
Abstract
The transmembrane DNA-binding protein CadC of E. coli, a representative of the ToxR-like receptor family, combines input and effector domains for signal sensing and transcriptional activation, respectively, in a single protein, thus representing one of the simplest signalling systems. At acidic pH in a lysine-rich environment, CadC activates the transcription of the cadBA operon through recruitment of the RNA polymerase (RNAP) to the two cadBA promoter sites, Cad1 and Cad2, which are directly bound by CadC. However, the molecular details for its interaction with DNA have remained elusive. Here, we present the crystal structure of the CadC DNA-binding domain (DBD) and show that it adopts a winged helix-turn-helix fold. The interaction with the cadBA promoter site Cad1 is studied by using nuclear magnetic resonance (NMR) spectroscopy, biophysical methods and functional assays and reveals a preference for AT-rich regions. By mutational analysis we identify amino acids within the CadC DBD that are crucial for DNA-binding and functional activity. Experimentally derived structural models of the CadC-DNA complex indicate that the CadC DBD employs mainly non-sequence-specific over a few specific contacts. Our data provide molecular insights into the CadC-DNA interaction and suggest how CadC dimerization may provide high-affinity binding to the Cad1 promoter.
Similar content being viewed by others


Introduction
Neutrophilic enterobacteria, such as Escherichia coli, Salmonella typhimurium, and Vibrio cholerae, possess different stress response systems to counteract a decrease of external pH by adjusting the pH to a physiological range. One system used by E. coli is the Cad-system1, that consists of the cytosolic protein CadA and the membrane proteins CadB and CadC (Fig. 1a). The lysine decarboxylase CadA converts lysine into cadaverine and CO2 under consumption of one proton. The antiporter CadB transports the basic product cadaverine in exchange for lysine out of the cell2, 3. The pH-receptor CadC senses a drop of the external pH and activates transcription of cadBA below pH 6.64, 5. This process requires the simultaneous presence of lysine, which is sensed by the co-sensor LysP, a lysine-specific transporter6, 7.

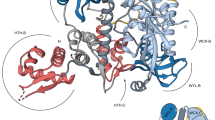
Crystal structure of the CadC DNA-binding domain (DBD). (a) Scheme showing the transcriptional activation of cadBA genes by CadC in dependence of low pH and increased lysine concentration. The mechanism requires dimerization and therefore two DBDs are shown to bind the cadBA promoter (PcadBA). (b) Scheme showing the domain organization of CadC from E. coli. The DNA-binding domain is shown in blue. Numbers indicate amino acid positions. TM, transmembrane domain. (c) Cartoon representation of the E. coli CadC1-107 DBD from two views. Selected secondary structure elements are annotated. (d) Superposition of CadC1-107 (blue) as in (c) with the bacterial transcriptional activator PhoB in complex with DNA (magenta)16. Note that only one PhoB monomer and only part of the DNA are shown for clarity. Secondary structure elements involved in DNA-binding by CadC are annotated. The PhoB transactivation loop necessary for binding to RNA polymerase is indicated. Pictures were created with PyMol (Delano Scientific).
CadC belongs to the family of ToxR-like receptors. This protein family is named after ToxR, a membrane-bound transcriptional regulator that controls virulence in V. cholerae 8. Other members of this protein family are TcpP, TfoS, and VttRA and VttRB in V. cholerae 9,10,11,12, PsaE in Yersinia pseudotuberculosis 13, WmpR in Pseudoalteromonas tunicata 14, and ArnR in Sulfolobus acidocaldarius 15. ToxR-like receptors are characterized by a conserved modular composition: external signals are sensed by a periplasmic sensor domain and are transduced through a single transmembrane helix to a cytoplasmic DBD of an OmpR type, which acts as effector domain to regulate gene expression. Hence, signal transduction appears to be independent of chemical modifications like phosphorylation. Despite their simple design principle, the exact mechanism of signal transduction has remained enigmatic for this receptor family. Mechanisms of transcription enhancement by OmpR-type effector domains of transcriptional activators have been studied biochemically for many bacterial response systems. However, structural information of these effector domains in complex with target DNA is limited to a few examples16,17,18 and no ToxR-like DBD structures are available to date.
Functional activity depends on CadC dimerization5, 19 (Fig. 1a). The structure of the periplasmic sensory domain of CadC and molecular features of the pH-sensing mechanism have been described previously5, 20. A patch of negatively charged amino acids located at the dimer interface is involved in pH sensing5. Protonation of these amino acids might reduce repulsing forces between the two monomers and thereby enable receptor activation. To investigate how the signal is processed in the cytoplasm, we have recently studied the role of the linker that connects the transmembrane helix and the DBD. We found that the linker is intrinsically disordered, but nevertheless required for proper positioning of the two DBDs for activation of transcription21. CadC-mediated stress response requires binding of the protein to two DNA-bindings sites, Cad1 and Cad2 within the cadBA promoter region22. It has been proposed that CadC targets a quasi-palindromic sequence within Cad122. However, the exact binding sites have not been mapped.
Here, we investigate the structural basis for DNA recognition by the CadC DBD. We present the crystal structure of the CadC DBD and analyse its interaction with DNA using NMR spectroscopy and complementary biophysical and functional techniques. We define the binding stoichiometry and affinities with the Cad1 DNA promoter and regions within. Mutational analysis identified amino acids in the CadC DBD that are important for DNA recognition and function. Analysis of the interaction of the CadC DBD with DNA demonstrates that CadC mainly utilizes non-sequence-specific and only few specific interactions for complex formation. NMR and in vitro binding data indicate a preference of CadC for binding to AT-rich regions within the Cad1 promoter. Finally, a structural model of the CadC DBD-DNA complex based on our experimental data provides a molecular rationale for the distinct binding preferences of the CadC and other DBDs found in bacterial response regulators.
Results
Crystal structure of the CadC DNA-binding domain
To characterize the CadC effector domain and the interaction with DNA, we first determined the crystal structure of the CadC DBD (amino acids 1–107, Fig. 1b and c) at 2.0 Å resolution (Supplementary Table S1 and Supplementary Fig. 1). Consistent with our recent study of the complete cytoplasmic part of CadC21 small angle X-ray scattering (SAXS) data indicate that the DBD is a monomer in solution (Supplementary Table S2). The overall fold and secondary structure is characteristic for the winged helix-turn-helix (HTH) motif found in members of the OmpR family effector domains. Thus, CadC comprises a recognition helix (α3) and a β-hairpin wing (β5-loop-β6) within a compact fold with the topology β1-β2-β3-β4-α1-α2-α3-β5-β6-β7. To our knowledge, the CadC DBD represents the first structure of a ToxR-like DBD.
We performed structural similarity searches using DALI23 (Supplementary Fig. S1). Comparing CadC1-107 with the eleven best-scored hits revealed overall highly similar folds. A unique feature of the CadC DBD is an additional short β-strand (β7) that extends the highly conserved C-terminal β-sheet (β1-β4) in OmpR-type DBDs. Consequently, the β-sheet in CadC is shifted towards the site of the β4 strand to maintain a compact globular fold. To rule out that these conformational differences reflect crystal-packing artefacts we analysed the CadC DBD in solution using heteronuclear NMR spectroscopy. Secondary 13C chemical shifts (Supplementary Fig. S1) show that the additional C-terminal β7-strand (residues 104–106) is also present in solution. This is confirmed by 15N, and 13C-edited nuclear Overhauser effect spectroscopy (NOESY) data, which show specific contacts between strands β1 and β7 (Supplementary Fig. S1). The additional strand β7 is important for the structural integrity of CadC, as a truncated variant (CadC1-102) was less soluble and unstable. Nevertheless, a CadC variant with the three amino acids V104, I105 and W106 replaced by alanine, supports stress-dependent signalling like wild-type CadC (data not shown). This implies that the integrity of the β7 strand is not critical for signal transduction. The highest structural similarity of CadC1-107 with an effector domain bound to DNA is found with PhoB. The structure of the PhoB DBD in complex with pho box DNA16 superimposes with the CadC DBD with a backbone coordinate RMSD (root-mean-square deviation) of 1.8 Å (Fig. 1d). Given the structural similarity, the CadC-DNA binding interface is expected to resemble the PhoB-DNA interaction.
DNA-binding of CadC to the cadBA promoter
CadC binding to the cadBA DNA promoter site depends on the complete Cad1 region22 (Fig. 2a and Supplementary Fig. S2). By comparing NMR spectra of the CadC DBD alone (CadC1-107) with a construct comprising the DBD and the cytoplasmic linker (CadC1-159), we have recently shown that amino acids within the flexible linker do not interact with the DBD and DNA21. We then defined the DNA-binding surface in the CadC DBD by following NMR chemical shift changes in 1H,15N correlation experiments upon titration of a double-stranded DNA oligonucleotide encompassing the Cad1 binding site (−153 to −113 bp) (Cad1 41-mer, Supplementary Fig. S2)22. Amide signals in CadC1-159 were monitored after titration of 41-mer DNA to a final protein:DNA ratio of 2:1. The spectrum shows severe line broadening, which reflects a significant increase in molecular weight upon complex formation and/or dynamics due to sliding of the protein on the DNA (Supplementary Fig. S2). The 2:1 complex was subsequently confirmed in static light scattering (SLS) and SAXS measurements (Supplementary Fig. S2). NMR signals that remain observable are located in loops of the DBD and the flexible CadC linker beyond residue 107. No chemical shift perturbations (CSPs) are observed for these residues indicating that these residues are not involved in DNA-binding.

NMR-based analysis of Cad1 sub-fragment binding to CadC1-107. (a) Top, scheme of the full cadBA promoter region between −175 bp and 25 bp relative to the transcription start with CadC-binding sites Cad1 and Cad2 and the RNA polymerase binding sites at −35 bp and −10 bp. Bottom, scheme of fragments Cad1A, B and C within the Cad1 41-mer sequence used in this study. Numbers represent the internal numbering along full-length Cad1 with the boundaries given. The previously suggested CadC target sequence22 (Cad1 17-mer) is shown for comparison (see Supplementary Fig. S4). (b–d) Imino proton spectra of fragments Cad1A, B and C alone (black) or in stoichiometric excess of CadC1-107 (red). Assignments are indicated. (e) Quantification of chemical shift perturbations (CSPs) from panels (b–d). The colour indicates categories of CSP effects, i.e. CS differences.
The role of AT-rich patches in Cad1
Previous studies22, 24 suggested that CadC preferentially binds to AT-rich sequence stretches of DNA. AT-rich regions of a DNA duplex exhibit a narrowing of the minor groove and thus may increase the contribution of specific contacts to the overall affinity25. To examine the cadBA promoter and map CadC binding sites in the Cad1 41-mer we studied three smaller regions, Cad1A (14-mer), Cad1B and Cad1C (13-mers) (Fig. 2a), all of which are of a length that is typically covered by DBDs16, 18. Notably, Cad1A and Cad1B together harbour a Cad1 17-mer sequence, which had been previously suggested as CadC target site 22. While the Cad1A and Cad1B DNA sequences are highly similar and exhibit a high AT-content with almost inverse/head-to-head sequence motifs, Cad1C lacks significant AT-patches. 1H NMR spectra of these DNAs with and without CadC show that all three DNAs bind to the protein (Fig. 2b–d). We assigned the imino proton NMR chemical shifts in both the DNA fragments alone and when in complex with CadC1-107 and analysed the effects of binding (Fig. 2e). An analysis of the CSPs reveals that CadC causes stronger effects upon binding to Cad1A and Cad1B than to Cad1C, indicating a preference of the CadC DBD for binding to AT-rich DNA.
We next quantified binding affinities of CadC to the three fragments using electromobility shift assays (EMSAs) (Fig. 3a and b). The two AT-rich fragments Cad1A and Cad1B show similar affinities with low micromolar dissociation constants while Cad1C is bound with a slightly lower affinity. These values are consistent with the binding of other DBDs to promoter half-sites17, 26. We then monitored the binding of these DNA regions to CadC in 1H,15N correlation experiments with 15N-labeled CadC1-107 (Supplementary Fig. S3). All NMR spectra show significant spectral changes, indicating binding of all three DNAs to CadC. Interestingly, line-broadening is observed for residues in the protein-DNA interface upon titration of Cad1C. In contrast, little line-broadening is observed during the titration of the AT-rich Cad1A and Cad1B DNA duplexes. This indicates that the interaction of Cad1C exhibits dynamics at μs-ms time scales. Possibly, the lack of AT-rich regions in Cad1C leads to sliding and is consistent with a reduced binding affinity to CadC compared to Cad1A and Cad1B. These data show that CadC recognizes shorter fragments in the Cad1 promoter with a preference for AT-rich regions.

EMSA-based analysis of Cad1 fragments. (a) Representative EMSA analysis of fragments Cad1A, B and C when titrated with CadC1-107 to ten-fold stoichiometric excess. Arrows indicate the complex species. (b) Quantification of EMSA experiments in (a). Binding constants are given as obtained from a one-site binding event and shown as mean ± standard deviation.
Interestingly, the Cad1A and Cad1B DNAs represent an AT-rich quasi-palindromic 17-mer motif (Figs 2a and 4a) which has been previously suggested to be the CadC interaction site within Cad122. We therefore studied binding of CadC1-107 to this Cad1 17-mer in NMR titrations. Spectral changes observed during the titration show binding kinetics in fast-exchange on the NMR chemical shift time scale27 (Fig. 4b), typically reflecting micromolar affinities (K D). Quantitative analysis of the 17-mer NMR titration data yields a K D = 67 ± 16 μM for the interaction (Fig. 4b, right panel), consistent with surface plasmon resonance data that indicate a steady state constant K D = 23 μM (Supplementary Fig. S4).

NMR analysis of CadC1-107 binding to Cad1 DNA. (a) Top, the scheme shows the full cadBA promoter region between −175 bp and 25 bp relative to the transcription start with CadC binding sites Cad1 and Cad2, and the RNA polymerase binding sites at −35 bp and −10 bp. Middle, web-logo showing the conservation of nucleotides in the Cad1 and Cad2 fragments of the cadBA promoter region for enteric bacteria according to NCBI blastn75. The loci of the Cad1 and Cad2 sites are indicated. Blue bars represent the Cad1 41-mer and the quasi-palindromic 17-mer core sequences used in this study. (b) Left, superposition of 1H-15N-HSQC spectra from CadC1-107 in the absence or presence of different amounts of Cad1 17-mer DNA (see colour codes) with selected residues depicted. The inset shows a magnification of selected amide resonances. Right, quantification of CSPs of Tyr 98 (see inset) by fitting relative CSPs to a 1:1 binding model. The given binding constant is calculated from all significantly shifting resonances and shown as mean ± standard deviation. (c) Plot of CSPs, i.e. the difference of chemical shifts of free CadC and the complex with maximum DNA (1:1.58). Gaps indicate prolines; negative bars are residues in which the assignment in either the bound or apo-form is missing. The scheme at the bottom of the diagram shows the secondary structure of the CadC DBD based on the crystal structure with elements annotated. Asterisks indicate positions that were used for mutational analyses at a later stage of the study. TL, transactivation loop; RH, recognition helix; wing, β-wing.
Based on the NMR spectral changes the binding interface with the 17-mer DNA (Fig. 4c) maps to two regions comprising the recognition helix α3 and the stretch between amino acids 90 and 100, i.e. the loop contacting the DNA minor groove in the hypothetical model with pho-box DNA (Fig. 1d). To map the binding interface from the side of the DNA we analysed chemical shift changes of the imino protons of the Cad1 17-mer DNA duplex upon addition of the CadC DBD (Supplementary Fig. S4). Interestingly, only thymine imino signals are perturbed upon addition of protein while no guanosine iminos are affected, demonstrating that the CadC DBD preferentially contacts A-T base pairs. This provides further evidence for the preference of CadC in binding to AT-regions within the cadBA promoter.
Stoichiometry of CadC-DNA complexes
Using NMR and SLS measurements we have previously shown that the cytoplasmic portion of CadC is monomeric independent of the concentration21. We then characterized the oligomeric state and binding stoichiometries of the CadC-DNA interaction (Supplementary Figs S2 and S4). SLS of the Cad1 41-mer with CadC1-107 clearly shows that the protein binds in a 2:1 ratio to the DNA (Supplementary Fig. S2). This is further supported by SAXS experiments (Supplementary Fig. S2; Supplementary Table S2). While SAXS data of CadC1-107 alone are in excellent agreement with the crystal structure (χ2 of 0.98), analysis of the CadC1-107 41-mer Cad1 DNA complex indicates the presence of two domains bound to the cadBA promoter DNA (Supplementary Fig. S2). In contrast, SAXS data demonstrate that the Cad1 17-mer DNA binds to CadC1-107 with a 1:1 stoichiometry (Supplementary Table S2). This is further supported by the tumbling correlation time of the protein-DNA complex derived from NMR relaxation data, which are in good agreement with the expected molecular weight of a 1:1 CadC DBD complex with 17-mer Cad1 DNA and indicate that the CadC DBD alone is monomeric (Supplementary Fig. S4). Thus, while the CadC DBD forms a 1:1 complex with the 17-mer harbouring an AT-rich region, the complete Cad1 41-mer DNA can adopt a 2:1 complex in vitro. In vivo this may allow the binding of two DBDs from a membrane-anchored CadC dimer with the promoter.
Identification of amino acids important for DNA recognition and function of CadC
The DNA-binding interface suggested by the superimposition with PhoB-pho was probed by replacement of selected amino acids in the CadC DBD. These were expected either to be involved in DNA contacts via the recognition helix (RH) and the β-wing or to be important for transcriptional regulation, e.g. through the transactivation loop (TL) (Fig. 1d). To test these CadC variants for cadBA promoter activation, we used the E. coli cadA::lacZ reporter strain EP31428. β-Galactosidase activities were determined for cells grown in minimal medium under cadBA-inducing (pH 5.8 and lysine) and non-inducing (pH 7.6 without lysine) conditions. We replaced selected charged amino acids and those that were affected in the NMR titration (Fig. 5 and Supplementary Fig. S5). Significantly impaired activity under stress conditions was observed for alanine replacements of Arg32, Arg50, Arg60, Val63, Thr64, His66, Thr69, Gln70, Ser73, Arg76, Lys95 and Arg96 (Fig. 5a). There is an excellent correlation with spatial proximity of the corresponding side chains to the DNA in the structural superimposition of CadC and PhoB that leads to the aligned CadC-pho-box DNA complex (Fig. 5b). Notably, these amino acid replacements in CadC abolished activation of the cadBA promoter under all tested conditions, demonstrating their functional significance (Supplementary Fig. S5).

Influence of amino acid substitutions within CadC on the expression of the cadBA operon. (a) Quantification of cadBA transcription in E. coli EP314 using the reporter gene lacZ. Cells were grown at inducing or non-inducing conditions, and the β-galactosidase activity was determined. The experiment was performed in triplicate, and error bars indicate standard deviations from the means. On top, a part of the CadC secondary structure is shown to give orientation along the elements that are essential for transcriptional regulation. See Supplementary Fig. S5 for all tested experimental conditions. (b) Visualization of mutation sites within CadC1-107. The structure shows the CadC DBD docked to pho-box DNA as in Fig. 1d. Selected side chains from amino acids with significant effect in (a) and from CSPs in Fig. 4c are shown as sticks and annotated. (c) CadC affinity for Cad1 41-mer DNA. Representative SPR sensorgrams obtained from CadC1-159 wild-type (WT) or the respective variants injected in different concentrations (see colour code) to immobilized Cad1 41-mer DNA. Sensorgrams representing real time binding are shown with steady-state dissociation constants. The figures represent each one characteristic of three independently performed experiments. Binding curves to derive the overall steady-state affinities are shown in Supplementary Fig. S5.
In NMR titrations of the CadC DBD with the 17-mer Cad1 DNA, these amino acid replacements strongly affect DNA-binding (Fig. 4b and c). It is noteworthy that the amide NMR signals of Asn65 and His66 could not be assigned in the apo-form of the CadC protein due to line-broadening suggesting that this region is conformationally flexible in the absence of DNA (Fig. 4c and Supplementary Fig. S4). The structural alignment of CadC with PhoB (Figs 1d and 5b) also explains the role of Val63 and Arg60. These two residues are located in the potential TL of CadC and might be involved in interactions with the RNAP. Amino acid replacements in this loop have been previously shown to suppress transcription activation by PhoB29.
Surprisingly, the position of Glu30 is extremely sensitive to amino acid substitutions. As Glu30 is expected to be in close proximity to the DNA backbone (Fig. 5b), the introduction of a positive charge (E30R) at this position can enhance the DNA-binding affinity by providing additional contacts to the DNA. This may explain why the E30R mutant induces constitutive cadBA promoter activation independent of pH and lysine (Supplementary Fig. S5).
To investigate whether the observed effects with the CadC variants are related to perturbed interactions with DNA we performed SPR (surface plasmon resonance, Biacore) experiments and compared the binding affinity of wild-type (WT) and variant CadC proteins (Fig. 5c). The overall affinity, determined from the steady state binding model, corresponds to an equilibrium dissociation constant K D = 380 nM (Supplementary Fig. S5), and is thus 60-fold stronger compared to the Cad1 derived 17-mer DNA (K D = 23 μM, Supplementary Fig. S4, see discussion). The R50A and R76A variants almost abolish, while the β-wing variant R96A reduces DNA-binding consistent with the importance of these residues for DNA-binding (Fig. 5a and c). Apparently, the decrease in affinities observed in vitro is sufficient to inhibit recruitment and activation of the RNAP on the cadBA promoter in cells where hierarchical binding of effector domains and polymerase has been found before30. The CadC E30R variant exhibits an almost 30-fold increased affinity (Fig. 5c, Supplementary Fig. S5) consistent with additional DNA contacts involving the arginine side chain.
Atomic Models of CadC-Cad1 DNA interactions
The mutational analysis of key residues in the CadC-DNA complex supports the protein-DNA interactions suggested by the superimposition of CadC with pho-box DNA shown in Fig. 5b. To obtain more detailed insight into specific interactions between the CadC DBD and the Cad1 DNA site we generated a docking model based on our experimental information and the structural similarity to PhoB (Figs 1d and 5b) using HADDOCK31 (Fig. 6; Supplementary Fig. S6). An overlay of the lowest energy model with the PhoB-DNA complex confirms the overall similarity of the protein-DNA-binding interface (Fig. 6b). A SAXS-derived ab initio model is consistent with the structural model of the CadC1-107/17-mer DNA complex (Fig. 6c).

HADDOCK model of the CadC1-107 structure with the Cad1 17-mer DNA. (a) Overlay of all 26 ensemble structures within the respective cluster from a HADDOCK modelling of CadC1-107 with Cad1 17-mer DNA (Supplementary Table S3). (b) Alignment of the lowest-energy model for the CadC1-107/Cad1 DNA complex from (a) with the structure of the PhoB-pho box DNA complex (PDB: 1gxp16) (see Fig. 1d). DNAs are coloured according to proteins. (c) Superposition of a SAXS-derived ab initio shape model of a 1:1 complex of CadC1-107 with 17-mer Cad1 DNA (grey, semi-transparent) with the lowest-energy structure from the cluster in (a) shown as blue cartoon. (d) Contacts of CadC1-107 in complex with Cad1 17-mer DNA. Red lines indicate sequence-specific interactions of protein side chains with DNA bases. Black lines are contacts to the DNA backbone, i.e. the phosphate or the sugar groups. Green lines show relevant, sequence-specific protein-protein contacts.
The analysis of the protein-DNA contacts in the docking model (Fig. 6d) highlights the predominant presence of non-sequence specific contacts over only a few sequence-specific interactions. Only the side chains of Lys95, His66 and Gln70 exhibit specific contacts with thymidines at position 77, 11/71, and 70 in the DNA, respectively (Fig. 6d and Supplementary Fig. S6). This is consistent with the importance of the AT-rich sequence in both the 17-mer and the 41-mer DNA ligands. Interestingly, the 12 base pairs in the 17-mer DNA sequence that are recognized by CadC correspond to the Cad1A region, indicating that CadC specifically recognizes a few identical nucleotides in the two DNA fragments. Altogether, the specific contacts observed suggest a consensus binding motif of 5′-T-T-A-x-x-x-x-T-3′. The Cad1 41-mer DNA harbours two of these consensus sites (Supplementary Fig. S6) in a head-to-head arrangement, which is in line with our biophysical analysis and the experimentally observed 2:1 stoichiometry. Depletion of these two sites leads to a reduced affinity of CadC in EMSA and ITC (isothermal titration calorimetry) experiments (Supplementary Fig. S7) consistent with a role for CadC dependent regulation. A more detailed analysis of the docking model also suggests the role of amino acids in CadC that are not directly contacting the DNA (Supplementary Fig. S6), but are still essential for expression of the target operon (Fig. 5). For example, an indirect contribution of Thr69 for DNA-binding is explained by hydrogen-bonds that arrange the His66 and Arg50 side chains, which both are crucial for DNA-binding (Supplementary Fig. S6). Thr69 replacement with alanine resulted in a loss of transcriptional activation of the cadBA operon by CadC (Fig. 5 and Supplementary Fig. S5).
Based on the presence of a weak double-consensus within the Cad1 41-mer we reasoned that CadC would reveal similar binding propensities for a minimal Cad1 26-mer that comprises the two fragments Cad1A and B (Cad1AB) (Fig. 7a and Supplementary Fig. S8). Indeed, SLS shows a clear 2:1 stoichiometry (Supplementary Fig. S8) and both by EMSA (Fig. 7b) and by ITC (Supplementary Fig. S8) we determined a binding constant identical to the 41-mer. Notably, an HSQC overlay of CadC1-107 alone and in complex with Cad1AB shows notable line-broadening (Supplementary Fig. S8). However, the effect is significantly less pronounced than in the NMR titration with Cad1 41-mer DNA. We conclude that the overall CadC1-107 binding affinity and stoichiometry to Cad1 41- and 26-mers is comparable but as the 26-mer DNA leaves less space for sliding we observe a more rigid complex as suggested by improved NMR spectral quality. We next aimed for defining imino proton CSPs of Cad1AB in order to use them as restraints in model building of CadC-Cad1AB (Supplementary Fig. S8). However, while the majority of NMR signals could be assigned in the free DNA, strong signal overlap (as a result of the high AT content) prohibits assignment of imino protons in the bound form. We used the protein CSPs to prepare a HADDOCK model using the same approach as described for the 17-mer complex, which yields a head-to-head arrangement of the two DBDs (Fig. 7c). This model is consistent with all available experimental data and also represented by a SAXS-derived overall shape (Supplementary Fig. S8). Interestingly, it does not indicate a direct interaction of the two DBDs, consistent with the lack of strongly enhanced DNA binding affinity compared to the 1:1 complex. Notably, the 2:1 model provides additional contacts, e.g. for Arg32 and Trp58 of CadC with the DNA that well resemble the mutational and NMR CS data and have not been observed in the model with 17-mer DNA (Fig. 7d).

HADDOCK model of the CadC1-107 structure with the Cad1AB 26-mer DNA. (a) The scheme shows the full Cad1 of the cadBA promoter. The new Cad1AB 26-mer spans the sub-fragments A and B. (b) Left, shown is a representative gel of Cad1AB 26-mer DNAs from panel (a) when titrated up to tenfold excess of CadC1-107. The band of free DNA was used for quantification of the complex formation (right). Binding constant is given as obtained from a triplicate dataset and shown as mean ± standard deviation. (c) Overlay of all ensemble structures within the respective cluster from a HADDOCK modelling of CadC1-107 with Cad1AB 26-mer DNA (see Supplementary Table S3). (d) Contacts of CadC1-107 in complex with Cad1AB 26-mer DNA. Red lines indicate sequence-specific interactions of protein side chains with DNA bases. Black lines are contacts to the DNA backbone, i.e. the phosphate or the sugar groups. Green lines show relevant, sequence-specific protein-protein contacts.
We then compared the CadC-DNA complex model with two recently reported complex structures: the PhoB (RMSD 1.8 Å) and PmrA (RMSD 2.7 Å) DBDs (Fig. 8a and b). A structure-based sequence alignment of the three proteins shows that equivalent positions are used for contacting DNA (Fig. 8c) including the highly conserved residues Arg50, Trp58, Arg76, Thr92, and Tyr98. The three side chains that mediate sequence-specific contacts to the DNA are different in all three proteins (i.e. His66, Gln70 and Lys95 in CadC, see Fig. 8c), which reflects the distinct DNA sequence preferences. Interestingly, the number of residues that mediate sequence-specific interactions is identical in all complexes. However, the PmrA and PhoB DBDs show additional non-sequence-specific contacts involving residues in regions 30–32 and 71–74. Notably, the amino acid sequence for residues 71–74 is distinct from the corresponding regions in PhoB and PmrA and its importance is demonstrated by the observation that single amino acid replacements in this region in CadC also abolish its functional activity.

Structural comparison of DBD - DNA complexes. (a) Superposition of CadC1-107 and PhoB16 as taken from their complex structures. Shown is the recognition helix and the β-wing with analogous residues that are involved in specific interactions with DNA in at least one of the complex structures/model. Residues were selected from the structure-based alignment shown in (c). (b) The same as in (a) but with PmrA as taken from its complex structure with DNA18. (c) Structure-based sequence alignment of the CadC DBD and the DBDs from PhoB and PmrA with residues contacting DNA as indicated. Arrows above the sequence show residues with a functional impact after mutation in CadC (see Fig. 5).
Discussion
Bacterial effector domains in both one- and two-component signalling systems show diverse structural topologies32. The crystal structure of the CadC DBD resembles OmpR effector domains, but, to the best of our knowledge, represents the first structure of an effector domain from the ToxR-like transcriptional regulator family. Available structural and biochemical data demonstrate that dimerization of CadC is essential to mediate its functional activity in the cell in response to acid stress. Dimerization is mediated by the periplasmic domain20 and the transmembrane region19, while the linker is required for transducing the pH-dependent response of the periplasmic sensor into a structural rearrangement that facilitates dimerization of the cytoplasmic CadC DBD21. We have recently shown that the CadC DBD is not per se dimeric. This is confirmed by the monomeric state of the DBD in the crystal and in solution as evidenced by our NMR and SAXS data.
Recent work has shown that ToxR-like transcriptional regulators can bind DNA variably via their effector domains dominated by non-specific interactions, often activating numerous promoter regions33, 34. The CadC-DNA interface determined by our NMR chemical shift perturbation analysis is very similar to other complexes, e.g. PmrA and its target DNA26. Affinities between HTH DNA-binding domains and DNA can span several orders of magnitude depending on the specificity and stoichiometry of interactions17, 18, 26, 34,35,36,37,38. While two-component systems involve dimers of DBDs mediated by their receiver domains, CadC will only act as a dimer when anchored in the membrane. In line with our previous data22, the interaction of CadC1-107 with single motifs in Cad1 fragments is of comparably low (micromolar) affinity. However, the binding affinity is increased with the full-length Cad1 41-mer DNA and a Cad1 26-mer fragment that includes the two AT-rich recognition sites. The affinity of the wild type CadC DBD to its cognate Cad1 DNA (K D = 380 nM as obtained by SPR) is in a typical range for two-site-binding26, 34, 37 although stronger interactions have been seen38. Notably, EMSAs and ITC revealed a 10-fold weaker K D of CadC for the 41-mer and 26-mer DNAs than SPR. Comparing only EMSA experiments, the increase in affinity from 17-mer to 41-mer binding is only about 5-10-fold, which may reflect a lower off-rate of CadC from the 41-mer considering sliding of the protein on the longer DNA. The lower apparent K D derived from the SPR data may be influenced by steady state conditions in the presence of immobilized DNA on a surface, which can overestimate binding affinities but consistently reports about relative affinities in our mutational studies (Fig. 5 and Supplementary Fig. 5). Nonetheless, we observed a comparable low affinity of CadC with Cad1 17-mer DNA by SPR (23 μM), NMR (67 μM) and EMSAs (60–67 μM). Altogether the data indicate that DNA affinity increases with the length of the DNA (due to lower off-rates and enhanced unspecific binding/sliding).
Dimerization of CadC enables the binding of two DBDs to the two Cad1 consensus target sites. Membrane-anchored CadC dimer will thus notably enhance the DNA-binding affinity which underlines an accepted hypothesis of transcriptional activation where multiple binding sites are the key to discrimination between transcription “on” and “off” states39. An affinity increase of CadC with full-length promoter DNA over individual sites was reported previously22 and is likely for other DBD-DNA complexes17, 26. Rearrangement of the transcription initiation complex may involve DNA bending by binding of multiple effector domains and was found for PhoB16, 30, 40. We propose a related mechanism for CadC: Our data show that the CadC DBD alone forms a 2:1 complex with 41-mer DNA increasing affinity. This affinity will be even more enhanced when a dimeric DBD is further stabilized by anchoring CadC in the membrane21.
For CadC in V. vulnificus, the presence of the leucine-responsive regulatory protein (Lrp) enhances the activation of the cadBA promoter through direct interaction of Lrp with DNA. DNA looping then enables protein-protein contacts between CadC and RNAP41. In a similar manner, the presence of multiple binding sites within the cadBA promoter in E. coli could promote cooperative DNA binding by CadC dimers in the membrane and thereby enhance the interaction with RNAP that, however, only takes full effect in stable CadC dimers.
Our structural and mutational analyses identified key residues in helix α3, the β-wing and the TL region to be important for interaction with the DNA and transcriptional initiation. Most surprisingly, charge inversion at residue Glu30 yields a constitutive complex with DNA and pH- and lysine-independent activity, which is expressed in its significantly higher DNA affinity. While CadC1-159-R96A has reasonable affinity for Cad1 in vitro, it fully fails to induce gene expression in vivo. Possibly, the loss in affinity is sufficient to overcome the threshold for transcription initiation by the RNAP. Not surprisingly, alanine replacement of the highly conserved residues Arg50 and Arg76 is not tolerated for DNA-binding or transcriptional activation. The ternary complex of CadC, DNA and RNAP likely requires a strict complex topology with key contacts present between enhancer and DNA.
Although structures of numerous effector domains are known and more than 60% of them have been shown to directly bind DNA32, high-resolution structural information of protein-DNA interactions has remained scarce16,17,18. We did not succeed in crystallizing CadC with Cad1 DNA and previous studies could not describe the target sequence in detail22, 24. Our two complex models provide insight into the DNA recognition by CadC and indicate a preference for AT-rich regions with a significant fraction of non-sequence-specific interactions compared to few sequence-specific ones. Predominantly non-specific interactions between transcriptional activators and DNA have been observed in other DBDs16, 26. The DNA consensus sequence 5′-T-T-A-x-x-x-x-T-3′ is present once in the quasi-palindromic Cad1 17-mer DNA, consistent with the formation of a 1:1 complex. However, a second consensus facilitates the formation of the 2:1 complex of CadC with Cad1 41-mer DNA as evidenced by the CadC model with the minimal Cad1 26-mer DNA that spans the two AT-rich regions, i.e. consensus sites. Therein, the two CadC DBDs form a dimeric head-to-head arrangement in line with the consensus site topology in Cad1. The sequence is also consistent with previous findings that CadC is able to replace the global repressor H-NS from AT-rich regions in the cadBA and cadC promoters42, 43. A sequence alignment of cadBA promoters shows that the AT-rich motif is also present in V. cholerae (Supplementary Fig. S6), consistent with a conserved DNA recognition by CadC in bacteria.
In summary, we propose that the presence of two DBDs by membrane-anchored CadC and a combination of AT-preference and non-sequence specific DNA contacts are important for cooperate DNA-binding and effective regulation of the cadBA promoter. In this respect it is noteworthy that cooperative DNA-binding has been proposed for ToxR-like proteins before17, 26, 37, 44. In addition, the architecture of the DNA-binding sites has a measurable influence on transcriptional control45. The presence of enhancer and promoter DNA guides RNAP to its point of action and thus provides spatiotemporal control of transcription initiation40, 41. Considering that the CadC dimer has a local cellular concentration at the membrane the regulation of cadBA expression may thus be less dependent on overall cellular CadC concentration and specificity-based affinity with target DNA. The formation of high-affinity complexes of CadC dimers with the promoter near the membrane may thus allow for a CadC-specific transcriptional regulation even though the recognition of the promoter does not involve high sequence specificity. Future structural and functional data will help to elucidate the molecular details and broaden our picture of CadC-DNA-RNAP interactions.
Materials and Methods
Cloning, production and purification of recombinant proteins
Expression vectors encoding the two CadC soluble variants CadC1-107 and CadC1-159 were generated by using standard procedures. We used the pETTrx1a vector provided by the Protein Expression and Purification Facility at the Helmholtz Zentrum München. All vectors encoded TEV protease recognition sites for subsequent proteolytic removal of the tags. The two proteins were produced as N-terminal His6-thioredoxin fusion proteins. A single fresh clone of E. coli BL21 DE3 cells was inoculated in lysogenic broth (LB)46 with 35 mg/l kanamycin and grown overnight. 1 ml was used to inoculate an expression culture. Cells were grown to an OD600 of 0.9 at 37 °C, induced with 0.5 mM IPTG and grown overnight at 20 °C before harvesting by centrifugation. Cells were resuspended in lysis buffer (50 mM Tris, 300 mM NaCl, 4 mM tris(2-carboxyethyl)phosphine (TCEP), 15 mM imidazole, 1 mg/ml lysozyme, 10 μg/ml DNaseI, and protease inhibitors, pH 8.0), incubated on ice for 30 min and sonicated. Cleared lysates were subjected to Ni2+-agarose beads. After intensive washing, beads were incubated with 500 µg/l TEV protease in lysis buffer under gentle shaking at room temperature for 3 h. Subsequently, the bead supernatant was collected, concentrated and gel-filtrated in 20 mM 2-[Bisamino]-2-1,3-propanediol (BisTris), 500 mM NaCl, pH 6.5. The respective protein monomer peak was pooled, and the salt concentration adjusted to 150 mM. CadC1-159 proteins were additionally purified via an anion exchange chromatography to remove proteolytic fragments. The protein was buffer exchanged to 20 mM Tris, 50 mM NaCl, and pH 8.0 and loaded onto a 1 ml MonoQ column. A gradient from 50–500 mM NaCl was applied over a volume of 10 ml. Fractions devoid of degradation were pooled and buffer exchanged to conditions as for CadC1-107. To obtain isotope-labelled proteins for NMR studies, cells were grown in M9 minimal medium supplemented with 0.5 g/l 15N ammonium chloride and 2 g/l unlabelled or [U-13C]-glucose. Amino acid replacements in CadC1-159 were introduced as described before21.
DNAs
All DNAs were obtained as HPLC-purified single-stranded oligomers from MWGoperon (Ebersberg, Germany) or Sigma Aldrich (Deisenhofen, Germany). Individual strands were dissolved in water and complement molecules annealed by mixing stoichiometric ratios to a final concentration of 2 mM each and boiling them at 95 °C for 5 minutes. Samples were then cooled down to room temperature over 30 minutes and finally frozen at −80 °C. Sense strand sequences were: 5′-TTGTAAACATTAAATGTTTATCTTTTCATGATATCAACTTG-3′ (comprising base pairs -153 to -113 of the cadBA promoter, designated as Cad1 41-mer WT, see nucleotide exchanges for Cad1 ABmut in Supplementary Fig. S7), 5′-TAAACATTAAATGTTTA-3′ (−144 to −128 bp of the cadBA promoter, designated as Cad1 17-mer), 5′-GTAAACATTAAATG-3′ (Cad1A, 14-mer), 5′-GTTTATCTTTTCA-3′ (Cad1B, 13-mer), 5′-GTAAACATTAAATGTTTATCTTTTCA-3′ (Cad1AB, 26-mer), and 5′-TGATATCAACTTG-3′ (Cad1C, 13-mer).
Crystallization, diffraction data collection and processing
Initial crystallization screening of CadC1-107 was set up at 292 K using 25 mg/ml of protein with a nanodrop dispenser in sitting-drop 96-well plates and commercial screens. Optimization was performed using the sitting-drop vapour-diffusion method at 292 K in 24-well plates. The crystals of CadC1-107 appeared after 14 days and required another 2 weeks to grow to the final size. The best diffracting crystals were obtained from 10 mM MES pH 6.5, 10 mM zinc sulphate, 27.5% (w/v) PEG550 MME. For the anomalous data collection, the native crystals were soaked with [Ta6Br12]2+ × 2 Br− (Jena Bioscience). Native diffraction data was collected on the ID29 beamline (ESRF, Grenoble, France) using a PILATUS 6 M detector at a wavelength of 0.99987 Å. A [Ta6Br12]2+ × 2 Br− derivatized crystal was used for the anomalous data collection at the tantalum absorption edge (1.25363 Å). Both data sets were collected at 100 K at the same beam line. All data sets were indexed and integrated using XDS 47 and scaled using SCALA 48, 49. Intensities were converted to structure factor amplitudes using the program TRUNCATE 49, 50. Supplementary Table S1 summarizes data collection and processing statistics for the two data sets.
Structure determination and refinement
The structure of CadC1-107 was solved using the SAD protocol of Auto-Rickshaw, the EMBL-Hamburg automated crystal structure determination platform51. The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs of the CCP4 suite49. FA values were calculated using the program SHELXC 52. Based on an initial analysis of the data, the maximum resolution for substructure determination and initial phase calculation was set to 2.6 Å. 15 heavy atom positions were located with the program SHELXD 52. The correct hand for the substructure was determined using the programs ABS 53 and SHELXE 52. The occupancy of all substructure atoms was refined using the program MLPHARE 49 and phases improved by density modification using the program DM 49, 54. The initial model was partially built using the program ARP/wARP 55, 56. Further model building and refinement using native data set (2.05 Å) was performed with COOT 57 and REFMAC5 58, respectively, using the maximum-likelihood target function including TLS parameters59. The final model is characterized by R and Rfree factors of 18.3% and 22.3% (Supplementary Table S1). Stereochemical analysis of backbone dihedral angles in the final model using PROCHECK 60 revealed 95.3% of all CadC amino acids in the most favoured region and the remaining 4.7% in allowed regions of the Ramachandran plot. The refined structure and the corresponding structure factor amplitudes were deposited with the PDB under the accession code 5ju7. The crystal contains one molecule of CadC1-107 in the asymmetric unit. Well-defined electron density map corresponds to the region comprising Ala2 – Tyr107. For further analysis and validation, structure based alignments were performed with the DALI server23 using the complete crystal structure. We used 95 protein structures from a total of 56 PDB entries with a Z-score of or higher than 7.2 (i.e. half the maximum Z-score found for the best hit). RMSD values were between 1.6 A and 3.9 A. The 11 best-scored hits (i.e. with a Z-score above 13, but excluding redundant protein species in multiple entries) are listed in Supplementary Fig. S1.
NMR spectroscopy
All NMR measurements of CadC1-107 and CadC1-159 were performed in 20 mM BisTris, 150 mM NaCl, pH 6.5 mixed with 10% (v/v) D2O. Backbone chemical shift assignments of both length versions and side chain assignments of CadC1-107 were recorded at protein concentrations of 1.2 mM (CadC1-107) and 600 μM (CadC1-159), respectively. HNCA, HNCACB, CBCAcoNH, HNCO, HNcaCO, HbHaNNH, HCCH-TOCSY and 3D 15N- and 13C-edited NOESY spectra61 were acquired at 298 K (RT) on BrukerAvance III spectrometers at field strengths corresponding to 600, 800 and 900 MHz proton Larmor frequency, equipped with TCI cryogenic probe heads. Amide 15N R 1 and R 2 relaxation data (e.g. to derive τC values) and steady-state heteronuclear {1H}-15N NOE experiments were performed as described62 at a temperature of 298 K and 600 MHz proton Larmor frequency. Spectra were processed with Topspin3.2 and analysed with CCPNMR Analysis63 and Sparky64.
Titrations of CadC1-107 or CadC1-159 with Cad1 DNA or fragments thereof were carried out at RT with protein concentrations of 100–300 μM in buffers as described above at molar ratios given in the respective figures. Chemical shifts were monitored using 1H-15N HSQC experiments and their perturbations (CSP) were calculated as described previously65. NMR relaxation data of the CadC1-107-Cad1 17-mer complex were measured at the final titration point. DNA imino proton resonances of Cad1 fragments alone and in complex with CadC1-107 were assigned using 2D imino NOESY spectra and SOFAST-HMQC experiments66. All 1D and 2D imino NOESY spectra were recorded using WATERGATE and water-flip-back pulses67, 68 at 600 MHz at a temperature range from 278 to 298 K and at 250 µM DNA concentrations. Spectra displayed in the respective imino proton 1D spectra were recorded at 283 K as unambiguous resonance assignments for DNA alone and in complex with CadC have only been obtained at this temperature. NOE cross peaks in 3D 15N- and 13C-edited NOESY spectra were automatically assigned with the program Cyana369 and manually confirmed.
CadC-DNA complex modelling
The complex of CadC1-107 with Cad1 17-mer DNA was modelled using the HADDOCK server31. We used the protein crystal structure as semi-flexible input. The Cad1-DNA was created with 3DNA70 using the DNA geometry from a PhoB-DNA complex structure16 and was otherwise kept rigid to avoid unfavourable strand melting at the DNA termini. However, with this we have automatically created a set of DNA restraints picked from the input structure (as listed in the HADDOCK dna_rna_restraints.def) including DNA base planarity (true for all), planarity in Watson-Crick base pairs (not restrained), phosphate backbone dihedral angles (restrained as from input), sugar pucker dihedral angles (DNA not restrained to A- or B-form) and Watson-Crick base pairing (true). Despite of limited flexibility we did not restrain the virtual C1’-C1’ bond length. Active nucleotides, i.e. those considered to be directly involved in CadC DBD interactions, were chosen based on the alignment of DNA promoter sequences with high ambiguity. I.e. multiple DNA nucleotides were considered for protein interaction (9, 11, 12, 13, 70, 71, 77, and 78) to allow the protein to move along the DNA double helix. The definition was also based on experimental chemical shift perturbations (CSPs) observed for the AT pairs, but not for the two GC pairs when adding CadC1-107 to Cad1 17-mer DNA (Supplementary Fig. S4). Ambiguous interaction restraints were derived from CSPs, i.e. the 10 most strongly affected amino acids used as active residues: Leu53, Val63, Val68, Thr69, Glu74, Arg76, Thr 92, Lys95, Tyr98, and Lys99. We also included the proximity of Glu30 to the DNA backbone. Water refinement by HADDOCK yielded 170 out of 200 structures in 12 clusters. The largest cluster (26 structures) which also showed least restraints violations and the lowest energy structure was chosen for further analysis. The first four structures in this cluster were among the eight lowest energy structures of all 200 models. The structural complex model was validated by mutational analysis and compared against all experimental CSPs. The complex of CadC1-107 with Cad1AB 26-mer DNA was modelled based on the DNA geometry from PDB entry 4kfc38 which was aligned to the final model of CadC1-107 with Cad1 17-mer DNA. While this arrangement was kept fixed the second CadC1-107 monomer was included with NMR-based restraints as mentioned above. The general settings for the HADDOCK run were identical with the CadC1-107-Cad1 17-mer complex modelling. HADDOCK yielded 9 clusters with 151 structures. The final cluster was ranked with the third best HADDOCK score and showed the lowest RMSD to the overall lowest-energy structure. It yielded the lowest overall enthalpy and contains the highest buried surface area and lowest AIR (ambiguous interaction restraints) violations. HADDOCK statistics for the two complexes are shown in Supplementary Table S3.
SAXS measurements
SAXS measurements were performed on a Rigaku BIOSAXS1000 instrument with a HF007 microfocus generator equipped with a Cu-target at 40 kV and 30 mA. Transmissions were measured with a photodiode beam stop; q-calibration was made by a silver-behenate measurement. Measurements were carried out in multiple 900-second-frames, checked for beam damage and averaged. Circular averaging and background subtraction was done with the Rigaku SAXSLab software v 3.0.1r1. The distance distribution functions P(r) were calculated with GNOM, 3D models were generated using the DAMMIF software within the ATSAS package v 2.5.0-271. Concentrations of samples were as follows: CadC1-107 (200 μM); CadC1-107 + Cad1 17-mer (each 200 μM); CadC1-107 + Cad1 41-mer (2:1: 267 μM + 134 μM); CadC1-107 + Cad1 41-mer (1:1: 160 μM + 162 μM); Cad1 41-mer alone (134 μM); and CadC1-107 + Cad1 26-mer (2:1: 200 μM + 100 μM). Buffers were identical to NMR conditions except for D2O. Molecular weights were calculated from Porod volumes. For complexes, the Porod volume of CadC was subtracted from the total volume and the protein weight calculated via its density. The remaining volume was processed using the density of DNA and finally both molecular weights combined again. CadC1-107-DNA ab initio shape models were created with dummy bead simulated annealing using the DAMMIF software within the ATSAS package v 2.5.0-271. For both the 1:1 model of CadC with Cad1 17-mer and the 2:1 model of CadC with Cad1AB 26-mer DNA we manually fitted the experimentally obtained or HADDOCK-derived structures into the SAXS-derived shape.
Static light scattering
Static Light Scattering (SLS) experiments of CadC1-107 were performed by connecting a Viscotek TDA 305 triple array detector to an Äkta Purifier equipped with an analytical size exclusion column at 4 °C. CadC samples in the presence of half-stoichiometric amounts of Cad1 41-mer or 26-mer DNA were run in gel filtration buffer on a GE Superdex200 10/300 column at 2 mg/ml protein concentration and a flow of 0.5 ml/min. The molecular masses of the samples were calculated from the experimentally determined refractive index and right-angle light scattering signals using Omnisec (Malvern Instruments). The SLS detector was calibrated with a 4 mg/ml BSA solution using 66.4 kDa for the BSA monomer and a dn/dc value of 0.185 ml/g for all protein samples.
Surface Plasmon Resonance spectroscopy (Biacore)
SPR assays were performed in a Biacore T200 (GE Healthcare) using Xantec SAD500-L carboxymethyl dextran sensor chips pre-coated with streptavidin (XanTec Bioanalytics GmbH, Düsseldorf, Germany). All experiments were carried out at a constant temperature of 25 °C using 10 mM HEPES pH 7.4; 150 mM NaCl; 3 mM EDTA; 0.05% (v/v) detergent P20 as running buffer. Before immobilizing the DNA fragments, the chip was equilibrated by three injections using 1 M NaCl/50 mM NaOH at a flow rate of 10 µl/min. Then, 10 nM of the respective double-stranded biotinylated DNA fragment was injected using a contact time of 420 s and a flow rate of 10 µl/min. As a final wash step, 1 M NaCl/50 mM NaOH/50% (v/v) isopropanol was injected. Approximately 400 RU of Cad1-DNA fragment was captured onto flow cell 2. The surface of flow cell 1 was immobilized with similar amounts of yhjX-DNA and used to obtain blank sensorgrams for subtraction of bulk refractive index background. Interaction studies of CadC1-159-WT or the corresponding variants with the Cad1-DNA fragments were performed at 25 °C at a flow rate of 30 µl/min. The proteins were diluted in running buffer and passed over all flow cells in different concentrations (1 nM–100 nM) using a contact time of 180 s followed by a 300 s dissociation time. After each cycle, the surface was regenerated by injection of 2.5 M NaCl for 60 s at 30 µl/min flow rate followed by a second regeneration step with 0.5% (w/v) SDS for 60 s at 30 µl/min at 25 °C. Sensorgrams were recorded and analysed with the Biacore T200 Control software 2.0 and T200 Evaluation software 2.0 (GE Healthcare). The referenced sensorgrams were normalized to a baseline of zero. Peaks in the sensorgrams at the beginning and the end of the injection emerged from the runtime difference between the flow cells of each chip. For the calculation of K D values, steady state affinity curves were used.
ß-galactosidase assays to monitor cadBA expression
The effects of all CadC variants on cadBA promoter activation were determined using a β-galactosidase-based reporter gene assay. For this purpose the reporter strain E. coli EP314 (cadC1::Tn10, cadA-lacZ transcriptional fusion28) was transformed with plasmids encoding CadC or a variant of CadC (pET16b-based). Cells from an overnight culture aerobically grown in KE minimal medium (pH 7.6) supplemented with 0.2% (w/v) glucose72 were inoculated in fresh KE minimal medium containing 0.2% glucose (w/v) and buffered to pH 5.8 or pH 7.6 (100 mM phosphate buffer), and the cell density was adjusted to an OD600 of 0.05. Where indicated, lysine was added to a final concentration of 10 mM. Bacteria were grown to an OD600 of 0.3–0.5 under microaerobic conditions, and harvested by centrifugation. β-Galactosidase assays were performed as described previously73. Enzyme activity was determined from at least three independent replicates and is given in Miller Units74.
EMSAs
Electrophoretic mobility shift assays (EMSAs) were performed by mixing 1.3 μg of Cad1 fragments or 1.9 μg of complete Cad1 with CadC1-107 in molar protein:DNA ratios of 0:1 to 10:1 in 20 mM BisTris and 150 mM NaCl, pH 6.5. Samples were incubated for 20 min at room temperature, mixed with native-PAGE loading buffer and loaded onto 15%- (fragments) or 12%- (Cad1 41-mer/26-mer) native polyacrylamide gels containing 5% (v/v) glycerol. Gels were run at conditions to avoid heating and maintain native complexes and finally stained with SYBRgold®. Quantification of complex formation was achieved by integration of bands with the Bio-Rad® gel documentation system.
ITC
Isothermal titration calorimetry (ITC) was performed with a MicroCal PEAQ-ITC device (Malvern, United Kingdom) in 20 mM BisTris and 150 mM NaCl, pH 6.5. In all experiments protein was titrated from a stock of 10-fold concentration excess to 40 µM Cad1 41-mer WT or ABmut or Cad1AB 26-mer DNA provided in the reaction cell. In a standard ITC run we used 19 injections of 2 µL with 120 seconds spacing at RT with a 750 rpm stirring speed. Raw data were analysed with the integrated software tool and heat production fitted to a one-site binding model. Cad1 41-mer DNA runs were performed in triplicate and the Cad1AB DNA in duplicate.
References
Foster, J. W. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol 2, 898–907 (2004).
Auger, E. A. et al. Construction of lac fusions to the inducible arginine- and lysine decarboxylase genes of Escherichia coli K12. Mol Microbiol 3, 609–20 (1989).
Soksawatmaekhin, W., Kuraishi, A., Sakata, K., Kashiwagi, K. & Igarashi, K. Excretion and uptake of cadaverine by CadB and its physiological functions in Escherichia coli. Mol Microbiol 51, 1401–12 (2004).
Watson, N., Dunyak, D. S., Rosey, E. L., Slonczewski, J. L. & Olson, E. R. Identification of elements involved in transcriptional regulation of the Escherichia coli cad operon by external pH. J Bacteriol 174, 530–40 (1992).
Haneburger, I., Eichinger, A., Skerra, A. & Jung, K. New insights into the signaling mechanism of the pH-responsive, membrane-integrated transcriptional activator CadC of Escherichia coli. J Biol Chem 286, 10681–9 (2011).
Fritz, G. et al. Induction kinetics of a conditional pH stress response system in Escherichia coli. J Mol Biol 393, 272–86 (2009).
Meng, S. Y. & Bennett, G. N. Regulation of the Escherichia coli cad operon: location of a site required for acid induction. J Bacteriol 174, 2670–8 (1992).
DiRita, V. J. & Mekalanos, J. J. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64, 29–37 (1991).
Dalia, A. B., Lazinski, D. W. & Camilli, A. Identification of a membrane-bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae. MBio 5, e01028–13 (2014).
Alam, A., Tam, V., Hamilton, E. & Dziejman, M. vttRA and vttRB Encode ToxR family proteins that mediate bile-induced expression of type three secretion system genes in a non-O1/non-O139 Vibrio cholerae strain. Infect Immun 78, 2554–70 (2010).
Merrell, D. S. & Camilli, A. Regulation of Vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcriptional regulators. J Bacteriol 182, 5342–50 (2000).
Hase, C. C. & Mekalanos, J. J. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci USA 95, 730–4 (1998).
Yang, Y. & Isberg, R. R. Transcriptional regulation of the Yersinia pseudotuberculosis pH6 antigen adhesin by two envelope-associated components. Mol Microbiol 24, 499–510 (1997).
Stelzer, S., Egan, S., Larsen, M. R., Bartlett, D. H. & Kjelleberg, S. Unravelling the role of the ToxR-like transcriptional regulator WmpR in the marine antifouling bacterium Pseudoalteromonas tunicata. Microbiology 152, 1385–94 (2006).
Lassak, K., Peeters, E., Wrobel, S. & Albers, S. V. The one-component system ArnR: a membrane-bound activator of the crenarchaeal archaellum. Mol Microbiol 88, 125–39 (2013).
Blanco, A. G., Sola, M., Gomis-Ruth, F. X. & Coll, M. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10, 701–13 (2002).
Li, Y. C. et al. Structural dynamics of the two-component response regulator RstA in recognition of promoter DNA element. Nucleic Acids Res 42, 8777–88 (2014).
Lou, Y. C. et al. Structure and dynamics of polymyxin-resistance-associated response regulator PmrA in complex with promoter DNA. Nat Commun 6, 8838 (2015).
Lindner, E. & White, S. H. Topology, dimerization, and stability of the single-span membrane protein CadC. J Mol Biol 426, 2942–57 (2014).
Eichinger, A., Haneburger, I., Koller, C., Jung, K. & Skerra, A. Crystal structure of the sensory domain of Escherichia coli CadC, a member of the ToxR-like protein family. Protein Sci 20, 656–69 (2011).
Buchner, S., Schlundt, A., Lassak, J., Sattler, M. & Jung, K. Structural and Functional Analysis of the Signal-Transducing Linker in the pH-Responsive One-Component System CadC of Escherichia coli. J Mol Biol (2015).
Kuper, C. & Jung, K. CadC-mediated activation of the cadBA promoter in Escherichia coli. J Mol Microbiol Biotechnol 10, 26–39 (2005).
Holm, L. & Rosenstrom, P. Dali server: conservation mapping in 3D. Nucleic Acids Res 38, W545–9 (2010).
Rhee, J. E., Kim, K. S. & Choi, S. H. CadC activates pH-dependent expression of the Vibrio vulnificus cadBA operon at a distance through direct binding to an upstream region. J Bacteriol 187, 7870–5 (2005).
Hancock, S. P. et al. Control of DNA minor groove width and Fis protein binding by the purine 2-amino group. Nucleic Acids Res 41, 6750–60 (2013).
Lou, Y. C. et al. Solution structure and tandem DNA recognition of the C-terminal effector domain of PmrA from Klebsiella pneumoniae. Nucleic Acids Res 42, 4080–93 (2014).
Gobl, C., Madl, T., Simon, B. & Sattler, M. NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog Nucl Magn Reson Spectrosc 80, 26–63 (2014).
Neely, M. N., Dell, C. L. & Olson, E. R. Roles of LysP and CadC in mediating the lysine requirement for acid induction of the Escherichia coli cad operon. J Bacteriol 176, 3278–85 (1994).
Makino, K. et al. DNA binding of PhoB and its interaction with RNA polymerase. J Mol Biol 259, 15–26 (1996).
Blanco, A. G., Canals, A. & Coll, M. PhoB transcriptional activator binds hierarchically to pho box promoters. Biol Chem 393, 1165–71 (2012).
Dominguez, C., Boelens, R. & Bonvin, A. M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125, 1731–7 (2003).
Gao, R. & Stock, A. M. Biological insights from structures of two-component proteins. Annu Rev Microbiol 63, 133–54 (2009).
DiRita, V. J. Co-ordinate expression of virulence genes by ToxR in Vibrio cholerae. Mol Microbiol 6, 451–8 (1992).
Gao, R. & Stock, A. M. Temporal hierarchy of gene expression mediated by transcription factor binding affinity and activation dynamics. MBio 6, e00686–15 (2015).
Aravind, L., Anantharaman, V., Balaji, S., Babu, M. M. & Iyer, L. M. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev 29, 231–62 (2005).
Santos-Beneit, F. The Pho regulon: a huge regulatory network in bacteria. Front Microbiol 6, 402 (2015).
Harlocker, S. L., Bergstrom, L. & Inouye, M. Tandem binding of six OmpR proteins to the ompF upstream regulatory sequence of Escherichia coli. J Biol Chem 270, 26849–56 (1995).
Narayanan, A., Kumar, S., Evrard, A. N., Paul, L. N. & Yernool, D. A. An asymmetric heterodomain interface stabilizes a response regulator-DNA complex. Nat Commun 5, 3282 (2014).
Diniz, M. M. et al. Fine-tuning control of phoBR expression in Vibrio cholerae by binding of PhoB to multiple pho boxes. J Bacteriol 193, 6929–38 (2011).
Blanco, A. G., Canals, A., Bernues, J., Sola, M. & Coll, M. The structure of a transcription activation subcomplex reveals how sigma(70) is recruited to PhoB promoters. EMBO J 30, 3776–85 (2011).
Rhee, J. E., Kim, K. S. & Choi, S. H. Activation of the Vibrio vulnificus cadBA Operon by Leucine-responsive regulatory protein is mediated by CadC. J Microbiol Biotechnol 18, 1755–61 (2008).
Shi, X., Waasdorp, B. C. & Bennett, G. N. Modulation of acid-induced amino acid decarboxylase gene expression by hns in Escherichia coli. J Bacteriol 175, 1182–6 (1993).
Krin, E., Danchin, A. & Soutourina, O. Decrypting the H-NS-dependent regulatory cascade of acid stress resistance in Escherichia coli. BMC Microbiol 10, 273 (2010).
Miller, V. L., Taylor, R. K. & Mekalanos, J. J. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48, 271–9 (1987).
Park, D. M. & Kiley, P. J. The influence of repressor DNA binding site architecture on transcriptional control. MBio 5, e01684–14 (2014).
Bertani, G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62, 293–300 (1951).
Kabsch, W. Xds. Acta Crystallogr D Biol Crystallogr 66, 125–32 (2010).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62, 72–82 (2006).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–42 (2011).
French, S. & Wilson, K. On the Treatment of Negative Intensity Observations. Acta Crystallogr A 34, 517–525 (1978).
Panjikar, S., Parthasarathy, V., Lamzin, V. S., Weiss, M. S. & Tucker, P. A. Auto-rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D Biol Crystallogr 61, 449–57 (2005).
Sheldrick, G. M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66, 479–85 (2010).
Hao, Q. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. J. Appl. Cryst. 37, 498–499 (2004).
Cowtan, K. D. & Zhang, K. Y. Density modification for macromolecular phase improvement. Prog Biophys Mol Biol 72, 245–70 (1999).
Morris, R. J. et al. Breaking good resolutions with ARP/wARP. J Synchrotron Radiat 11, 56–9 (2004).
Perrakis, A., Harkiolaki, M., Wilson, K. S. & Lamzin, V. S. ARP/wARP and molecular replacement. Acta Crystallogr D Biol Crystallogr 57, 1445–50 (2001).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–55 (1997).
Winn, M., Isupov, M. & Murshudov, G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57, 122–133 (2000).
Laskowski, R., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 (1993).
Sattler, M., Schleucher, J. & Griesinger, C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progress in Nuclear Magnetic Resonance Spectroscopy 34, 93–158 (1999).
Farrow, N. A. et al. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 (1994).
Vranken, W. F. et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–96 (2005).
Goddard, T. D. & Kneller, D. G. SPARKY 3. University of California, San Francisco.
Schlundt, A. et al. Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat Struct Mol Biol 21, 671–8 (2014).
Schanda, P., Kupce, E. & Brutscher, B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR 33, 199–211 (2005).
Grzesiek, S. & Bax, A. The importance of not saturating H2O in protein NMR - application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 115, 12593–12594 (1993).
Piotto, M., Saudek, V. & Sklenář, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661 (1992).
Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol Biol 278, 353–78 (2004).
Zheng, G., Lu, X. J. & Olson, W. K. Web 3DNA–a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res 37, W240–6 (2009).
Petoukhov, M. V. et al. New developments in the ATSAS program package for small-angle scattering data analysis. Journal of Applied Crystallography 45, 342–350 (2012).
Epstein, W. & Kim, B. S. Potassium transport loci in Escherichia coli K-12. J Bacteriol 108, 639–44 (1971).
Tetsch, L., Koller, C., Haneburger, I. & Jung, K. The membrane-integrated transcriptional activator CadC of Escherichia coli senses lysine indirectly via the interaction with the lysine permease LysP. Mol Microbiol 67, 570–83 (2008).
Miller, J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY (1972).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Res 36, W5–9 (2008).
Acknowledgements
We acknowledge the use of the X-ray crystallography platform and the protein expression and purification facility of the Helmholtz Zentrum München, the Bavarian NMR Center, and SAXS measurements at the facility of the SFB1035, Technische Universität München. SPR analyses were performed in the Bioanalytics core facility of the LMU Biocenter. This work was supported by the Deutsche Forschungsgemeinschaft (Exc114/1 and Exc114/2). S.B. obtained financial support by the Women program of the Center for Integrated Protein Science Munich. In addition, A.S. and K.J. acknowledge support by the Deutsche Forschungsgemeinschaft (SCHL2062/1-1) and TRR174/1, project 9.
Author information
Authors and Affiliations
Contributions
A.S. and T.H. performed protein expression, NMR experiments and crystallisation screens. R.J. collected X-ray data and determined the structure. A.S. performed EMS assays and HADDOCK modelling. S.B. and J.L. carried out reporter assays. R.H. accomplished SPR experiments. R.S. collected SAXS data. A.G. and A.S. performed SLS runs. A.S., S.B., K.J., and M.S. designed experiments and wrote the manuscript. A.S. prepared the figures. All authors discussed data.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession Numbers: Atomic coordinates of CadC1-107 are deposited in the protein data bank under PDB entry 5JU7. NMR chemical shifts of CadC1-159 have been deposited in the BMRB under accession code 25417.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schlundt, A., Buchner, S., Janowski, R. et al. Structure-function analysis of the DNA-binding domain of a transmembrane transcriptional activator. Sci Rep 7, 1051 (2017). https://doi.org/10.1038/s41598-017-01031-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01031-9
This article is cited by
-
Division of labor and collective functionality in Escherichia coli under acid stress
Communications Biology (2022)
-
Acid resistance system CadBA is implicated in acid tolerance and biofilm formation and is identified as a new virulence factor of Edwardsiella tarda
Veterinary Research (2021)
-
The enigma of environmental pH sensing in plants
Nature Plants (2021)
-
Disulfide-compatible phage-assisted continuous evolution in the periplasmic space
Nature Communications (2021)
-
Proline codon pair selection determines ribosome pausing strength and translation efficiency in bacteria
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
