
Abstract
The Cape platanna, Xenopus gilli, an endangered frog, hybridizes with the African clawed frog, X. laevis, in South Africa. Estimates of the extent of gene flow between these species range from pervasive to rare. Efforts have been made in the last 30 years to minimize hybridization between these two species in the west population of X. gilli, but not the east populations. To further explore the impact of hybridization and the efforts to minimize it, we examined molecular variation in one mitochondrial and 13 nuclear genes in genetic samples collected recently (2013) and also over two decades ago (1994). Despite the presence of F 1 hybrids, none of the genomic regions we surveyed had evidence of gene flow between these species, indicating a lack of extensive introgression. Additionally we found no significant effect of sampling time on genetic diversity of populations of each species. Thus, we speculate that F 1 hybrids have low fitness and are not backcrossing with the parental species to an appreciable degree. Within X. gilli, evidence for gene flow was recovered between eastern and western populations, a finding that has implications for conservation management of this species and its threatened habitat.
Similar content being viewed by others
Introduction
Gene flow (introgression) between species may facilitate adaptive evolution through the exchange of beneficial genetic variation. This expedites the colonization of specialized ecological niches1,2,3, and affects future adaptive potential by increasing genetic and phenotypic variation2, 4,5,6,7. However, gene flow between species also poses risks by eroding species boundaries8, disrupting adaptively evolved complexes of alleles9, 10, promoting the exchange of genetic variation associated with disease11, influencing pathogen emergence12, and facilitating species invasion13, 14. As such, hybridization has important implications for biodiversity conservation.
Hybridization in African clawed frogs
Hybridization features prominently in the evolutionary history of African clawed frogs (genus Xenopus); 28 of 29 species are polyploid, and all of these are probably allopolyploid15, 16. When backcrossed in the laboratory, there is variation among F 1 X. gilli-laevis hybrid females with respect to whether or not their progeny are polyploid17. Laboratory studies indicate that in some crosses (X. gilli-X. laevis and X. laevis-X. muelleri) F 1 hybrid males are sterile, but female F 1 hybrids are fertile17, 18. F 1 X. gilli-X. laevis hybrid females are capable of backcrossing with either parental species, and both sexes of the F 2 backcross generation can be fertile17. Thus there exists the possibility that gene flow among Xenopus species could occur in nature. At least three Xenopus hybrid zones are thought to exist19,20,21, and hybrids in each of these zones may have the same ploidy level as the parental species (pseudotetraploid; ref. 22).
The X. gilli/X. laevis hybrid zone
Classified by the IUCN as Endangered23, X. gilli 24 occurs in southwestern Western Cape Provence, South Africa25,26,27,28. Xenopus gilli is a found in seasonal ponds in lowland coastal fynbos habitat, a component of the Cape Floristic Region, which is a biodiversity hotspot29 with an extreme level of plant endemism30. These ponds have high concentrations of humic compounds derived from the surrounding fynbos vegetation, and a characteristic dark color and low pH25, 31, 32. The range of X. gilli is disjunct and includes the Cape of Good Hope section of Table Mountain National Park (CoGH), habitat near the town of Kleinmond, and habitat near the town of Pearly Beach (refs 25,26,27,28; Fig. 1). These three localities are interrupted by unsuitable, highly modified habitat that may impede contemporary gene-flow26. As with many amphibians33, habitat degradation is a major threat to X. gilli 25, 26.

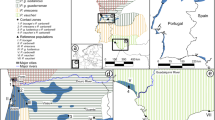
Sampling locations. For each species, numbers indicate the sum of number of individuals from each locality sampled in 1994 and 2013. An inset indicates the study area in southern Africa and altitude in meters is indicate on the scale. The map was made using the R package Marmap81 using topographic data from the National Oceanic and Atmospheric Administration, USA.
In contrast, X. laevis 34, is found throughout southern Africa, in both natural and disturbed areas of South Africa and Malawi35, 36. Xenopus laevis is syntopic throughout the range of X. gilli 25,26,27,28 and can tolerate a broad spectrum of environmental challenges including extremes of desiccation, salinity, anoxia, and temperature37. Picker et al. 32proposed that there may be an ecological basis for speciation of X. laevis and X. gilli centered on higher tolerance of X. gilli embryos to low pH, allowing for habitat specialization.
Several aspects of external morphology readily distinguish these species, including smaller size of X. gilli, the presence of longitudinal dorsal mottling that does not connect over the midline in X. gilli only (for example, see Fig. 1 of ref. 19), and orange and black vermiculation on the venter of X. gilli. F 1 hybrids between X. gilli and X. laevis are readily identified based on individuals that are morphologically intermediate with respect to size and coloration, and this identification has been confirmed by molecular tests19, 28, 38, 39. The reported abundance of F 1 hybrids varies from relatively common26, 38, 39, to rare27, 28. Morphological variation of some individuals has been previously interpreted as being derived from backcrosses of F 1 hybrids with each parental species39.
The western extent of the X. gilli distribution occurs within the CoGH25, 26. Following reports of hybrids and expansion of X. laevis populations, steps were taken in the mid-1980s to minimize co-occurence of these two species within the CoGH which included removal of X. laevis from X. gilli ponds, translocation of X. gilli to new sites40, and construction of a wall around a known X. gilli pond25, 41. The hope was to minimize hybridization and resource competition, for example, if larger X. laevis individuals are able to outcompete X. gilli for food39, 42. With some interruptions, these efforts have continued for the last 30 years in the CoGH. Similar efforts have not been made for eastern populations of X. gilli which are located on private property, and in some ponds in these areas where X. gilli had been found in the past, now only X. laevis are found26, 41.
To further investigate the effect of hybridization on gene flow between X. gilli and X. laevis, we examined DNA sequence variation in these species from one mitochondrial DNA (mtDNA) marker and 13 nuclear DNA (nDNA) markers. Genetic samples were collected from within managed (west) and unmanaged (east) portions of the range of X. gilli. Samples were analyzed from both locations that were collected shortly after management began, and then in the same areas again 20 years later. We expected that if introgression was occurring in the populations during this time period, it would be more pronounced in the east population. If efforts to minimize hybridization in the west were successful, we expected more evidence of gene flow in the samples collected soon after management began as compared to more recently. However, in both localities and both sampling times, we found no evidence of shared mitochondrial haplotypes or nuclear alleles between these species, suggesting that the F 1 hybrids have low fitness and are not backcrossing with the parental species to an appreciable degree, despite potential fertility of F 1 females17. Within X. gilli, we recovered evidence of gene flow between east and west populations, and found genetic diversity to be higher in the unprotected eastern population. These findings have implications for management and conservation of this endangered habitat specialist.
Materials and Methods
Genetic samples analyzed in this study were collected either in 1994 or in 2013. Some of the samples from the earlier collection were also analyzed in two earlier studies27, 28. The 2013 collection included X. gilli and X. laevis individuals from the same or geographically close (within 5 km) sites as the 1994 collection, and both sampling efforts used funnel traps. Animal sampling protocols approved by the Institutional Animal Care and Use Committee at Columbia University and work was performed in accordance with all relevant guidelines and regulations for animal experimentation, in accordance with laws for studying wildlife in South Africa and with appropriate collection permits from the Chief Directorate of Nature Conservation and Museums, and was approved by the Animal Ethics Committee at the University of Cape Town and the Stellenbosch University Research Ethics Committee: Animal Care and Use. Samples were obtained east and west of False Bay for both species and for both time periods (Fig. 1). We assigned individuals to species (X. gilli or X. laevis) based on dorsal and ventral patterning, shape of head, and overall size43, 44. Because this study aimed to explore genetic effects of backcrossed hybrids, for both time points, we intentionally excluded individuals whose intermediate morphology (and genetic analysis in the case of the 1994 individual28) indicated that they were F 1 hybrids (1 individual from 1994 and 9 from 2013).
DNA was extracted from tissue samples using Qiagen DNEasy tissue extraction kits (Qiagen, Inc), following the manufacturer’s protocol, or a phenol-chloroform protocol. A fragment of the mtDNA genome was amplified and sequenced for 36 and 33 X. gilli and X. laevis individuals, respectively, using primers from ref. 45 that target a portion of the 16S ribosomal RNA gene (16S). Exons of 13 nDNA genes ranging from 333–770 bp in length were sequenced for 20–41 X. gilli and 11–31 X. laevis individuals using paralog specific primers (primers are from ref. 46). These exons came from the genes B-Cell CLL/Lymphoma 9 (BCL9), BTB domain containing 6 (BTBD6), Chromosome 7 Open Reading Frame 25 (C7orf25), Fem-1 Homolog C (FEM1C), Microtubule Associated Serine/Threonine Kinase Like (MASTL), Mannosyl-oligosaccharide glucosidase (MOGS-1), Nuclear Factor, Interleukin 3 Regulated (NFIL-3), protocadherin 1 (PCDH1), phosphatidylinositol glycan anchor biosynthesis class O (PIGO), protein arginine methyltransferase 6 (PRMT6), Ras association domain family member 10 (RASSF10), SURP and G-patch domain containing 2 (SUGP2), and zinc finger BED-type containing 4 (ZBED4). A table of sample IDs and which loci were amplified for which samples is available in the Appendix. In the phylogenetic analysis of individual genes (discussed below), we used as an outgroup a sequence from X. tropicalis from the genome assembly version 9.0 on Xenbase47. When possible, we also included orthologous and homeologous sequences from X. laevis from the genome assembly version 9.1 on Xenbase47, which was identified using BLAST48; this was not possible when a homeologous sequence was not identified, which could be due to gene loss or missing data in the genome sequence. Sequence data were aligned using MAFFT49 and corrected by eye. Coding frame was estimated using the ‘minimize stop codons’ option in Mesquite v.3.0450, and alignments were trimmed to begin at the first position and end at the third position of the reading frame.
We calculated the phase of nDNA alleles (i.e. haplotypes) using the ‘best guess’ option of PHASE51, 52 with default parameters. Each individual’s allelic sequences for each locus were used in subsequent population genetic, clustering, and gene tree analyses. Thus, for each nuclear locus, an individual frog was represented by two sequences, each corresponding to one allele.
Gene trees
Gene trees were estimated for each phased nDNA exon and the mtDNA alignment using BEAST v1.8.353. Substitution models were selected based on the Akaike Information Criterion using MRMODELTEST v.254, and xml files were prepared for BEAST using BEAUTI (part of the BEAST package). For each nDNA locus, we ran two Markov chain Monte Carlo runs for 25 million generations. For the mtDNA, the model selected by MRMODELTEST2 (GTR+Γ) failed to converge on stable parameter estimates, and we therefore instead used the simpler HKY+Γ model, and ran two chains for 50 million generations. For each analysis, convergence of parameter estimates on the posterior distribution was assessed using TRACER v.1.555 based on an effective sample size (ESS) value >200 and inspection of the trace of parameter estimates against the MCMC generation number. Based on this, for all phylogenetic analyses the first 25% of the posterior distribution was discarded as burn-in. Then, using TREEANNOTATOR, we produced consensus trees from the post-burn-in posterior distribution of trees.
Species tree
We also estimated a species tree (with the nuclear sequences used in the STRUCTURE analysis, see below) using the multi-species coalescent model of *BEAST56. We trimmed the dataset to include only nDNA genes with all populations sampled (see Genetic clusters section for details) and included only individuals sampled for all genes. All X. laevis individuals were considered to be the same species (17 individuals), and we separated the east and west X. gilli populations into separate species (10 and 11 individuals, respectively), and X. tropicalis was considered its own species. We set a simple HKY+Γ model joined for all data partitions (so that convergence of parameter estimates could be reached), assumed a strict molecular clock joined for all data partitions, and allowed the underlying gene tree structure to vary across data partitions. We ran the 8 chains for 170 million generations and removed 50 million generations as burn-in.
Genetic clusters
We used STRUCTURE v.2.3.457 to estimate individual assignment probabilities to genetic clusters using best-guess phased nDNA alleles on a subset of individuals. Three loci lacked data from the east X. gilli 1994 population (exons of the genes MOGS-1, PCDH1 that also lacked data from this exon for X. laevis east 1994 samples, and PIGO), so we excluded them from STRUCTURE analysis. We also excluded individuals with >50% missing data for the remaining 10 loci. This resulted in a dataset of 13, 8, 11, and 6 X. gilli individuals from the following localities and years respectively: east 1994, east 2013, west 1994, and west 2013, where east and west refer to the sampling locations relative to False Bay. This analysis also included 9, 12, 6, 4 X. laevis individuals from east 1994, east 2013, west 1994, and west 2013 respectively. We used the admixture model of STRUCTURE and assumed no correlation between alleles at different loci. We ran the Markov chain Monte Carlo for 20 million generations, following a two million generation burn-in. We tested a number of clusters (K) ranging from 1–8, with 5 replicate analyses for each setting of K. To correct for label switching and to average assignment probabilities across runs, we used CLUMPP v.1.1.258. We first computed the D statistic, following recommendations in the CLUMPP manual58, to decide on the particular algorithm to employ for maximizing similarity across runs. We then used the ad hoc method of ref. 57 and the method described by ref. 59 to evaluate the most likely number of genetic clusters (K).
Evolutionary models
As discussed below, our analyses did not detect mitochondrial DNA haplotypes or nuclear alleles that were shared between X. laevis and X. gilli but did detect shared haplotypes and alleles between populations of X. gilli and between populations of X. laevis. Because X. gilli is of conservation concern, we evaluated the fit of data from this species to three evolutionary models (Fig. 2). In the first (isolation) model, divergence of two X. gilli populations was followed by no migration between each population. Under the isolation model, all shared alleles between these populations would be due to incomplete lineage sorting (ILS). In the second (ongoing migration) model, X. gilli population divergence was followed by ongoing symmetrical migration between the populations. Under the ongoing migration model, shared alleles would be due to ILS or migration, and some of the alleles shared due to migration could have been exchanged millions of years ago. In the third (secondary contact) model, X. gilli population divergence was followed by a period of no migration and then by a period during which symmetrical migration occurred between east and west populations. Under the secondary contact model, shared alleles would again be due to ILS or migration, but alleles shared due to migration could only have been exchanged recently.

Evolutionary models considered for X. gilli sequence data from east and west populations included (a) population division without subsequent gene flow, (b) separation followed by ongoing gene flow, and (c) separation followed by secondary contact after a period of no gene flow. Model parameters include the population polymorphism parameter θ, which is assumed to be constant in the ancestral and both descendant populations, the time of speciation T, the amount of migration m, and the time of secondary contact τ.
All models include a parameter T, which is the time of separation between the X. gilli populations and a parameter θ, which is the population polymorphism parameter of the ancestral and both descendant populations. The second and third models have an additional parameter m, which is the number of individuals in each population that are replaced per generation by individuals from the other population (east vs west), divided by the product of four times the effective population size of each population. The third model includes another parameter τ, which is the proportion of T going back in time from the present that secondary contact began. Thus, the ongoing migration and isolation models are special cases of the secondary contact model in which τ = 1, or τ = 1 and m = 0, respectively. We note that several assumptions of these models are undoubtedly violated (e.g., constancy of population size over time, equivalent population size of both descendant and the ancestral populations) but we made them nonetheless so we could complete the simulations (see below) within a reasonable amount of time, and because of the relatively small size of the dataset.
The approximate likelihood of combinations of values for these parameters was estimated using rejection sampling60. In this approach, the likelihood is approximated by the natural logarithm of the number of simulations for which the sum of four summary statistics from a simulation (discussed next) were within ±ε % of the sum of the observed four summary statistics from actual sequence data, divided by the number of simulations, where ε = 25. The value of ε determined how close the simulations must match the observed data in order to contribute to the likelihood, and was selected based on a compromise between the computational efficiency of the likelihood estimation and the accuracy of the estimate60. For the ongoing migration model and the secondary contact model, 40,000 simulations were performed for each combination of parameter values we considered. For the isolation model, no simulations had summary statistics within ±ε % of the observed; thus, 1,000,000 simulations were performed in order to achieve an upper bound for the likelihood estimate. The likelihood of the data over all combinations of the following parameter value intervals were estimated: T: every 1,000,000 generations in the interval of 0–20,000,000 generations; θ: every 0.001 units in the interval of 0.001–0.01 and every 0.01 units in the interval of 0.01–0.1; τ: every 10% in the interval of 10–100%; m every 0.1 units in the interval of 0–1 and every integer in the interval of 1–10.
We used the sum across loci of four summary statistics described by ref. 61 for these likelihood calculations, and simulations were performed using the program mimarsim62. These four summary statistics include the number of sites with a derived polymorphism (i) in the west population of X. gilli only, (ii) in the east population of X. gilli only, (iii) shared between the west and east populations of X. gilli, or (iv) fixed in either the west or in the east population of X. gilli. The simulations used a fixed value for the mutation rate equal to 2.69e −9 substitutions per site per generation, which was estimated based on the average synonymous divergence between a randomly selected X. gilli sequence and an orthologus sequence from X. tropicalis, and assuming a divergence time of 65 million years for the separation of these lineages63, and a generation time of one year. Each locus had a mutation rate scalar based on synonymous divergence to X. tropicalis that accommodated variation among loci in the rate of evolution. To minimize the influence of natural selection on the polymorphism data, summary statistics and likelihood calculations were based only on variation at synonymous positions. Confidence intervals were estimated using the profile likelihood method (i.e., that the 95% confidence interval is defined by the two points that are 1.92 log-likelihood (lnL) units from the maximum).
Population dynamics over time and space
We performed various analyses to assess whether the genetic diversity varied among these species, over time, or among populations east and west of False Bay. Pairwise F ST (with significance computed by a permutation test) was quantified for the same data used in the STRUCTURE analysis using ARLEQUIN v3.5.2.264. Nucleotide diversity (π) of each locus was calculated using the pegas package in R65, 66. We then calculated a mean value of π across loci for each of the eight populations, weighting the estimate by gene length for each locus. Confidence intervals were obtained by bootstrapping the weighted π values 5000 times.
Because allelic diversity is influenced by sample size, we using the program HP-RARE to calculate rarefied estimates of allelic diversity67, which involves downsampling data to the smallest number of samples in each population across all nuclear loci for which there were data. This analysis was performed with the same data as used in the STRUCTURE analysis. For X. laevis populations there was one exception; the prmt6 locus had only four sampled alleles for the X. laevis west 1994 population, thus we did one run with all of the data (using four as the smallest number of sampled alleles) and another run excluding prmt6 (in which case, eight was the smallest number of sampled alleles). For all X. gilli populations, the smallest number of sampled alleles was eight. We generated confidence intervals by bootstrapping of the allelic diversity measurements 5000 times.
To statistically evaluate differences in genetic diversity over time, location and species, we constructed linear mixed models using the R package lme468. We built models for the estimated values of nucleotide diversity (π) and allelic diversity independently with diversity values measured for each locus, using time (1994 or 2014), location (east or west) and species (X. gilli or X. laevis) as fixed effects (all additive, no interaction terms) and considering locus as a random effect. For each parameter of both models, we also used lme4 to compute confidence intervals with the confint function.
Results
Molecular polymorphism and gene trees
In the mitochondrial and 13 nuclear gene trees, alleles from X. gilli and X. laevis clustered in reciprocally monophyletic clades (Fig. 3, Fig. S1). No individuals were found to have introgressed loci, which would have been evidenced by an allele in one species having a closer relationship to the alleles of the other species (i.e. a paraphyletic relationship). Similar to previous studies26, 27, the mtDNA gene tree identified divergence between the east and west populations of X. gilli (Fig. 3). We identified one individual (Sample ID: XgUAE_08) from the west population of X. gilli that carried a mtDNA haplotype that was more closely related to haplotypes that were carried by individuals from the east population. This observation was also reported previously, from different samples26, 27. The *BEAST analysis recovered the expected species tree of these three species with posterior probabilities of one (Fig. S2). This analysis estimated the divergence time of X. gilli and X. laevis at about 14.05 my and divergenc of the east and west X. gilli populations at about 1 my (0.51–1.36 my 95% HDP; when a calibration point of 65 my from X. tropicalis is assumed63).

Representative gene trees that collectively provide no evidence of genetic exchange between X. gilli and X. laevis. The phylogeny on the left illustrates divergence between 16S rDNA mitochondrial sequences in the east and west populations of X. gilli, and with one shared sequence (indicated with an arrow) that occurred on both sides of False Bay. The nuclear phylogenies in the center and right provide examples of no shared alleles and shared alleles between the east and west X. gilli populations, respectively. Gene name acronyms are described in the Materials and Methods section. These and other phylogenies are depicted with sample labels in Fig. S1.
Genetic clusters
STRUCTURE analyses assigned each individual to groups that corresponded with species assignment (Fig. 4a). All X. laevis individuals were assigned to a single genetic cluster at K = 2–8, indicating a lack of allele frequency clustering, which is consistent with gene flow across the population range. The X. gilli samples were assigned to two clusters corresponding to sampling location (east and west) at K = 3–8, indicating differences in allele frequencies, which is consistent with restricted gene flow between them (Fig. 4a). Assignment of individuals to clusters stabilized at K = 3, with no new clusters being detected at higher values of K (Fig. 4a). The likelihood plot plateaus at K = 3 (Fig. 4b); the Evanno method59 supports K = 2 and the ad hoc method of ref. 57 supports K = 3.

(a) Structure analyses for 10 loci, which had sequence data for all populations. (b) Likelihood for each value of K.
Evolutionary models
Using simulations and summary statistics, we evaluated the fit of the X. gilli data to evolutionary models with no migration after speciation, with ongoing migration after speciation, or with secondary contact after speciation. The lnL of the secondary contact model was −8.032, the ongoing migration model was −8.987, and the isolation model was <−13.815. We were not able to more precisely estimate the likelihood of the isolation model because no simulations under this model resulted in data whose four summary statistics were within ±ε of the observed values (see Methods).
Nested models can be compared by assuming that twice the difference between the lnL of each model follows a χ 2 distribution with degrees of freedom equal to the difference in the number of parameters in each model (denoted \({\chi }_{1}^{2}\) for comparison between models that differ in one parameter). However, because comparison between these successively more complex models involves a boundary condition on one parameter (τ = 1 for the ongoing migration model, m = 0 for the isolation model), this difference in model likelihoods follows a mixture of \({\chi }_{0}^{2}\) and \({\chi }_{1}^{2}\) distributions69. The secondary contact model is thus not supported over the ongoing migration model (p = 0.08), but the ongoing migration model is supported over the isolation model (p = 0.009). Overall then, these results support an inference of gene flow between X. gilli populations, but fail to discern substantial temporal heterogeneity in the level of gene flow.
The maximum likelihood parameter estimates and 95% confidence intervals for the ongoing migration model were θ: 0.002 (0.001–0.003) and m: 0.7 (0.1–2) individuals/generation. The maximum likelihood estimate for T was 8,500,000 generations; the 95% CI was unable to be estimated because it exceeded the boundaries we tested (1,000,000–20,000,000), suggesting low statistical power to estimate this parameter. Comparisons to similar parameters estimated for African clawed frogs by other studies using other methods16, 36, 70 suggest that these estimates are biologically plausible. Our intuition that the shared identical alleles between east and west X. gilli populations are due to ongoing migration is thus supported, with caveats that several model assumptions, discussed below, are violated to some degree.
Population dynamics over time and space
In line with results from STRUCTURE analysis, a high F ST was measured in all pairwise comparisons of the east and west populations of X. gilli (comparing within the same year 2013 east to 2013 west, and between years 1994 east to 2013 west and 2013 east to 1994 west; range: 0.55–0.60, p < 0.05). For X. gilli, between time points within each location (east or west), F ST was not significantly different from zero (east 1994 to east 2013 and west 1994 to west 2013; p > 0.05, F ST < 0.02). For X. laevis, pairwise comparisons of east and west populations, within the same year (1994 east to 1994 west or 2013 east to 2013 west) and between time points (1994 east to 2013 west and 2013 east to 1994 west), had intermediate F ST values that departed significantly from zero (p < 0.05, F ST = 0.07–0.16). But within locations comparing time points (1994 east to 2013 east and 1994 west to 2013 west), F ST was not significantly different from zero (p > 0.05, F ST = 0.04 for both comparisons).
Both nucleotide diversity and allelic diversity did not change drastically over time, but within species, both statistics were higher in the east population than the west (Fig. 5). In the linear mixed model analysis of π, the effect of species was significant with X. laevis higher than X. gilli by 0.00073 substitutions per site (95% CI: 0.00027–0.00119). The effect of location was significant with π lower in the west than the east population by 0.00091 (95% CI: 0.00045–0.00137). The effect of time of sampling was not significant, with the 2013 samples being lower by 0.00016 but the 95% CI of this difference spanning zero (−0.00062–0.00030). Similar results were recovered for allelic diversity, with X. laevis having higher allelic diversity than X. gilli (0.59, 95% CI: 0.21–0.97), the west being less diverse than the east (−0.77, 95% CI: −1.14–−0.40), and no significant effect of sampling time (−0.15, 95% CI: −0.52–0.21).

Genetic diversity statistics including rarefied estimates of allelic diversity (top panels) and nucleotide diversity (π) weighted by length of sequence; bottom panels). For allelic diversity, the analysis considered the same 10 loci as were analyzed by the STRUCTURE analysis (see Materials and Methods). Allelic diversity for X. laevis did not include PRMT6 locus because this locus only had four alleles for the west 1994 population.
Discussion
Gene Flow between X. laevis and X. gilli
Previous investigations of the genetic consequences of hybridization between X. laevis and X. gilli found no evidence of widespread genetic introgression27, 28, a result that seemed to be at odds with the incidence of morphologically and genetically identified hybrids in this and other studies19, 26, 38, 39, 71. In this study, we analyzed many of the samples from refs 27, 28 and also genetic samples that were collected more recently. Evidence of introgression between X. laevis and X. gilli was not detected in mitochondrial DNA or in any of 13 nuclear loci (Fig. 3; Fig. S1). Furthermore, each species formed separate genetic clusters with no evidence for similarities in allele frequencies (Fig. 4a). These findings were consistent in both sampling efforts examined here, which included targeting both populations of X. gilli and sampling time points separated by about two decades. Previous investigations into the extent of genetic introgression28, used two nuclear loci that were not used in this study. Combining that study with ours brings the total number of genomic regions studied to 15, and includes variation from 6 of the 18 chromosome pairs based on gene location in the X. laevis version 9 genome, on Xenbase. This expanded sampling is thus consistent with the interpretation by ref. 28 that genomic introgression is not extensive.
The lack of introgression is despite the continued identification (based on morphology) of a low frequency of putative F 1 hybrids in both localities. Though there could be an adaptive benefit for hybridization because X. gilli embryos can tolerate ponds with higher pH levels than X. laevis, which perhaps could allow for invasion of X. gilli habitat, we found no evidence that hybridization has led to gene flow of the genetic basis of this or other ecological adaptations that evolved after these two species diverged from their most recent common ancestor. Although not the focus of this study, the relatively low abundance of F1 hybrids argues against the possibility that a new species of hybrid origin is evolving in this zone of sympatry between X. laevis and X. gilli. Reproductive isolation in amphibians has been shown to happen in a few million years for some lineages72, 73. In Xenopus species, female individuals respond to species-specific calls evoked by males (phonotaxis)74 and this presumably acts to some degree as a prezygotic barrier to hybridization. However, an observation is that at high densities, Xenopus individuals amplex indiscriminately (G. J. Measey, personal observation), potentially overriding some prezygotic barriers. In some ponds, X. gilli individuals can be outnumbered by X. laevis 3:141, and indiscriminate amplexus could mean X. gilli males (which are also smaller) are outcompeted for access to females. This may be why hybrids are occasionally seen, but the extended period of divergence between these species (~14 my) appears to have resulted in strong post-zygotic barriers preventing introgression.
Hybridization followed by back-crossing is expected to generate a mosaic of introgressed and non-introgressed genomic regions. Variation among genomic regions in the extent of introgression can be further augmented by natural selection favoring or disadvantaging genetic variants from one species in the genomic background of the other5. In California tiger salamanders (Ambystoma californiense), for example, some loci are fixed for foreign alleles from the introduced barred tiger salamander (A. mavortium), whereas other loci exhibit no sign of introgression6. That the barred tiger salamander was introduced only 60 years ago suggests that mosaicism of genomic introgression arose rapidly (in ~20 generations; ref. 6). In this study it is therefore possible that we failed to identify some introgressed regions of the genome due to the relatively sparse sampling of genomic regions. Future studies that survey variation across the entire genome, such as RAD-Seq75, could more precisely quantify the extent of gene flow between these species, if it occurs.
Population structure in X. gilli and change over time
Analysis of mtDNA26, 27, 45 and skin peptides secreted by these populations76 support the existence of at least two distinct populations in X. gilli in the western and eastern portion of its range. Our mitochondrial analysis, STRUCTURE analysis, and some of the gene trees reported here (such as MOGS-1 and RASSF10) also exhibit substantial geographic differences in X. gilli allele frequencies between these populations (Figs 3 and 4a, Fig. S1). In contrast, genetic diversity in X. laevis has minimal geographic structure, with most alleles occurring on both sides of False Bay, and STRUCTURE analyses assigning all X. laevis individuals to a single genetic cluster (Fig. S1, Fig. 4a). This is similar to findings reported by ref. 27.
When and why did population structure arise in X. gilli? Using mtDNA sequence data and a relaxed molecular clock45, estimated that the divergence between X. gilli populations occurred 8.5 million years (my) (95% CI: 4.8–13.4), which is the same as the estimate obtained here using our coalescent modeling approach. This estimate is older than the 1 my divergence time estimated by the *BEAST analysis (Fig. S2), but this is not unexpected because *BEAST does not incorporate gene flow after divergence in its model. Using similar data and a coalescent modeling approach ref. 26 recovered a somewhat more recent divergence time of 4.63 my, but with confidence intervals that overlapped with the previous estimate (95% CI: 3.17–6.38). Evans et al. 27 proposed that the two populations split following inundation of the Cape Flats. Fogell et al. 26 pointed out that marine inundation probably occurred multiple times in the last few million years and that cycles of aridification also likely influenced the costal fynbos habitat, on which X. gilli relies. Our finding of gene flow after divergence supports the idea that these populations have been periodically reconnected, allowing exchange of migrants. Therefore, whatever the cause of divergence was, it was demonstrably not a permanent barrier.
Of note is that the evolutionary models we tested are almost certainly violated by the system we explored in many ways, including variation over time and among populations in population size, mutation rate, and migration rate. Although we do not anticipate that these violations are influential enough as to negate the rejection of the isolation model, a larger dataset might provide statistical power with which to better evaluate more complex scenarios, such as the secondary contact model.
The F ST and linear mixed model analyses suggest that allele frequencies have not changed substantially in the last 20 years, though there is a trend of decreasing diversity (Fig. 5 and from values obtained in linear mixed models indicated a non- significant decline in diversity from 1994 to 2013). If generation time is about one year or less (which is based on laboratory studies and could be an underestimate, ref. 35), this represents 20 generations. Changes in allelic diversity may signal population declines earlier than nucleotide diversity, because loss of rare alleles (which happens during population declines) would have a greater impact on count based metrics, such as allelic diversity, than they would on frequency based metrics such as π 77. Thus, though not significant, a declining trend seen for allelic diversity (Fig. 5) may be an early indication of population declines. Linear mixed models allowing for independent changes in diversity for each locus over time revealed declining genetic diversity (except for two loci in the π models; results not shown).
Management
Hybridization and introgression has the potential to threaten species survival10. In an attempt to reduce gene flow between species, three conservation actions were implemented in the mid-1980s. A wall was built around one impoundment in CoGH25, populations of pure X. gilli were translocated to areas without X. laevis 40, and X. laevis were manually removed from CoGH25, 41, 78. Removal of X. laevis ceased in 2000, but resumed in 201125, 41, 78. The same management efforts have not been conducted for the population of X. gilli east of False Bay, most of which resides in non-protected areas26, 41.
Interestingly, the CoGH has greater juvenile recruitment of X. gilli 41 and fewer hybrids (2.5% vs 8–27% of individuals in ponds in the west and east respectively, ref. 26). Our results suggest that these hybrids are not producing successful offspring via backcrossing with either parental species frequently enough to produce large scale genomic impacts. These results suggest that the genomes of X. gilli and X. laevis are largely genetically distinct. Thus, the major benefits to X. gilli of removal of X. laevis from habitat shared with X. gilli probably stem from minimizing competition for ecological resources between these species25, 41, 42.
For X. gilli and X. laevis, east populations harbor more genetic diversity than the west populations (Fig. 5). Allelic diversity and heterozygosity reflect a population’s ability to respond to selection79, and thus from a genetic perspective conservation of east populations of X. gilli is paramount.
This study suggests that patterns of gene flow within X. gilli included genetic exchange between populations in the east and west. The ancestral distribution of X. gilli was likely patchy to begin with and has contracted considerably in the last several decades, including in locations now occupied only by X. laevis or X. laevis and hybrids25, 26. Ancestral patterns of gene flow are presumably imperiled by further habitat fragmented by human activity, including habitat altering effects of invasive species such as Acacia saligna (Port Jackson Willow), Acacia mearnsii (Black Wattle), and Hakea sericea (Silky Hakea)80. Continued efforts to conserve and restore coastal fynbos habitat both inside and outside of protected areas43, such as removal of invasive vegetation, restoration of native vegetation, and removal of X. laevis, stands to benefit X. gilli. This is particularly important in the east population of X. gilli near Kleinmond where genetic diversity is highest and the population resides on private land.
References
Anderson, E. & Hubricht, L. Hybridization in tradescantia. iii. the evidence for introgressive hybridization. Am. J. Bot. 396–402 (1938).
Dowling, T. E. & Secor, C. L. The role of hybridization and introgression in the diversification of animals. An. Rev. Ecol. Sys. 593–619 (1997).
Rieseberg, L. H. et al. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301, 1211–1216, doi:10.1126/science.1086949 (2003).
Stelkens, R., Brockhurst, M., Hurst, G., Miller, E. & Greig, D. The effect of hybrid transgression on environmental tolerance in experimental yeast crosses. J. Evol. Biol. 27, 2507–2519 (2014).
Arnold, M. L. & Martin, N. H. Adaptation by introgression. J. Biol. 8, 1 (2009).
Fitzpatrick, B. M. et al. Rapid fixation of non-native alleles revealed by genome-wide snp analysis of hybrid tiger salamanders. BMC Evol. Biol. 9, 176 (2009).
Anderson, E. Introgressive hybridization. Introgressive hybridization (1949).
Arnold, M. L. Evolution through genetic exchange, vol. 3 (Oxford University Press Oxford, 2006).
Gilk, Sara E. et al. Outbreeding depression in hybrids between spatially separated pink salmon, Oncorhynchus gorbuscha, populations: marine survival, homing ability, and variability in family size. In Genetics of Subpolar Fish and Invertebrates 287–297 (Springer, Netherlands, 2004).
Rhymer, J. M. & Simberloff, D. Extinction by hybridization and introgression. An. Rev. Ecol. Sys. 83–109 (1996).
Simonti, C. N. et al. The phenotypic legacy of admixture between modern humans and Neandertals. Science 351, 737–741 (2016).
Stukenbrock, E. H. The role of hybridization in the evolution and emergence of new fungal plant pathogens. Phytopathology 106, 104–112 (2016).
Figueroa, M. et al. Facilitated invasion by hybridization of Sarcocornia species in a salt-marsh succession. J. Ecol. 91, 616–626 (2003).
Blair, A. C., Blumenthal, D. & Hufbauer, R. A. Hybridization and invasion: An experimental test with diffuse knapweed (Centaurea diffusa Lam). Evol. Appl. 5, 17–28 (2012).
Evans, B. J. Genome evolution and speciation genetics of clawed frogs (Xenopus and Silurana). Front. Biosci. 13, 4687–4706 (2008).
Evans, B. J. et al. Genetics, morphology, advertisement calls, and historical records distinguish six new polyploid species of African clawed frog (Xenopus, Pipidae) from west and central Africa. PLoS One 10, e0142823 (2015).
Kobel, H. R. Reproductive capacity of experimental Xenopus gilli x X. l. laevis hybrids. In The Biology of Xenopus (eds. Kobel, H. R. & Tinsley, R. R.) 73–80 (Oxford University Press, Oxford, 1996).
Malone, J. H., Chrzanowski, T. H. & Michalak, P. Sterility and gene expression in hybrid males of Xenopus laevis and X. muelleri. PLoS One 2, e781 (2007).
Kobel, H. R., Pasquier, L. D. & Tinsley, R. C. Natural hybridization and gene introgression between Xenopus gilli and Xenopus laevis laevis (Anura: Pipidae). J. Zool. 194, 317–322 (1981).
Fischer, W., Koch, W. & Elepfandt, A. Sympatry and hybridization between the clawed frogs Xenopus laevis laevis and Xenopus muelleri (Pipidae). J. Zool. 252, 99–107 (2000).
Yager, D. D. Sound production and acoustic communication in Xenopus borealis. In The Biology of Xenopus (eds. Kobel, H. R. & Tinsley, R. R.) 121–140 (Oxford University Press, Oxford, 1996).
Tymowska J. Polyploidy and cytogenetic variation in frogs of the genus Xenopus. In Amphibian Cytogenetics and Evolution (eds. Green D. S. & Sessions S. K.) 259–297 (Academic Press, San Diego 1991).
South African Frog Re-assessment Group (SA-FRoG), IUCN SSC Amphibian Specialist Group. Xenopus gilli. The IUCN Red List of Threatened Species 2010: e.T23124A9417597. http://dx.doi.org/10.2305/IUCN.UK.2004.RLTS.T23124A9417597.en Accessed: 2016-09-29 (2010).
Rose, W. & Hewitt, J. Description of a new species of Xenopus from the Cape Peninsula. Trans. Royal Soc. South Africa 14, 343–346 (1926).
Picker, M. D. & de Villiers, A. L. The distribution and conservation status of Xenopus gilli (Anura: Pipidae). Biol. Conserv. 49, 169–183 (1989).
Fogell, D. J., Tolley, K. A. & Measey, G. J. Mind the gaps: Investigating the cause of the current range disjunction in the Cape Platanna, Xenopus gilli (Anura: Pipidae). PeerJ 1, e166 (2013).
Evans, B. J., Morales, J. C., Picker, M. D., Kelley, D. B. & Melnick, D. J. Comparative molecular phylogeography of two Xenopus species, X. gilli and X. laevis, in the south-western Cape Province, South Africa. Mol. Ecol. 6, 333–343 (1997).
Evans, B. J., Morales, J. C., Picker, M. D., Melnick, D. J. & Kelley, D. B. Absence of extensive introgression between Xenopus gilli and Xenopus laevis laevis (Anura: Pipidae) in Southwestern Cape province, South Africa. Copeia 1998, 504–509 (1998).
Myers, N., Mittermeier, R. A., Mittermeier, C. G., Da Fonseca, G. A. & Kent, J. Biodiversity hotspots for conservation priorities. Nature 403, 853–858 (2000).
Kier, G. et al. A global assessment of endemism and species richness across island and mainland regions. PNAS 106, 9322–9327 (2009).
Mitchell, D., Coley, P., Webb, S. & Allsopp, N. Litterfall and decomposition processes in the coastal fynbos vegetation, south-Western Cape, South Africa. T. J. Ecol. 977–993 (1986).
Picker, M. D., McKenzie, C. & Fielding, P. Embryonic tolerance of Xenopus (Anura) to acidic blackwater. Copeia 1072–1081 (1993).
Marsh, D. M. & Trenham, P. C. Metapopulation dynamics and amphibian conservation. Conserv. Biol. 15, 40–49 (2001).
Daudin, F. M. Histoire naturelle des rainettes, des grenouilles et des crapauds. Ouvrage orné de 38 planches représentant 54 espéces peintes d’aprés nature (Levrault, 1802).
Kobel, H. R. & Tinsley, R. C. (eds.) The Biology of Xenopus. (Oxford University Press, 1996).
Furman, B. L. S. et al. Pan-African phylogeography of a model organism, the African clawed frog Xenopus laevis. Mol. Ecol. 24, 909–925 (2015).
Measey, G. et al. Ongoing invasions of the African clawed frog, Xenopus laevis: a global review. Biol. Invasions 14, 2255–2270 (2012).
Picker, M. D. Hybridization and habitat selection in Xenopus gilli and Xenopus laevis in the South-Western Cape Province. Copeia 574–580 (1985).
Picker, M. D., Harrison, J. & Wallace, D. Natural hybridization between Xenopus laevis laevis and X. gilli in the south-western Cape province, South Africa. In The Biology of Xenopus (eds. Kobel, H. R. & Tinsley, R. R.) 61–70 (Oxford University Press, Oxford, 1996).
Measey, G. J., de Villiers, A. L. & Soorae, P. Conservation introduction of the Cape Platanna within the Western Cape, South Africa. Global Re-introduction Perspectives 91–93 (2011).
de Villiers, F. A., de Kock, M. & Measey, G. J. Controlling the African clawed frog Xenopus laevis to conserve the Cape Platanna Xenopus gilli in South Africa. Conserv. Evi. 13, 17 (2016).
Vogt, S., de Villiers, F.A., Ihlow, F., Rödder, D. & Measey, J. Competition and feeding ecology in two sympatric Xenopus species (Anura: Pipidae). PeerJ, 5, e3130 (2017).
de Villiers, A. Species account: Xenopus gilli (Rose & Hewitt, 1927). In Atlas and red data book of the frogs of South Africa, Lesotho and Swaziland (eds. Minter, L., Burger, M., Harrison, J., Bishop, P. & Braack, H.) 260–263 (Smithsonian Institution Press, 2004).
Kobel, H. R., Loumont, C. & Tinsley, R. C. The extant species. In The Biology of Xenopus (eds. Kobel, H. R. & Tinsley, R. R.) 9–33 (Oxford University Press, Oxford, 1996).
Evans, B. J., Kelley, D. B., Tinsley, R. C., Melnick, D. J. & Cannatella, D. C. A mitochondrial DNA phylogeny of African clawed frogs: Phylogeography and implications for polyploid evolution. Mol. Phylogenet. Evol. 33, 197–213 (2004).
Bewick, A. J., Anderson, D. W. & Evans, B. J. Evolution of the closely related, sex-related genes DM-W and DMRT1 in African clawed frogs (Xenopus). Evolution 65, 698–712 (2011).
Bowes, J. B. et al. Xenbase: gene expression and improved integration. Nucleic Acids Res. gkp953 (2009).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–80 (2013).
Maddison, W. P. & Maddison, D. R. Mesquite: A modular system for evolutionary analysis. Version 3.04, http://mesquiteproject.org (2015).
Stephens, M. & Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. T. Am. J. Hum. Genet. 73, 1162–1169 (2003).
Stephens, M. & Scheet, P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. T. Am. J. Hum. Genet. 76, 449–462 (2005).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–73 (2012).
Nylander, J. MrModeltest v2 distributed by author. Evolutionary Biology Center, Uppsala University (2004).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer v1.6. http://beast.bio.ed.ac.uk/Tracer (2014).
Heled, J. & Drummond, A. J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580 (2010).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Jakobsson, M. & Rosenberg, N. A. Clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Weiss, G. & von Haeseler, A. Inference of population history using a likelihood approach. Genetics 149, 1539–1546 (1998).
Becquet, C. & Przeworski, M. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 17, 1505–1519 (2007).
Becquet, C. & Przeworski, M. Learning about modes of speciation by computational approaches. Evolution 63, 2547–2562 (2009).
Bewick, A. J., Chain, F. J. J., Heled, J. & Evans, B. J. The pipid root. Syst. Biol. 61, 913–926 (2012).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Paradis, E. Pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420 (2010).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, URL https://www.R-project.org (2015).
Kalinowski, S. T. hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 5, 187–189 (2005).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Self, S. G. & Liang, K.-Y. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J. Am. Stat. Assoc. 82, 605–610 (1987).
Evans, B. J., Bliss, S. M., Mendel, S. A. & Tinsley, R. C. The Rift Valley is a major barrier to dispersal of African clawed frogs (Xenopus) in Ethiopia. Mol. Ecol. 20, 4216–4230 (2011).
Rau, R. E. The development of Xenopus gilli Rose & Hewitt (Anura, Pipidae). T. Ann. South African Mus. 76, 247–263 (1978).
Dufresnes, C. et al. Timeframe of speciation inferred from secondary contact zones in the european tree frog radiation (Hyla arborea group). BMC Evol. Biol. 15, 155 (2015).
Colliard, C. et al. Strong reproductive barriers in a narrow hybrid zone of West-Mediterranean green toads (Bufo viridis subgroup) with Plio-Pleistocene divergence. BMC Evol. Biol. 10, 232 (2010).
Picker, M. D. Xenopus laevis (Anura: Pipidae) mating systems: a preliminary synthesis with some data on the female phonoresponse. African Zool. 15, 150–158 (1980).
Davey, J. W. et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 12, 499–510 (2011).
Conlon, J. M. et al. Evidence from peptidomic analysis of skin secretions that allopatric populations of Xenopus gilli (Anura: Pipidae) constitute distinct lineages. Peptides 63, 118–125 (2015).
Greenbaum, G., Templeton, A. R., Zarmi, Y. & Bar-David, S. Allelic richness following population founding events – A stochastic modeling framework incorporating gene flow and genetic drift. PLoS One 9, e115203 (2014).
Measey, J. et al. Invasive amphibians in southern Africa: a review of invasion pathways. Bothalia-Applied Biodiv. Conserv. 47, a2117.
Caballero, A. & Garca-Dorado, A. Allelic diversity and its implications for the rate of adaptation. Genetics 195, 1373–1384 (2013).
Wilson, J. R. et al. Biological invasions in the cape floristic region: history, current patterns, impacts, and management challenges. Fynbos: Ecology, Evolution, and Conservation of a Megadiverse Region 273 (2014).
Pante, E. & Simon-Bouhet, B. Marmap: a package for importing, plotting and analyzing bathymetric and topographic data in R. PLoS One 8, e73051 (2013).
Acknowledgements
We thank Mike Picker for collaboration on early fieldwork, advice on X. gilli and for initiating a long-standing legacy of conservation of this species. We also thank Brian Golding for access to computational resources, Jonathan Dushoff for statistical advice, and two annonymous reviewers for helpful comments on an earlier version of this manuscript. We thank the staff at the CoGH for the efforts in preserving X. gilli, both genetically and its habitat. This work was supported by support to B.J.E. from the Natural Science and Engineering Research Council of Canada (RGPIN/283102-2012) and the Museum of Comparative Zoology, Harvard University, G.J.M. was funded by the National Research Foundation of South Africa (NRF Grant No. 87759) and the DST-NRF Centre of Excellence for Invasion Biology at Stellenbosch University. Research permission came from the Chief Directorate of Nature Conservation and Museums (2009/94), CapeNature (AAA007-01867), and SANParks.
Author information
Authors and Affiliations
Contributions
B.J.E. and G.J.M. collected samples. G.A.C. and B.L.S.F. generated sequence data. Analyses were performed by B.L.S.F., C.M.S.C., and B.J.E. B.L.S.F. and B.J.E. wrote the manuscript and all authors provided edits.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession codes: Alignment for each locus, along with the corresponding BEAST XML files, resulting gene trees, and data for Structure analyses have been deposited in a Dryad repository (doi:10.5061/dryad.g6g2r). Genbank accession number for the sequences include the following: Rassf10: HQ221332–HQ221356, KY824194–KY824236 Sugp2: HQ221211–HQ221235, KY824150–KY824193 c7orf25: HQ220710–HQ220732, KY824433–KY824476 fem1c: HQ221309–HQ221331, KY824395–KY824432 mastl: HQ221046–HQ221070, KY824354–KY824394 mogs: HQ221071–HQ221092, KY824311–KY824353 nfil3: HQ221093–HQ221120, KP344016, KY852029–KY852072 BTBD6/p7e4: HQ220685–HQ220709, KY824477–KY824517 pigo: HQ221144–HQ221167, KY824237–KY824280 prmt6: HQ221168–HQ221178, HQ221180–HQ221190, zbed4: HQ221262–HQ221284, KY824107–KY824149, bcl9: KP345721, KP345621, KY851962–KY852028 pcdh1: HQ221129, KY824281–KY824310 16s: KP345307, KP345309–KP345313, KP345315–KP345318, KY852073–KY852133.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Furman, B.L.S., Cauret, C.M.S., Colby, G.A. et al. Limited genomic consequences of hybridization between two African clawed frogs, Xenopus gilli and X. laevis (Anura: Pipidae). Sci Rep 7, 1091 (2017). https://doi.org/10.1038/s41598-017-01104-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01104-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
