
Abstract
COX-1/PGE2 is an important protective mediator in ulcerative colitis (UC). β-arrestin1 (β-arr1), which acts as a scaffold protein, is involved in PGE2-mediated signaling pathways. However, the interaction between PGE2 and β-arr1 in maintaining mucosal barrier integrity remains unexplored. In this study, we demonstrated that COX-1 and PGE2 were significantly decreased, and EP4 mRNA was downregulated in both UC patients and mice during the injury phase. PGE2 treatment was found to alleviate mucosal injury and induce EP4 expression during dextran sulfate sodium (DSS)-induced colitis in wild-type (WT) mice. Following DSS-induced injury, β-arr1 deficient mice showed increased signs of colitis compared to β-arr1 WT mice, and the expression of PI3K and p-Akt were remarkably downregulated in β-arr1 deficient mice. In parallel, HCT116 cells transfected with β-arr1 siRNA were examined in the presence or absence of PGE2 in vitro. PGE2 treatment in the β-arr1 WT/KO DSS model and β-arr1 siRNA transfection of HCT116 cells confirmed that PGE2 upregulated β-arr1 in vivo and in vitro. Collectively, our results indicate that COX-1/PGE2/EP4 upregulates the β-arr1 mediated Akt signaling pathway to provide mucosal protection in colitis. Thus, these findings provide support for the future development and clinical application of COX-1/PGE2 in UC.
Similar content being viewed by others

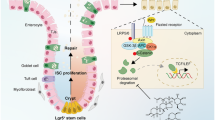

Introduction
Inflammatory bowel disease (IBD), a multifactorial disease perpetuated by a dysregulated immune response, includes ulcerative colitis and Crohn’s disease. The etiology of IBD involves a complex interaction of genetic predisposition, environmental triggers, microbial factors and immune responses1. Ulcerative colitis is a chronic, idiopathic, and inflammatory disease of the rectal and colonic mucosa. The incidence and prevalence of UC are increasing worldwide2. No innovative treatment has been developed, although progress has been made in the overall management of the disease. Conventional nonsteroidal anti-inflammatory drugs (NSAIDs) are occasionally involved in the development of de novo colitis, although they are more frequently implicated in the aggravation of pre-existing intestinal diseases3. This is most obviously demonstrated in patients with inflammatory bowel disease, who frequently require anti-inflammatory analgesics due to peripheral arthritis, sacroiliitis, ankylosing spondylitis and osteoporosis-related fractures4. The administration of NSAIDs is one of the major risk factors in triggering the clinical relapse of IBD5. Cyclooxygenase (COX) has two isoforms, COX-1 and COX-26. COX-1 is expressed constitutively in epithelial cells in the crypt with the exception of the villi and it has been proposed to maintain the cell integrity of the gastrointestinal tract7. In the normal intestine, COX-2 is not expressed in appreciable amounts in epithelial cells but is expressed in colonic adenomas, carcinomas and IBD8,9,10. Cyclooxygenase catalyzes the production of prostanoids, which are a group of eicosanoids consisting of four types of prostaglandins (PGs) and thromboxanes: PGE2, PGD2, PGF2α, PGI2 and TXA2. The PGE2 receptor is EP11. EP consists of four subtypes: EP1, EP2, EP3 and EP4. All of these subtypes respond to a naturally occurring agonist, PGE2, but differ in their actions and responses to various analogues12. The EP4 receptor has been shown to stimulate cAMP/protein kinase A (PKA) signaling through the activation of Gsα and adenylate cyclase in the regulation of growth and proliferation13. Furthermore, EP4 also elicits the activation of phosphoinositide3-kinase (PI3K) via the β-arrestin (β-arr) pathway14.
β-arr1 and β-arr2, two of the four members of the arrestin family, are scaffolding proteins that modulate GPCR signaling pathways and signal transduction. β-arrs are involved in several types of diseases, including Parkinson’s disease, multiple sclerosis, cardiovascular conditions, and colitis by serving as a scaffold protein in various signaling pathways, including ERK1/2, Wnt/β-catenin and c-Src15,16,17. Previously, we discovered that β-arr2 contributed to the development of experimental colitis and mucosal repair in the process of UC18, 19. In addition to their role in receptor downregulation, β-arrs also act as scaffold proteins to accelerate signal transduction in the EP4 pathway. The physical association between EP4 and β-arr1 was demonstrated using bioluminescence resonance energy transfer20. β-arrs also have been reported to play a role in cell proliferation, apoptosis and differentiation via several signaling pathways21. Additional studies have shown that β-arr1 was involved in the activation of the PI3K/Akt pathway and affects apoptosis or cell survival22.
In the present study, we found that PGE2 and the PGE2 receptor, EP4, were downregulated in both ulcerative colitis patients and mice with DSS-induced experimental colitis. In addition, treatment with PGE2 alleviated mucosal injury in colitis. We also found that expression of β-arr1 decreased in both human and mice colitis. Targeted deletion of β-arr1 increased signs of colitis compared to β-arr1 wildtype (WT) mice following DSS-induced injury by activating PI3K/Akt signaling. Moreover, PGE2 contributed to the preservation of epithelial proliferation of experimental colitis by mainly enhancing EP4/β-arr1/p-Akt signaling. Taken together, these findings demonstrated the pivotal role of β-arr1 in the integrity of the PGE2-mediated colonic epithelial barrier and provided sufficient scientific evidence to establish EP4/β-arr1/p-Akt signaling as a new therapeutic target of UC.
Results
Expression of COX, prostaglandins and prostaglandin receptors in colitis
To examine the role of COX in UC, colon mucosal specimens from colitis patients and healthy volunteers were analyzed. The expression pattern of COX-1 mRNA was markedly suppressed in UC patients, whereas COX-2 was significantly increased in the patients’ mucosa in the injury phase. Western blotting revealed that colonic specimens obtained from patients with UC displayed reduced COX-1 protein expression, whereas the expression of COX-2 protein was increased (Fig. 1A). Similar results were found in animal experiments, in which COX-1 mRNA levels and protein levels decreased in DSS-treated mice. When DSS was withdrawn, the expression of COX-1 nearly returned to the levels observed in untreated controls (Fig. 1B). To further investigate the expression of prostaglandins in colitis, colon mucosal specimens from colitis patients and healthy volunteers were analyzed. As shown in Fig. 1C, the concentrations of PGE2, PGD2, PGF2α, and PGI2 were measured in biopsies of rectal mucosa using an ELISA kit. The PGE2 concentration in the control group was 207.27 ± 6.8 pg/mg of protein, while PGE2 concentrations of the patients’ mucosa in the injury phase revealed decreased concentrations (127.38 ± 4.9 pg/mg of protein), and these differences were significant (p < 0.05). In contrast, the PGE2 concentration (213.78 ± 8.7 pg/mg of protein) of the patients’ mucosa in the repaired phase showed significant differences compared with the in the injury phase. No significant changes in the expression of other prostaglandins were observed (Fig. 1C). To further investigate the expression of PGE receptors, EP1, EP2, EP3 and EP4 mRNA levels were analyzed using real-time PCR in human normal colon tissue and colitis colon tissue (injury and repaired phases). There were no apparent differences in the levels of EP1, EP2, and EP3 mRNA between the normal colon tissue and the colitis colon tissue (injury and repaired phases), but a significant difference in the level of EP4 was observed (Fig. 1D). Furthermore, EP4 mRNA revealed a decrease in colitis during the injury phase. To further study the role of prostaglandins and receptors in UC, DSS was used to induce colitis in mice, and similar results were found in animal experiments (Fig. 1E,F).

Expression of COX, prostaglandins and prostaglandin receptors in colitis. (A) COX-1 and COX-2 expression in colon tissues were determined using RT-PCR and Western blotting in the non-UC group (human normal colon tissue), injury group (colitis colon tissue in the injury phase) and repair group (colitis colon tissue in the repair phase); β-actin was used as a loading control (n = 6 per group). *P < 0.05, # P < 0.05 non-UC versus injury. (B) COX-1 and COX-2 expression in mouse colon tissue was determined using RT-PCR and Western blotting analyses in the vehicle group, injury group and repair group. (n = 4 per group) *P < 0.05, # P < 0.05 vehicle versus injury (C) Concentrations of mucosal prostaglandins PGE2, PGD2, PGF2α and PGI2 in of three pairs of representative human specimens. Values are expressed as the mean ± SD (n = 6 per group). *P < 0.05, non-UC versus injury phase. # P < 0.05 injury phase versus repair phase (D) mRNA expression of EP1, EP2, EP3 and EP4 in the colonic mucosa were evaluated using real-time PCR in the indicated group. Values are expressed as the mean ± SD (n = 6 per group). *P < 0.05, non-UC versus injury phase. # P < 0.05 injury phase versus repair phase (E) Concentrations of mucosal prostaglandins PGE2, PGD2, PGF2α and PGI2 in the mouse vehicle group, injury group and repair group; The values are expressed as the mean ± SD (n = 4 in each group). *P < 0.05 versus vehicle mice, # P < 0.05 injury group versus repair group. (F) EP1, EP2, EP3 and EP4 mRNA expression in the colonic mucosa of mice were evaluated by real-time PCR in the indicated group. The values are expressed as the mean ± SD (n = 4 in each group). *P < 0.05 versus vehicle mice, # P < 0.05 injury group versus repair group.
PGE2/EP4 alleviates mucosal injury in colitis
To examine whether PGE2/EP4 signaling contributed to injury in colitis, we employed colitis mouse models. Mice were randomly divided into the control group, UC model group, indomethacin group (DSS treatment administered with indomethacin) and PGE2 group (DSS treatment administered with PGE2). The macroscopic finding of the mice treated with 5% DSS and sacrificed on day 7 showed that the intestines of the mice had edema and hemorrhagic redness all throughout the colon and cecum (Supplementary Fig. S1). In response to DSS treatment, mice displayed features of colitis characterized by a loss in body weight, loose stools and occult blood in the feces. Colonic mucosa suffered from crypt destruction, goblet cell loss and inflammatory cell infiltration. The indomethacin group developed worsened symptoms while the PGE2 group exhibited clinical symptoms, such as diarrhea and an attenuation of hemoccult. Consequently, PGE2 significantly suppressed the histological injury and the disease activity index scores (Fig. 2A). Immunostaining showed that EP4 and PCNA were increased in mice in the PGE2 group (Fig. 2B). Quantification of PCNA-positive cells demonstrated that the proliferation index was higher in the PGE2 group. Furthermore, epithelial apoptosis, which was stained by TUNEL, was significantly reduced in the PGE2 group (Fig. 2C). Expectedly, double labeling of cytokeratin and PCNA, PAS and PCNA revealed that PGE2 increased the regeneration of epithelial cells and goblet cells. However, double staining of cytokeratin and TUNEL, and PAS and TUNEL demonstrated that indomethacin attenuated the regeneration of epithelial cells and goblet cells (Fig. 2D). Taken together, these findings further confirm that PGE2 alleviates mucosal injury in colitis.

PGE2/EP4 alleviates mucosal injury in colitis. (A) The disease activity index was determined at the indicated time points as described in the Methods. Histological damage after DSS treatment for 7 days was scored after H&E staining as described in the Methods. *P < 0.05 compared with control group mice. # P < 0.05 versus vehicle group (n = 4 in each group). (B) Representative photomicrographs of H&E staining, TUNEL staining (brown, ×200), immunostaining of EP4 (brown, ×200) and PCNA (brown, ×200) in colonic sections of WT littermates in the indicated group. (n = 4 in each group). (C) The apoptotic index was measured by quantifying TUNEL signals in 100 random fields per section. The percentage of PCNA-positive cells is represented graphically. Values are expressed as the mean ± SD. n = 6 in each group, * P < 0.05 versus control mice, # P < 0.05 versus vehicle group. (D) Double stain for PAS and PCNA and PAS and TUNEL in four groups. PAS for goblet cells is pink (×200). Immunostaining of PCNA and TUNEL are shown in brown. Double immunofluorescence stain for cytokeratin and PCNA, cytokeratin and TUNEL in the indicated group (×400).Nuclei are stained with DAPI in blue. Localization of PCNA and TUNEL are visualized in green and cytokeratin is stained in red. The merging positive signals of PCNA or TUNEL and cytokeratin are visualized in yellow. (n = 4 in each group).
β-arr1 is downregulated in active colitis
Immunohistochemistry studies were performed to confirm β-arr1 expression in normal colon tissues and colitis tissues. Moreover, β-arr1 expression was decreased in colonic specimens in UC patients (Fig. 3A). At both the mRNA and protein levels, β-arr1 expression was examined in normal colon tissues and colitis colon tissues. These results showed that β-arr1 expression was significantly downregulated in colitis colon tissue compared with normal colon tissues (Fig. 3C). To further study the role of β-arr1 in UC, DSS was used to induce colitis in mice. Interestingly, both β-arr1 mRNA and protein levels decreased in the DSS-induced intestinal mucosa (Fig. 3D). Simultaneously, immunostaining results of β-arr1 showed little positive signal in the ulceration area compared with the vehicle. However, there were more signals in the non-ulcerative area compared with the ulcerative area (Fig. 3B). These results suggest that β-arr1is downregulated in colitis in both humans and mice.

β-arr1 is downregulated in active colitis. (A) Immunostaining of β-arr1 in human colonic mucosa in the healthy volunteer group and UC group (brown, ×200). (n = 4 in each group). (B) Immunostaining of β-arr1 in mouse colonic mucosa in the control group, and ulcer sections and non-ulcer sections in the DSS group (brown, ×200). (n = 6 in each group). (C) The expression of intestinal mucosal β-arr1 mRNA and protein was evaluated in human colonic mucosa in the healthy volunteer group and UC group using real-time PCR and western blotting. Values are expressed as the mean ± SD. (n = 6 in each group). *P < 0.01 versus control group. (D) The expression of intestinal mucosal β-arr1 mRNA and protein was evaluated in the mouse colonic mucosa in the control group and DSS group using real-time PCR and western blotting. Values are expressed as the mean ± SD. (n = 6 in each group). *P < 0.01 versus vehicle mice. DSS: dextran sulfate sodium; Non-UC: healthy volunteers; UC: ulcerative colitis.
Targeted deletion of β-arr1 exacerbates DSS-induced colitis in mice
The above observations prompted us to use β-arr1-gene-deficient models to further investigate the role of β-arr1 in colitis. To assess the extent and severity of colonic pathological changes, we measured the length of colons from mice subjected and not subjected to colitis. Although DSS-induced colitis resulted in colon shortening in both β-arr1 WT and KO mice, the colon was still significantly longer in β-arr1 WT mice than in the KO mice (Supplementary Fig. S2). Disease activity index scores were significantly lower in β-arr1 WT mice compared with KO mice (Fig. 4D). Fewer and smaller colonic ulcers were also detected in β-arr1 WT mice compared with KO mice after DSS treatment (Supplementary Fig. S2). Consistent with the overall phenotypic differences in ulcer status, clinical signs and total colon morphology, H&E-stained microscopic sections of the colon revealed marked differences between β-arr1 WT mice and KO mice. In addition, histological analysis revealed substantially less epithelial damage and disruption of crypt architecture in β-arr1 WT mice (Fig. 4A,E). Simultaneously, immunostaining showed highly positive TUNEL signals in β-arr1 KO mice after DSS treatment (Fig. 4B,F). Cytokeratin and PAS immunostaining indicated that targeted deletion of β-arr1 exacerbated the regeneration of epithelial cells and goblet cells (Supplementary Fig. S2). Immunostaining studies also showed that the expression of PCNA decreased during colitis periods, and β-arr1 KO mice exhibited significantly decreased levels compared with WT mice (Fig. 4C,G). These results reveal that the critical role of β-arr1 in colitis is associated with epithelial cell apoptosis.

Targeted deletion of β-arr1 exacerbates DSS induced colitis in mice. (A) Representative photomicrographs of H&E staining in colonic sections of β-arr1 WT and KO mice in the control group and the ulcer section and non-ulcer section of the DSS group (×200, n = 4 per group). (B) TUNEL staining revealed apoptotic induction in intestinal epithelial cells of β-arr1 WT and KO mice (brown, ×200, n = 4 per group). (C) Immunohistochemical staining for PCNA in colonic sections of β-arr1 WT and KO mice in the control group and the ulcer section and non-ulcer section of the DSS group. (brown, ×200, n = 4 per group). (D) The disease activity index of mice treated with or without DSS of WT and β-arr1 KO mice was measured at the indicated time points. *P < 0.01 compared with the WT mice (n = 4 per group). (E) Histological damage in colonic tissues obtained from the mice treated with DSS or without DSS of β-arr1 WT and KO mice was scored after H&E staining. *P < 0.05 compared with the control mice, # P < 0.05, β-arr1 WT mice versus KO mice. n = 6 per group. (F) Apoptotic index was measured by quantifying TUNEL signals in 100 random fields per section. Values are expressed as the mean ± SD. n = 6 in each group, *P < 0.05 compared with the control mice, # P < 0.05, β-arr1 WT mice versus KO mice. (G) The percentage of PCNA-positive cells is represented graphically. Values are expressed as the mean ± SD. n = 6 in each group, *P < 0.05 compared with the control mice, # P < 0.05, β-arr1 WT mice versus KO mice.
β-arr1 activates PI3K/Akt signaling in colitis
Previous reports have shown that β-arr1 is an important signaling scaffold that facilitates the activation of numerous effector pathways regulating cellular proliferation, differentiation, and apoptosis, such as the MAPK and PI3K signaling pathways. The PI3K-Akt pathway plays an important role in many diseases15. To confirm whether PI3K/Akt is targeted downstream by β-arr1 in colitis, we used β-arr1 WT and β-arr1 KO mice to induce colitis via DSS. Western blots showed that PI3K and Akt phosphorylation were remarkably downregulated in DSS-induced colitis in β-arr1 WT and KO mice compared with the control group. However, this decrease was markedly exacerbated in β-arr1 KO mice compared with WT mice (Fig. 5A,B). Similar results were also observed in immunohistochemical staining (Fig. 5C,D). These results suggest that β-arr1 activates PI3K/Akt signaling in colitis.

β-arr1 actives PI3K/Akt signaling in colitis. (A) Western blot of PI3K, p-Akt and Akt in colonic mucosa of β-arr1 WT and KO mice with or without DSS treatment at the indicated time points. All western blot images are representative images from five independent experiments, and β-actin was used as a loading control. (B) Quantitative analysis of p-Akt/Akt as measured by densitometry scanning of Western blots. Values are expressed as the mean ± SD of five independent experiments, *P < 0.05 versus vehicle group, # P < 0.05 versus β-arr1 KO mice after 5% DSS treatment. (C) Immunohistochemical staining for PI3K in colonic sections of β-arr1 WT and KO mice in the control group, and the ulcer section and non-ulcer section in the DSS group (brown, ×200, n = 4 per group). (D) Immunohistochemical staining for p-Akt in colonic sections of β-arr1 WT and KO mice in the control group, and the ulcer section and non-ulcer section in the DSS group (brown, ×200, n = 4 per group).
PGE2/EP4 upregulats β-arr1 mediated Akt signaling in vivo and in vitro
Current views indicate that PGE2 promotes epithelial proliferation after mucosal injury, and we also know that targeted deletion of β-arr1 exacerbates DSS-induced colitis in mice. Based on this knowledge, we investigated whether PGE2 could improve symptoms in mice with established colitis after DSS treatment. A western blot and immunohistochemical staining showed that Akt phosphorylation was remarkably upregulated in β-arr1 WT mice with established colitis after PGE2 treatment. In contrast, there was no change in the expression of p-Akt in β-arr1 KO mice after PGE2 treatment (Fig. 6A–C). However, in both β-arr1 WT and KO mice, the expression of EP4 significantly increased during colitis periods after PGE2 treatment as detected by Western blot (Fig. 6A).

PGE2/EP4 upregulates β-arr1 mediated Akt signaling in vivo and in vitro. (A) Western blotting of EP4, p-Akt and Akt in colonic mucosa of β-arr1 WT and KO mice treated with DSS with or without PGE2 at the indicated time points. All western blot images are representative images from three independent experiments, and β-actin was used as a loading control. (B) Quantitative analysis of p-Akt/Akt ratio as measured by densitometry scanning of Western blots. Values are expressed as the mean ± SD of three separate experiments, *P < 0.05 versus vehicle. (C) Immunohistochemical staining for p-Akt staining in colonic sections of β-arr1 WT and KO littermates under DSS with or without PGE2 treatment at the indicated time points (brown, ×200, n = 4 per group). (D) Expression of p-Akt in HCT116 cells transfected with control or β-arr1 siRNA after PGE2 treatment. Nuclei were stained with DAPI in blue. Localization of p-Akt was visualized in green. (E) HCT116 cells were stimulated with or without PGE2 (20 μm/ml) for 8 h in the presence of control siRNA or β-arr1 specific siRNA. The lysates were subjected to Western blotting analysis for β-arr1, p-Akt and Akt. All western blot images are representative images from three independent experiments, and β-actin was used as a loading control. (F) Quantitative analysis of the p-Akt/Akt ratio as measured by densitometry scanning of Western blotting. Values are expressed as the mean ± SD of three experiments, *P < 0.05 versus control siRNA group.
To further confirm these results, the effects of β-arr1 siRNA on EP4 signaling were also examined. Transfection of β-arr1 siRNA reduced the level of β-arr1 protein by 63% compared with that of non-targeting control siRNA. Similarly, EP4 expression was unaffected by β-arr1 downregulation. After β-arr1 siRNA treatment, the expression of p-Akt was downregulated as shown by cell immunofluorescence and Western Blotting, indicating that silencing of β-arr1 gene expression markedly attenuated the activation of p-Akt and suppressed cell proliferation (Fig. 6D–F). In summary, these data demonstrates that PGE2/EP4 upregulates β-arr1-mediated Akt signaling in vivo and in vitro.
Discussion
PGE2 is present throughout the gastrointestinal tract and is known to elicit a variety of actions in the gut23. However, the role of PGE2 in the onset or alleviation of mucosal injury is not clearly known. In this study, we demonstrated that PGE2 treatment alleviated mucosal injury in colitis. We also revealed the role of EP4 receptors and PGE2-induced β-arr1/p-Akt signaling in maintaining epithelial barrier integrity.
PGs can protect the gastrointestinal (GI) tract24, 25. PGE2, the most abundant gastrointestinal prostaglandin, regulates many of the normal homeostatic functions of the gut, including motility and secretion25. The versatility of PGE2 is attributed to its differential local production and its selectivity for EP receptor subtypes (EP1, EP2, EP3 and EP4). Prostaglandins are synthesized through COX-1 and COX-2 and exhibit both pro- and anti-inflammatory effects. COX-1 maintains the integrity of the GI mucosa under physiological conditions, and COX-2 has been implicated in invasive events, such as inflammation6, 7, 10. In the present study, DSS treatment resulted in a loss of COX-1 expression in the crypt epithelium and an increase in COX-2 expression10, 26. A subsequent study showed that administration of a selective COX-2 inhibitor has no effect on either apoptosis or crypt survival; similarly, radiation of COX-1 deficient mice results in increased apoptosis and decreased crypt survival compared to WT mice27. These findings show that PGE2 produced through COX-1 plays an important role in epithelial homeostasis. The role of PGE2 in UC is still controversial. Previous study showed that COX-2 derived PGE2 played the pro-inflammatory role in UC28, 29. However, ours and other plenty of evidence showed that PGE2 release was suppressed in irritable bowel syndrome, radiation-induced small intestine injury, diverticular disease and so on30,31,32, and PGE2 protected the gastrointestinal tract through maintaining the mucosal integrity, limiting the inflammatory response and promoting the wound healing25, 33, 34. What is interesting, the local PGE2 level within tissues is difficult to measure due to the short half-life of PGE2 and the level of intestinal PGE2 changed dynamicly in mice experimental colitis35,36,37. Moreover, the reported contradictory mucosal PGE2 data may be due to the difference in the position of biopsy specimen, scored degree of mucosal injury and the phase of colitis in different researches28, 29, 38. In our study, we focused on the COX-1 derived-PGE2 in the epithelial cells and the biopsy samples were taken from the ulcer edge including the ulcerative tissue and the uninvolved tissue, while other researchers collected tissue samples from the most active inflammatory part of the colon to test the PGE2 as a surrogate marker28. But the cellular origins of these PGs and the relative contributions of COX-1 and COX-2 to their production have not been fully investigated and the precise underlying mechanism of these differences will be explored in our future study. Next, we found that PGE2 concentration decreased in the injury phase of human colitis, which was the only prostaglandin to show a significant change among the analyzed prostaglandins. In addition, there was a significant change in the EP4 mRNA levels among the analyzed EP receptors. Current views indicate that the PGE2 pathway has a specific physiological significance in humans, which was highlighted by the finding that the PTGER4 gene, which encodes the PGE2 receptor EP4, demonstrates a strong genetic association with both forms of IBD39. Zhang et al.40 discovered that inhibiting 15-hydroxyprostaglandin dehydrogenase, an enzyme that physiologically oxidizes PGE2 to prevent it from binding to prostaglandin receptors, led to improvements in hematopoietic stem cell transplants and colitis recovery during injury. The PGE2 level has dynamic change process in mice experimental colitis. In a study by Tessner, TG et al.26, PGE2 levels in cecal tissue were diminished after 5 days of DSS treatment, and PGE2 levels were dramatically increased during recovery from DSS-induced injury. Similar results were found in other reasearches35,36,37. Kandil et al.41 suggested that low endogenous levels of PGE2 predisposed relapses in a rat model of enterocolitis, and supported the anti-inflammatory activity of PGE2. A subsequent study showed that mPGES-1 deficient mice (PGES-1 is the terminal synthase for inducible PGE2 formation) had a greater susceptibility to acute DSS-induced injury, indicating a specific protective role for PGE2 42. Taken together, these published studies corroborate our finding that COX-1/PGE2/EP4 contributes to the maintenance of mucosal integrity to maintain GI homeostasis.
The EP4 receptor is one of four known G-protein-coupled receptor subtypes for prostaglandin E2, which was a Gsα-coupled and adenylyl cyclase stimulating receptor43. EP4 signaling plays a variety of roles through cAMP effectors11. However, abundant evidence from studies using pharmacological approaches and genetically modified mice suggests that EP4 can also be coupled to Giα, phosphatidylinositol3-kinase (PI3K), β-arr or β-catenin44. In our study, we found that EP4 receptor expression was downregulated in colitis, but increased after the treatment of PGE2 in DSS-induced experimental colitis, which contributed to the protection of PGE2. Our results showed that EP4 upregulated β-arr1, which protected the colon tissue. It has been shown that EP4 receptors play an important role in suppressing colitis and downregulating immune responses in DSS-induced colitis. Dey I et al.30 demonstrated that the role of EP4 signaling was different between early onset (pro-inflammatory on colonic epithelial cells) and late progressive stages of colitis (anti-inflammatory on immune cells in the lamina propria). Another study found that the EP4 agonist was effective in the treatment of IBD by enhancing epithelium survival and regeneration after DSS challenge45. However, it has been reported that the PGE2-EP4 system is the major PG system in preventing mucosal intestinal inflammation caused by DSS46. In our study, we demonstrated that human EP4 receptor expression was decreased in colitis. These results support the concept that EP4 contributes to ulcer healing.
β-arr1, which serves as a multifunctional adaptor and scaffold that mediates GPCRs, is expressed ubiquitously in mammalian tissues and mediates many signaling functions15. Moreover, it serves as a mediator for various E3 ubiquitin ligases and a transducer of numerous cellular functions, such as membrane-, cytosolic- and nuclear-associated signaling to regulate cellular responses to external stimuli47, 48. In addition to β-arr1 mediation of receptor internalization and degradation, previous studies have established that β-arr1 is involved in cell proliferation, apoptosis and differentiation by mediating PI3K/AKT signaling in response to a wide range of stimuli in different cell types49. It is now evident that β-arr1 is a critical cell signaling regulator in many inflammatory diseases, including sepsis, arthritis, autoimmune encephalomyelitis, and inflammatory bowel diseases50,51,52,53. However, the role of β-arr1 in intestinal epithelial cell apoptosis in colitis remains controversial. A recent study demonstrated that β-arr1 deficiency protected mice from experimental colitis53, while another study showed that β-arr1 in the non-hematopoietic compartment (likely epithelial cells) is protective during colitis54. The most recent studies have shown that β-arr1 exerts a protective effect in epithelial cells and stem cells55, 56. In our study, β-arr1 was found to be decreased in colonic specimens obtained from UC patients and in a mouse colitis model as determined by immunostaining and Western blotting. These findings further confirm that β-arr1 is involved in human and mice colitis and has a protective role. In our study, we found that deletion of β-arr1 exacerbated DSS-induced colitis in mice. Furthermore, PGE2 protection was only observed in β-arr1 WT mice. These findings demonstrated that β-arr1 plays a crucial role in the signaling process of cell proliferation.
Among the signaling pathways, PI3K/AKT signaling has been proposed to induce proliferative signals in IEC57,58,59. Previous reports have indicated that PI3K-mediated generation of PI-3, 4, 5-triphosphate recruits Akt to activate proliferation and survival signaling59. To investigate the involvement of PI3K signaling in colitis, activation of PI3K, p-Akt and Akt were assessed by western blot analysis. Immunohistochemical staining of PI3K and p-Akt found that β-arr1 activated PI3K/Akt signaling in colitis. To investigate the involvement of β-arr1-mediated PI3K signaling during PGE2-induced differentiation, activation of EP4, p-Akt and Akt were assessed by western blot analysis both in vivo and in vitro. We demonstrated that β-arr1 interacted with Akt, which provides a strong basis for PGE2/EP4 up-regulation of β-arr1 mediated Akt signaling. In conclusion, the present study revealed that PGE2 treatment helps to maintain colonic epithelial barrier integrity, and recovers normal expression and distribution of proteins by COX-1/PGE2/EP4 up-regulation of β-arr1/p-Akt signaling. Thus, these results suggest that PGE2 has a favorable therapeutic effect on DSS-induced ulcerative colitis, supporting its future development and clinical application in inflammatory bowel disease.
Materials and Methods
Ethics
All methods described in this study were performed in accordance with the approved guidelines by Research Ethics Committee of The Third Affiliated Hospital of Sun Yat-Sen University.
Tissue samples
Frozen specimens from patients with UC and normal colon tissue samples were obtained from the Endoscopy Center of The Third Affiliated Hospital of Sun Yat-Sen University according to the ethical and legal standards. Our study was approved by the Research Ethics Committee of The Third Affiliated Hospital of Sun Yat-Sen University. Written informed consent was obtained from every patient prior to his/her inclusion in the study.
Mice and treatment
All mice used in the animal experiments were approved by the Institutional Animal Care and Use Committee at the Sun Yat-Sen University. The experimental protocols in the present study, including all surgical procedures and animal usages, were performed according to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (NIH) and approved by the Animal Care and Use Committee of Sun Yat-Sen University. The original β-arr1 heterozygous mice in the C57BL/6 background were obtained from Dr. Robert J. Lefkowitz (Duke University Medical Center, Durham, NC, USA) as a generous gift. The mice were housed in micro-isolator cages containing wood shavings (3–6 mice per cage) and kept in a temperature-controlled (23 ± 5 °C) and humidity-controlled (50% ± 15%) environment with a 12:12 h light–dark cycle. Eight- to ten-week-old mice were used for all experiments. For colitis induction, mice were exposed to 5% DSS (MP Biomedical, LLC, Solon, OH) in their drinking water. Control mice were allowed to drink water without DDS concurrently. For indomethacin induction, mice were allowed to drink water with indomethacin (Sigma, St. Louis, MO, USA) at a dose of 4 mg/kg/day, which was administered to the animals during the entire experimental period. For PGE2 induction, mice received PGE2 (Cayman Chemical, Ann Arbor, MI) at a dose of 200 μg/150 μl/20 g body weight during DSS treatment.
Determination of Disease Activity Index
Disease activity index scores were determined daily during the experiment, as previously described60. The extent of colitis, body weight, stool consistency and occult blood in the stool were monitored daily. Body weight was scored as follows: no weight loss was scored as 0; loss of 1–5% weight was registered as 1; loss of 6–10% weight was registered as 2; loss of 11–20% weight was registered as 3; and loss of greater than 20% weight was registered as 4. Stool consistency was scored as follows: 0 was scored for well-formed stool pellets, 2 was scored for pasty and semi-formed stools that did not adhere to the anus and 4 was scored for watery diarrhea that adhered to the anus. Bleeding, which was analyzed by the Hemoccult fecal occult blood test, was scored as follows: 0 was assigned for no blood, 2 was assigned for positive hemoccult, and 4 was assigned for gross bleeding. All of the scores were blindly confirmed by three independent individuals.
Analysis of histology
Tissue sections (4 μm) of the colon were subjected to hematoxylin-eosin (H&E) for histological analysis. Histological scores were determined blindly based on previously described criteria60.
Immunohistochemical and Immunofluorescence Staining
For immunohistochemical staining, 4 mm paraffin-embedded colon sections were deparaffinized in xylene and rehydrated in graded alcohol and then treated with 3% hydrogen peroxide, followed by antigen retrieval in boiling 0.1 M citrate (pH 6.0) buffer. Next, sections were then blocked with 10% goat serum (Zymed, Carlsbad, CA, USA) for 10 min and stained with antibodies directed against β-arr1 (kindly provided by Dr. Robert J Lefkowitz), EP4, Ki-67, and PCNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and Cytokeratin-18 (Abcam, Cambridge, MA, USA), respectively. For immunofluorescence (IF) staining, the targeted protein was detected by the corresponding secondary antibody. Antibody-antigen complexes were visualized by incubation with biotin-conjugated secondary antibody and streptavidin Alexa 488 or 594 (Molecular Probes, China), and the nuclei were counterstained with 4′6-diamidino-2-phenylindole dihydrochloride (DAPI, Molecular Probes, Eugene, OR, USA). For double staining, a secondary targeted protein was detected after the initial protein-detection stage on the same slides. For cells IF staining, when the cells after the indicated treatment, they immediately fixed in 4% paraformaldehyde prior to the abovementioned procedure. The proliferative index was measured by quantifying a minimum of 20 randomly chosen fields following p-Akt (Cell Signaling Technology, Danvers, MA, USA) IF staining. The index was acquired by determining the number of p-Akt-positive cells against the total number of cells.
Apoptosis Assays
TUNEL staining was performed using the ApopTag kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Six mice from each group were examined. TUNEL-positive cells were counted in 100 randomly selected TUNEL-positive crypts as previously reported60.
Real-time Polymerase Chain Reaction (Real-time PCR)
The mice were sacrificed after different treatments. We collected a piece of distal colon approximately 12 mm in length from the same location in all mice. Colonic mucosa was isolated by careful scraping and total RNA was isolated from the colon tissue using ISOGEN (Nippon Gene, Toyama, Japan). First strand cDNA synthesis was performed from 2 μg of total RNA using the Superscript Reverse Transcriptase (Toyobo Co., Ltd., Osaka, Japan) and random primers according to the manufacturer’s instructions in a final volume of 20 μl. Real-time RT-PCR was performed using the SYBR Greenmaster mixture (Thermo Fisher Scientific, USA) on an HT7500 system (Life Technologies, USA), and the reaction mixtures were incubated at 95 °C for 10 min, followed by 45 cycles of 95 °C for 15 s and 60 °C for 32 s. Melting curve analysis was performed to validate the generation of the expected PCR products. Each sample was analyzed in triplicate. The expression levels of each RNA were normalized to that of β-actin. The relative transcription expression of the mRNAs was calculated using the 2−ΔΔCt method. The expression levels of β-arr1, EP1, EP2, EP3, EP4, COX-1, COX-2 and β-actin in human or DSS-treated mice were determined by real-time PCR using specific primers. Primer sequences are listed in Supplementary Table 1.
Enzyme-linked immunosorbent assay (ELISA)
The PGE2, PGD2, PGF2α and PGI2 concentration in biopsies of rectal mucosa were determined quantitatively using human and mice ELISA kits and performed in strict accordance with the manual of the experimental kit. Briefly, the ELISA kit (all from Cusabio Biotech Co., China) was equilibrated at room temperature for at least 30 min prior to preparation of the experimental solutions. A volume of 100 μl of standard solution was added into the reaction plate to produce the standard curve after the standard was dissolved. Next, a volume of 100 μl sample solution was added into each well, and the plate was incubated at 37 °C for 120 min. After washing the plate, 100 μl of freshly made working solution containing biotinylated antibodies was added into the wells and incubated at 37 °C for 60 min. After the second washing, 100 μl of freshly made solution containing horseradish peroxidase avidin was added and incubated at 37 °C for 60 min. The plate was then washed three times consecutively, and 100 μl of substrate solution was added and incubated in the dark at 37 °C for 15 to 30 min. Finally, stop solution was quickly added into the plate to terminate the reactions, and within 5 min. the optical density (OD) values were measured at a wavelength of 450 nm using a multifunction plate reader (Synergy 2; Bio Tek Instruments, Inc., Winooski, VT, USA). The standard curve was generated based on the measured OD values, and the levels of PGE2, PGD2, PGF2α and PGI2 were calculated from the standard curve.
Cell Culture and Transfection
The human colorectal cancer cell line HCT116 was routinely cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum, 30 Uml−1 penicillin and 30 mg ml−1 streptomycin at 37 °C under 5% CO2. Small interfering RNA (siRNA) was performed according to the manufacturer’s instructions. The cells were transfected with 20 μM β-arr1 using an RNA oligo kit (GenePharma, Shanghai, China) according to the manufacturer’s instructions, and the most effective sequence was selected to achieve a transfection efficiency of >90%. After incubation for 24 h, the transfection medium was replaced with regular culture medium before PGE2 administration.
SDS-PAGE and Western Blotting
Proteins extracted from colonic mucosa and cell lysates were analyzed by SDS-PAGE. Separated proteins were transferred to nitrocellulose membranes, then blocked at room temperature for 1.5 hours in Tris- buffered saline with 0.1% Tween20 containing 5% skim milk and probed with primary antibodies at 4 °C overnight, and then, incubated with the appropriate peroxide-conjugated secondary antibody. Finally, images were acquired using ECL detection in a darkroom. The following antibodies were used: β-arr1 (kindly provided by Dr. Robert J Lefkowitz, Duke University Medical Center), p-Akt, Akt, PI3K (all from Cell Signaling Technology), EP4, COX-1, COX-2 (Santa Cruz) and β-actin (Sigma). The blots were detected with enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and exposed to X-ray film (Fuji Hyperfilm, Tokyo, Japan).The mean pixel density of the blots was analyzed by Quantity One softerware 4.6.2 (BioRad). β-actin was used as a loading control.
Statistical Analysis
Statistical comparisons were made using SPSS20.0. All data are reported as means ± SD. To compare multiple groups, differences between groups were evaluated using parametric analysis of variance (ANOVA), followed by the Bonferroni’s post test. The differences between two groups were determined using Student’s t test. P < 0.05 was considered statistically significant.
References
Ordas, I., Eckmann, L., Talamini, M., Baumgart, D. C. & Sandborn, W. J. Ulcerative colitis. Lancet. 380, 1606–1619, doi:10.1016/S0140-6736(12)60150-0 (2012).
Molodecky, N. A. et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54, e30 (2012).
Puspok, A., Kiener, H. P. & Oberhuber, G. Clinical, endoscopic, and histologic spectrum of nonsteroidal anti-Inflammatory drug-Induced lesions in the colon. Dis Colon Rectum. 43, 685–691 (2000).
Smale, S., Natt, R. S., Orchard, T. R., Russell, A. S. & Bjarnason, I. Inflammatory bowel disease and spondylarthropathy. Arthritis Rheum. 44, 2728–2736 (2001).
Bonner, G. F., Fakhri, A. & Vennamaneni, S. R. A long-term cohort study of nonsteroidal anti-inflammatory drug use and disease activity in outpatients with inflammatory bowel disease. Inflamm Bowel Dis. 10, 751–757 (2004).
Herschman, H. R. Regulation of prostaglandin synthase-1 and prostaglandin synthase-2. Cancer Metastasis Rev. 13, 241–256 (1994).
Cohn, S. M., Schloemann, S., Tessner, T., Seibert, K. & Stenson, W. F. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest. 99, 1367–1379 (1997).
DuBois, R. N., Radhika, A., Reddy, B. S. & Entingh, A. J. Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors. Gastroenterology 110, 1259–1262 (1996).
Eberhart, C. E. et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 107, 1183–1188 (1994).
Singer, I. I. et al. Cyclooxygenase 2 induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 115, 297–306 (1998).
Narumiya, S., Sugimoto, Y. & Ushikubi, F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 79, 1193–1226 (1999).
Legler, D. F., Bruckner, M., Uetz-von, A. E. & Krause, P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 42, 198–201 (2010).
Okuyama, T. et al. Activation of prostaglandin E2-receptor EP2 and EP4 pathways induces growth inhibition in human gastric carcinoma cell lines. J Lab Clin Med. 140, 92–102 (2002).
Buchanan, F. G. et al. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA 103, 1492–1497 (2006).
Shukla, A. K., Xiao, K. & Lefkowitz, R. J. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 36, 457–469 (2011).
Wood, H. Alzheimer disease: Arrestin’ Alzheimer disease progression? beta-arrestin 2 is a potential therapeutic target. Nat Rev Neurol. 9, 60 (2013).
Ohguro, H. et al. Beta-arrestin and arrestin are recognized by autoantibodies in sera from multiple sclerosis patients. Proc Natl Acad Sci USA 90, 3241–3245 (1993).
Zeng, L. X. et al. β-Arrestin2 encourages inflammation-induced epithelial apoptosis through ER stress/PUMA in colitis. Mucosal Immunol. 8, 683–695 (2014).
Chen, T. et al. Insulin-like growth factor-1 contributes to mucosal repair by β-Arrestin2–mediated extracellular signal-related kinase signaling in experimental colitis. Am J Pathol. 185, 2441–2453 (2015).
Leduc, M. et al. Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J Pharmacol Exp Ther. 331, 297–307 (2009).
Schulte, G. & Shenoy, S. K. β-Arrestin and dishevelled coordinate biased signaling. Proc Natl Acad Sci USA 108, 19839–19840 (2011).
Hu, S. et al. Involvement of β-arrestins in cancer progression. Mol Biol Rep. 40, 1065–1071 (2013).
Kawahara, K., Hohjoh, H., Inazumi, T., Tsuchiya, S. & Sugimoto, Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta. 1851, 414–421 (2015).
Halter, F., Tarnawski, A. S., Schmassmann, A. & Peskar, B. M. Cyclooxygenase 2-implications on maintenance of gastric mucosal integrity and ulcer healing: controversial issues and perspectives. Gut. 49, 443–453 (2001).
Stenson, W. F. Prostaglandins and epithelial response to injury. Curr Opin Gastroenterol. 23, 107–110 (2007).
Tessner, T. G., Cohn, S. M., Schloemann, S. & Stenson, W. F. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology 115, 874–882 (1998).
Houchen, C. W., Stenson, W. F. & Cohn, S. M. Disruption of cyclooxygenase-1 gene results in an impaired response to radiation injury. Am J Physiol Gastrointest Liver Physiol. 279, G858–G865 (2000).
Wiercińska-Drapało, A., Flisiak, R. & Prokopowicz, D. Plasma and mucosal prostaglandin E2 as a surrogate marker of ulcerative colitis activity. Rocz Akad Med Bialymst. 46, 60–8 (2001).
Wiercińska-Drapa, O. A., Flisiak, R. & Prokopowicz, D. Effects of ulcerative colitis activity on plasma and mucosal prostaglandin E2 concentration. Prostagothlipidm 58, 159 (1999).
Dey, I., Lejeune, M. & Chadee, K. Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol. 149, 611–623 (2006).
Casellas, F. et al. Intraluminal colonic release of immunoreactive tumor necrosis factor in chronic ulcerative colitis. Clin Sci 90, 453–8 (1994).
Dai, L. et al. Inverse Expression of prostaglandin E2-related enzymes highlights differences between diverticulitis and inflammatory bowel disease. Dig Dis Sci. 60, 1236–1246 (2015).
Miyoshi, H. et al. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 36, 5–24 (2017).
Ferrer, R. & Moreno, J. J. Role of eicosanoids on intestinal epithelial homeostasis. Biochem Pharmacol. 43, 1–8 (2010).
Tanaka, K. et al. Inhibition of both COX-1 and COX-2 and resulting decrease in the level of prostaglandins E2 is responsible for non-steroidal anti-inflammatory drug (NSAID)-dependent exacerbation of colitis. Eur J Pharmacol. 603(1–3), 120–32 (2009).
Melgar, S., Drmotova, M., Rehnstrom, E., Jansson, L. & Michaelsson, E. Local production of chemokines and prostaglandin E2 in the acute, chronic and recovery phase of murine experimental colitis. Cytokine. 35, 275–283 (2006).
Yamashita, S. Studies on changes of colonic mucosal PGE2 levels and tissue localization in experimental colitis. Gastroenterol Jpn. 28, 224–35 (1993).
Zifroni, A., Treves, A. J., Sachar, D. B. & Rachmilewitz, D. Prostanoid synthesis by cultured intestinal epithelial and mononuclear cells in inflammatory bowel disease. Gut. 24, 659–664 (1983).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Zhang, Y. et al. Tissue regeneration. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 348, a2340 (2015).
Kandil, H. M., Argenzio, R. A. & Sartor, R. B. Low endogenous prostaglandin E2 predisposes to relapsing inflammation in experimental rat enterocolitis. Dig Dis Sci. 45, 2091–2099 (2000).
Hara, S. et al. Prostaglandin E synthases: understanding their pathophysiological roles through mouse genetic models. Biochimie. 92, 651–659 (2010).
Fujino, H. & Regan, J. W. EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol Pharmacol. 69, 5–10 (2006).
Yokoyama, U., Iwatsubo, K., Umemura, M., Fujita, T. & Ishikawa, Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 65, 1010–1052 (2013).
Jiang, G. L. et al. The prevention of colitis by E prostanoid receptor 4 agonist through enhancement of epithelium survival and regeneration. J Pharmacol Exp Ther. 320, 22–28 (2007).
Zimecki, M. Potential therapeutic interventions via EP2/EP4 prostaglandin receptors. Postepy Hig Med Dosw (Online). 66, 287–294 (2012).
Shukla, A. K. et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 (2014).
Shenoy, S. K. Arrestin interaction with E3 ubiquitin ligases and deubiquitinases: functional and therapeutic implications. Handb Exp Pharmacol. 219, 187–203 (2014).
Kovacs, J. J., Hara, M. R., Davenport, C. L., Kim, J. & Lefkowitz, R. J. Arrestin development: emerging roles for beta-arrestins in developmental signaling pathways. Dev. Cell. 17, 443–458 (2009).
Sharma, D., Malik, A., Lee, E., Britton, R. A. & Parameswaran, N. Gene dosage-dependent negative regulatory role of beta-arrestin-2 in polymicrobial infection-induced inflammation. Infect Immun. 81, 3035–3044 (2013).
Li, J. et al. Deficiency of beta-arrestin1 ameliorates collagen-induced arthritis with impaired TH17 Cell differentiation. Proc Natl Acad Sci USA 110, 7395–7400 (2013).
Shi, Y. et al. Critical regulation of CD4+ T Cell survival and autoimmunity by beta-arrestin 1. Nat Immunol. 8, 817–824 (2007).
Lee, T. et al. Beta-arrestin-1 deficiency protects mice from experimental colitis. Am J Pathol. 182, 1114–1123 (2013).
Lee, T., Lee, E., Arrollo, D., Lucas, P. C. & Parameswaran, N. Non-hematopoietic beta-arrestin1 confers protection against experimental colitis. J Cell Physiol. 231, 992–1000 (2016).
Tan, S. et al. β-arrestin-1 protects against endoplasmic reticulum stress/p53-upregulated modulator of apoptosis-mediated apoptosis via repressing p-p65/inducible nitric oxide synthase in portal hypertensive gastropathy. Free Radical Bio Med. 87, 69–83 (2015).
Zhan, Y. et al. β-Arrestin1 inhibits chemotherapy-induced intestinal stem cell apoptosis and mucositis. Cell Death Dis 7, e2229 (2016).
Sheng, H., Shao, J., Townsend, C. J. & Evers, B. M. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut. 52, 1472–1478 (2003).
He, X. C. et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 39, 189–198 (2007).
Engelman, J. A., Luo, J. & Cantley, L. C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 7, 606–619 (2006).
Qiu, W. et al. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest. 121, 1722–1732 (2011).
Acknowledgements
We are grateful to Professor Robert J. Lefkowitz at Duke University for providing the β-arr1-KO mice and the β-arr1 antibody, Professor Pei G. at the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences for providing the β-arr1 plasmids. This work was supported in part by grants from the National Natural Science Foundation of China (U1501224, 81370511), the Natural Science Foundation of Guangdong Province (S2013010016541), the Science and Technology Planning Projects of Guangdong Province (2016A020216014), the Science and Technology Planning Projects of Guangzhou City (201604020118, 201604020002), the Fundamental Research Funds for the Central Universities (15ykpy24).
Author information
Authors and Affiliations
Contributions
Study concept and design (X.P., J.L. and B.W.); acquisition of data (X.P., J.L., S.T., M.X., J.T., J.J., H.L., and B.W.); analysis and interpretation of data (X.P., J.L., S.T., M.X., J.T., J.J., H.L., and B.W.); drafting of the manuscript (X.P. and B.W.); critical revision of the manuscript for important intellectual content (X.P., J.L., S.T., M.X. and B.W.); statistical analysis (X.P., J.L. and B.W.); obtained funding (B.W.); technical or material support (J.J. and H.L.); study supervision (B.W.).
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, X., Li, J., Tan, S. et al. COX-1/PGE2/EP4 alleviates mucosal injury by upregulating β-arr1-mediated Akt signaling in colitis. Sci Rep 7, 1055 (2017). https://doi.org/10.1038/s41598-017-01169-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01169-6
This article is cited by
-
Network pharmacology and in vivo experiments reveal the pharmacological effects and molecular mechanisms of Simiao Powder in prevention and treatment for gout
BMC Complementary Medicine and Therapies (2022)
-
Aspirin sensitivity of PIK3CA-mutated Colorectal Cancer: potential mechanisms revisited
Cellular and Molecular Life Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
