. Chem. Phys. 11, 11617–11629 (2011).
Linsebigler, A. L., Lu, G. & Yates, J. T. Jr. Photocatalysis on TiO2 surfaces: principles, mechanisms, and selected results. Chem. Rev. 95, 735–758 (1995).
Baergen, A. M. & Donaldson, D. J. Photochemical renoxification of nitric acid on real urban grime. Environ. Sci. Technol. 47, 815–820 (2013).
Hanisch, F. & Crowley, J. N. Ozone decomposition on Saharan dust: an experimental investigation. Atmos. Chem. Phys. 3, 119–130 (2003).
Dentener, F. J., Carmichael, G. R., Zhang, Y., Lelieveld, J. & Crutzen, P. J. Role of mineral aerosol as a reactive surface in the global troposphere. J. Geophys. Res: Atmos 101, 22869–22889 (1996).
Ji, Y. M. et al. Can Silica Particles Reduce Air Pollution by Facilitating the Reactions of Aliphatic Aldehyde and NO2. J Phys. Chem. A. 119, 11376–11383 (2015).
Dutta, P. K. et al. Interaction of carbon monoxide with anatase surfaces at high temperatures: optimization of a carbon monoxide sensor. J. Phys. Chem. B. 103, 4412–4422 (1999).
Zhang, X. T. et al. Self-cleaning particle coating with antireflection properties. Chem. Mater. 17, 696–700 (2005).
Kasanen, J., Suvanto, M. & Pakkanen, T. T. Self-cleaning, titanium dioxide based, multilayer coating fabricated on polymer and glass surfaces. J. Appl. Polym. Sci. 111, 2597–2606 (2009).
Bang, J. J. & Murr, L. E. Atmospheric nanoparticles: preliminary studies and potential respiratory health risks for emerging nanotechnologies. J. Mater. Sci. Lett 21, 361–366 (2002).
Beaumont, S. K., Gustafsson, R. J. & Lambert, R. M. Heterogeneous photochemistry relevant to the troposphere: H2O2 production during the photochemical reduction of NO2 to HONO on UV-illuminated TiO2 surfaces. Chem. Phys. Chem. 10, 331–333 (2009).
Monge, M. E., D’Anna, B. & George, C. Nitrogen dioxide removal and nitrous acid formation on titanium oxide surfaces–an air quality remediation process? Phys. Chem. Chem. Phys. 12, 8991–8998 (2010).
Monge, M. E. et al. Ozone formation from illuminated titanium dioxide surfaces. J. Am. Chem. Soc. 132, 8234–8235 (2010).
Pradhan, M. et al. Heterogeneous uptake of gaseous hydrogen peroxide by Gobi and Saharan dust aerosols: a potential missing sink for H2O2 in the troposphere. Atmos. Chem. Phys. 10, 7127–7136 (2010).
Chen, H., Navea, J. G., Young, M. A. & Grassian, V. H. Heterogeneous photochemistry of trace atmospheric gases with components of mineral dust aerosol. J. Phys. Chem. A. 115, 490–499 (2011).
Bedjanian, Y. & El Zein, A. Interaction of NO2 with TiO2 surface under UV irradiation: products study. J. Phys. Chem. A. 116, 1758–1764 (2012).
Styler, S. A. & Donaldson, D. J. Heterogeneous photochemistry of oxalic acid on Mauritanian sand and Icelandic volcanic ash. Environ. Sci. Technol. 46, 8756–8763 (2012).
Bedjanian, Y., Romanias, M. N. & El Zein, A. Interaction of OH radicals with Arizona test dust: uptake and products. J. Phys. Chem. A. 117, 393–400 (2013).
El Zein, A., Romanias, M. N. & Bedjanian, Y. Kinetics and products of heterogeneous reaction of HONO with Fe2O3 and Arizona Test Dust. Environ. Sci. Technol. 47, 6325–6331 (2013).
Styler, S. A., Myers, A. L. & Donaldson, D. J. Heterogeneous photooxidation of fluorotelomer alcohols: a new source of aerosol-phase perfluorinated carboxylic acids. Environ. Sci. Technol. 47, 6358–6367 (2013).
George, C. et al. Emerging areas in atmospheric photochemistry. Top. Curr. Chem. 339, 1–53 (2014).
Wang, H. H., Ji, Y. M., Chen, J. Y., Li, G. Y. & An, T. Ch. Theoretical investigation on the adsorption configuration and ·OH-initiated photocatalytic degradation mechanism of typical atmospheric VOCs styrene onto (TiO2)n clusters. Sci. Rep 5, 15059 (2015).
Bonsen, E. M., Schroeter, S., Jacobs, H. & Broekaert, J. A. C. Photocatalytic degradation of ammonia with TiO2 as photocatalyst in the laboratory and under the use of solar radiation. Chemosphere. 35, 1431–1445 (1997).
Kim, S. & Choi, W. Kinetics and mechanisms of photocatalytic degradation of (CH3)nNH4-n +(0 ≤ n ≤ 4) in TiO2 suspension: The role of OH radicals. Environ. Sci. Technol. 36, 2019–2025 (2002).
Vanini, A. S., Audouard, J. P. & Marcus, P. The role of nitrogen in the passivity of austenitic stainless steels. Corros. Sci. 36, 1825–1834 (1994).
Baltrusaitis, J., Jayaweera, P. M. & Grassian, V. H. XPS Study of nitrogen dioxide adsorption on metal oxide particle surfaces under different environmental conditions. Phys. Chem. Chem. Phys. 11, 8295–8305 (2009).
Nanayakkara, C. E., Jayaweera, P. M., Rubasinghege, G., Baltrusaitis, J. & Grassian, V. H. Surface photochemistry of adsorbed nitrate: the Role of adsorbed water in the formation of reduced nitrogen species on α-Fe2O3 particle surfaces. J. Phys. Chem. A. 118, 158–166 (2014).
Rousset, J. L. Practical surface analysis, volume 2-ion and neutral spectroscopy. Appl. Catal. A: Gen 98, N19–N22 (1993).
Cwiertny, D. M., Young, M. A. & Grassian, V. H. Chemistry and photochemistry of mineral dust aerosol. Annu. Rev. Phys. Chem. 59, 27–51 (2008).
Miller, T. M. & Grassian, V. H. Heterogeneous chemistry of NO2 on mineral oxide particles: Spectroscopic evidence for oxide-coordinated and water-solvated surface nitrate. Geophys. Res. Lett. 25, 3835–3838 (1998).
Su, F. et al. Spectroscopic and kinetic studies of a new metastable species in the photooxidation of gaseous formaldehyde. Chem. Phys. Lett. 65, 221–225 (1979).
Neeb, P., Sauer, F., Horie, O. & Moortgat, G. K. Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour. Atmos. Environ. 31, 1417–1423 (1997).
Gaya, U. I. & Abdullah, A. H. Heterogeneous photocatalytic degradation of organic contaminants over titanium dioxide: a review of fundamentals, progress and problems. J. Photoch. Photobio. C: Photochem. Rev 9, 1–12 (2008).
Chen, H. H., Nanayakkara, C. E. & Grassian, V. H. Titanium dioxide photocatalysis in atmospheric chemistry. Chem. Rev. 112, 5919–5948 (2012).
Haynes, W. M. CRC Handbook of Chemistry and Physics; 91st Edition (CRC Press: Boca Raton, 2010).
Ndour, M., Conchon, P., D’Anna, B. & Ka, O. Photochemistry of mineral dust surface as a potential atmospheric renoxification process. Geophys. Res. Lett. 36, L05816 (2009).
Cantrell, C. A. & Johnston, H. S. et al. Kinetic study of the nitrate free radical (NO3)-formaldehyde reaction and its possible role in nighttime tropospheric chemistry. J. Phys. Chem. 89, 139–146 (1985).
Magnotta, F. & Johnston, H. S. Photodissociation quantum yields for the NO3 free radical. Geophys. Res. Lett. 7, 769–772 (1980).
Gankanda, A. & Grassian, V. H. Nitrate photochemistry on laboratory proxies of mineral dust aerosol: Wavelength dependence and action spectra. J. Phys. Chem. C. 118, 29117–29125 (2014).
Sun, Y. L. et al. Chemical composition of dust storms in Beijing and implications for the mixing of mineral aerosol with pollution aerosol on the pathway. J. Geophys. Res. Atmos 110, 1064–1067 (2005).
Hu, W. et al. Insights into a dust event transported through Beijing in spring 2012: Morphology, chemical composition and impact on surface aerosols. Sci.Total Environ. 565, 287–298 (2016).
Finlayson-Pitts, B. J. & Pitts, J. N. Jr. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications. (Academic Press, 2000).
Acknowledgements
The authors are grateful to the financial support provided by National Natural Science Foundation of China (21577003, 21277004, 41421064), the national key Research and Development Program of China (2016YFC0202200), the Marie Curie International Research Staff Exchange project AMIS (295132) and the Special Funds for State Key Joint Laboratory of Environmental Simulation and Pollution Control, Peking University, China.
Author information
Authors and Affiliations
Contributions
J. Shang, W. Xu and C. George designed and supervised experiments, analysis and preparation of the manuscript. Ch. Ye and T. Zhu participated in the preparations of manuscript, Figures and Tables.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shang, J., Xu, W.W., Ye, C. et al. Synergistic effect of nitrate-doped TiO2 aerosols on the fast photochemical oxidation of formaldehyde. Sci Rep 7, 1161 (2017). https://doi.org/10.1038/s41598-017-01396-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01396-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
