
Abstract
Retinitis pigmentosa (RP) is highly heterogeneous in both clinical and genetic fields. Accurate mutation screening is very beneficial in improving clinical diagnosis and gene-specific treatment of RP patients. The reason for the difficulties in genetic diagnosis of RP is that the ethnic-specific mutation databases that contain both clinical and genetic information are largely insufficient. In this study, we recruited 98 small Han Chinese families clinically diagnosed as RP, including of 22 dominant, 19 recessive, 52 sporadic, and five X-linked. We then used whole exome sequencing (WES) analysis to detect mutations in the genes known for RP in 101 samples from these 98 families. In total, we identified 57 potential pathogenic mutations in 40 of the 98 (41%) families in 22 known RP genes, including 45 novel mutations. We detected mutations in 13 of the 22 (59%) typical autosomal dominant families, 8 of the 19 (42%) typical autosomal recessive families, 16 of the 52 (31%) sporadic small families, and four of the five (80%) X-linked families. Our results extended the mutation spectrum of known RP genes in Han Chinese, thus making a contribution to RP gene diagnosis and the pathogenetic study of RP genes.
Similar content being viewed by others

Introduction
Retinitis pigmentosa (RP, OMIM#268,000) is caused by abnormalities of the photoreceptors (rods and cones) or the retinal pigment epithelium (RPE) of the retina, and results in progressive vision loss1. RP is an inherited degenerative eye disease that causes severe vision damage and often results in blindness1. Affected individuals may experience difficulties in light-to-dark and dark-to-light adaptation or night blindness at the early stage of RP. RP is likely the most common type of retinal dystrophy. The worldwide prevalence of non-syndromic RP is approximately 1 in 40002. The prevalence of non-syndromic RP in China had been reported at 1 in 38003.
RP exhibits autosomal dominant (adRP), autosomal recessive (arRP), or X-linked (xlRP) models. In very rare cases, the cause is a digenic pattern of inheritance. Non-systemic RP represents about 70–80% of all cases4. Autosomal dominant, autosomal recessive, and X-linked account for approximately 30–40%, 50–60%, and 5–15% respectively of patients with RP2. Approximately 30% are sporadic cases4, most of which may belong to the autosomal recessive inheritance group.
RP genetics are complicated and heterogeneous. To date, 27 autosomal dominant, 58 autosomal recessive, and three X-linked RP genes have been identified in the RetNet database (http://www.sph.uth.tmc.edu/retnet/). Among these genes, six—BEST1, NR2E3, NRL, RHO, RP1, and RPE65—can cause both autosomal dominant and autosomal recessive RP. In addition, mutations in several genes, including ABCA4 5, PROM1 6, PRPH2 7, C8orf37 8, and PRPF31 9, can cause both RP and macular degeneration.
Because of the highly genetic heterogeneity of RP, an accurate genetic diagnosis is needed to improve clinical diagnosis10. In recent years, whole exome sequencing (WES) has been used for the molecular diagnosis of Mendelian diseases11. Although similar studies in RP have been published in the last few years, most of these reports were focused on the Caucasian population. Published RP mutations in the Chinese population are rare in the Human Gene Mutation Database (HGMD, http://www.hgmd.org/) and the Online Mendelian Inheritance in Man (OMIM, http://omim.org/). Different populations may have different mutation spectra, which is very important in studying the origin and pathogenesis of heterogeneous diseases such as RP. In this study, we investigated the mutations of known RP genes in 101 patients in 98 small Han Chinese RP families, which is beneficial for RP gene diagnosis and the pathogenic study of RP.
Methods
Ethics statement
This project was approved by the Ethics Committee of the Hospital of the University of Electronic Science and Technology of China and Sichuan Provincial People’s Hospital, Chengdu, Sichuan, China; and the ethics committee of Xinhua Hospital, Shanghai Jiao Tong University, Shanghai, China. Written informed consent was obtained from all participants involved in this study. All experiments were performed in accordance with relevant guidelines and regulations, including any relevant details.
Study subjects
Complete histories, pedigree analysis, and ophthalmic examinations were performed when sampling. Eye exams consisted of cycloplegic refractions, fixation testing, Snellen visual acuities (when possible), pupillary responses, slit lamp exams, dilated fundus exam by indirect ophthalmoscopy, retinal photography, and Goldmann visual field analyzer and Humphrey field analyzer testing (when possible).
Electroretinograms (ERGs) were performed according to International Society for Clinical Electrophysiology of Vision standards. Patients with a rod-specific b wave that was reduced or undetectable and a color fundus photo of the eye with intraretinal pigment migration were included in this study. The criteria for defining RP in the families were based on the probands’ descriptions of the features of their family members, such as poor vision and night blindness, and then confirmed by clinical examination. DNA samples were extracted from whole blood. The concentration of DNA was determined by using a Nano Drop spectrophotometer.
WES experiments and data analysis
We sequenced 101 RP patients using the Illuminan Truseq Enrichment System Capture, following the manufacturer’s instructions on the Illumina HiSeq 2500 platform. The sequencing reads were mapped against UCSC hg19 (http://genome.ucsc.edu) by BWA (http://bio-bwa.sourceforge.net/). Individual sample single nucleotide polymorphisms (SNPs) and insertion or deletion events (indels) were detected by SAMTOOLS (http://samtools.sourceforge.net/). After generating initial single non-synonymous variant (SNV) calls, we performed further filtering to identify high-confidence variants that (i) had quality >Q30 and depth of ≥5× and (ii) were not located in the major histocompatibility complex homologous sequence.
WES data from 1000 Genomes (http://browser.1000genomes.org/index.html), dbSNP135-common, the ExAC database (http://exac.broadinstitute.org/), and unrelated individuals of 2020 in-house non-RP controls were used as reference data for variant filtering. Prediction of potential functional consequences of variants was conducted using SIFT and PROVEAN (http://sift.jcvi.org/www/SIFT_chr_coords_submit.html)12, and Polymorphism Phenotyping v2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/)13.
Autosomal recessive, autosomal dominant, X-linked, and digenic heredity models were all considered in this study. The mutations were filtered with the following multiple-step bioinformatics analysis: (1) the SNPs and short indels in the exome region were filtered against data from 1000 genome, dbSNP135-common, ExAC and unrelated individuals of 2020 in-house non-RP controls, removing minor allele frequency (MAF) values that were greater than 0.005 for recessive model and any frequency for dominant model; (2) excluded non-coding variants without altering splicing sites; (3) excluded the synonymous variants without altering splicing sites in the genes; (4) excluded missense variants predicted to be Neutral/Tolerated /Benign by PROVEAN, SIFT and Polyphen-2 simultaneously.
Polymerase chain reaction and direct Sanger sequencing for variant confirmation
If pathogenic mutations were found, further Sanger validation and segregation were carried out. Primers were designed (Primer3, http://biotools.umassmed.edu/bioapps/primer3_www.cgi) to use polymerase chain reaction (PCR) amplification on the 400–500 bp region flanking the mutation. To ensure high-quality Sanger sequencing, the amplification was designed to have a boundary at least150 bp away from the mutation base. The amplification was then Sanger sequenced on an Applied BioSystems 3730 capillary sequencer (Waltham, MA, USA). The Sanger sequencing results were analyzed with Applied BioSystems’s Sequencer software. The samples of RP family members were also sequenced by Sanger sequencing to perform segregation analysis, and clinical reevaluation (if necessary) were carried out after mutations found. We defined variants as “compound heterozygous” in a patient when we detected the patient’s father and mother each carrying a heterozygous mutation or the direct relatives without RP only carrying a heterozygous mutation. Variants were excluded when the exactly same variants were detected in the relative who did not diagnosed with RP phenotype. When RP patients’ mutations were not detected in their biological parents, we defined these mutations as “de novo”. We defined a variant as “novel” if it had not been reported in the literature or registered in HGMD and OMIM databases.
Results
Whole exome sequencing of 101 RP patients in 98 RP families
We recruited 98 families clinically diagnosed with RP, which included 22 adRP families, 19 arRP families, 52 sporadic small families, and five xlRP families. To obtain all the variants in the coding regions, we performed WES for 101 patients (two samples were sequenced in three families, and one sample was sequenced in each of the remaining 95 families), with an average of 9.6 Gb of sequence with 96× coverage of average throughput depth of exome target regions for each individual, leading to a total of 101 paired-end, base pair reads. The summary of the average output of each sample is shown in Supplementary Table 1.
Since RP is a rare Mendelian disease, variants with a frequency <0.5% for the recessive model10 and no frequency for the dominant model were kept for further consideration. We first searched for variants of the 77 known RP genes in the RetNet database of autosomal dominant, autosomal recessive, and X-linked RP genes. Then, to identify potential pathogenic mutations among rare variants in each patient, we searched for variants that matched the reported inheritance pattern of the respective genes: novel or previously reported heterozygous mutations in dominant RP genes, homozygous or compound heterozygous variants in recessive RP genes, and homozygous or heterozygous variants in X-linked RP genes. Missense variants predicted to be pathogenic by at least one predictor were kept for segregation analysis: deleterious by PROVEAN, damaging by SIFT or probably damaging by Polyphen-2. All identified potential pathogenic variants were then validated by Sanger sequencing within families. Segregation analysis was performed within family members.
Putative pathogenic mutations in known RP genes
We identified possible pathogenic mutations for 40 of the 98 (41%) families, which contained 57 mutations in 22 of the 77 known RP genes (Table 1, Fig. 1). The 57 mutations included 12 reported mutations: p.Asn247Ile14, p.Glu1122Lys15, 16, p.Ala1773Val17, and p.His55Arg18 in ABCA4; p.Arg526*19 in CRB1; p.Cyc2139Tyr20 in EYS; p.Gly122Asp21 in CRX; p.Asp311Asn22 in IMPDH1; p.Pro347Leu23 in RHO; p.Arg1653*24, c.8559-2A > G)25, and p.Cys934Trp26 in USH2A (Table 1). Thirteen of the 22 (59%) dominant families, eight of the 19 (42%) autosomal recessive families, 16 of the 52 (31%) of the sporadic small families, and four of the five (80%) of the X-linked families had identified mutations. A summary of the patients’ clinical diagnoses and presentations is shown in Supplementary Table 2.

Mutation proportions of individual genes in the 98 small RP families. Mutation proportions of individual genes in the 98 small RP families were shown. The proportion of uncertain genes (with conserved amino acid prediction results and with low frequencies, Table 2), other retinal genes (mutations in other retinopathy genes, Table 3) and unknown genes were also shown.
In the 22 known RP genes, there were six families in which compound heterozygous mutations were detected in USH2A (6%) (Table 1, Fig. 1), which was the most frequently mutated gene in the families under study. They had all been diagnosed as simple RP patients before we found mutations. Among these six families, family RP-028 had exon 44:c.8559-2A > G25 and p.Val4171Gly mutations; family RP-033 had p.Gly4384Val and p.Val2228Glu mutations; family RP-116 had p.Gln4541* and p.Arg4137* mutations; patient RP-063 had p.Arg1653* and p.Cys934Trp26 mutations; patient RP-076 had c.6326_6331delATTTAG (p.Asp2109_Leu2110del) and p.Pro2730Ser mutations; and patient RP-166 had p.Ser1136Asn and p.Gly2317Asn mutations. After mutations were detected, we revisited and re-diagnosed patient RP-116 as Usher type II, while the other patients had no obvious hearing problems.
Mutations in RP1 accounted for 4%; families RP-008 and RP-153 showed heterozygous mutations in the RP1 gene (p.Thr1922Ala and p.Ser1771Pro respectively). No parents in RP-008 and RP-153 carried the mutations, and all biological parents were excluded as RPs by eye examination, suggesting that these two mutations were de novo. Patient RP-012 showed homozygous c.788_790delTAA and p.Ile263del mutations, and patient RP-096 showed compound heterozygous mutations of p.Gln732* and p.Asn494Lys.
RPGR accounted for 3% of all the probands. Three families displayed hemizygote mutations in the RPGR gene of which RP-054 and RP-138 showed frameshift deletion mutations p.Glu746Argfs*23 and p.Gly494Glufs*7 respectively.
ABCA4 showed a 3% frequency in the investigated families. Patients RP-047, RP-070, and RP-134 showed compound heterozygous mutations in the ABCA4 gene. For patient RP-047, p.Asn247Ile14 and p.Thr2028Ile mutations were detected. Patient RP-070 had p.Trp499* and p.Glu1122Lys15, 16 mutations, and patient RP-134 had p.Ala1773Val17 and p.His55Arg18 mutations; these two patients were reassessed as cases of Stargardt macular dystrophy (STGD).
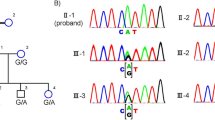
PRPF3 showed a 3% frequency in the investigated families. In PRPF31, the same mutation p.Gln411Glyfs*63 was identified in two families (RP-098 and RP-126). This mutation causes the protein to be pre-stopped at amino acid 474 (499 AAs for the wild-type PRPF31 protein), which might result in nonsense mediated decay. The clinical fundus pictures, DNA sequencing tracing, and analysis of amino acid conservation for this mutation are shown in Fig. 2. This frameshift deletion is believed to be the causative mutant of adRP.

The phenotypes and mutation of patient RP-126 who carries a heterozygous mutation in the PRPF31 gene. (A,B) Color fundus photograph (A) and while and black fundus photograph (B) of patient RP-126 shows a prominent multilobulated central atrophic maculopathy surrounded by concentric rings of black deposits. (C) Optical coherence tomography images of patient RP-126. (D) Vision field diagram of patient RP-126 shows the obvious vision loss (Humphrey automated threshold perimetry, Program 30-2). (E) ERG recording of A and B waves of patient RP-126 (30 µV/D, 25 ms/D). (F) Pedigree of RP-126 family. (G) The Sanger sequencing tracing of the mutation detected in the RP-126 family (PRPF31: NM_015629:c.1231_1232delCA, p.Gln411Glyfs*63).
Mutations in PROM1, PRPF6, RDH12, EYS, and SNRNP200 showed a 2% frequency in the families studied. The mutations in the rest of the genes—BEST1, CRB1, CRX, IMPDH1, MERTK, RHO, C8orf37, RP2, FSCN2, and ROM1—showed only a 1% frequency in the 98 investigated families.
Of the 98 families, only RP-62 exhibited two novel mutations in two RP genes (RDH12, p.Val41Leu and SNRNP200, p.Arg2009Cys) (Table 1). Amino acid conservation analysis by PROVEAN, SIFT, and Polyphon-2 suggests that the p.Arg2009Cys is more likely to be the causative mutation in this family; however, we could not exclude the possibility of the two mutations working together to cause the disease phenotype in this family at this stage.
We may exclude the dominant mutations when we filter the variants in our control datasets if the non-RP controls show RP onset at the late adult stage after sampling. To avoid excess exclusion of possible causative mutations, we analyzed the heterozygous variants in sporadic RPs when the minor allele frequency was less than 0.5% in our in-house dataset; we then analyzed the amino acid conservation feature by using the PROVEAN and SIFT prediction methods. Next, we looked up the detected variants’ frequencies in the ExAC database of 60,706 unrelated individuals. We found three variants in PRPF4 and RDH12 genes with conserved amino acid prediction results and low frequencies (Table 2); these three variants may also be causative.
We also analyzed the non-synonymous variants’ spectrum in the 77 genes known to be responsible for RP in the 101 RP samples and the 2020 in-house controls and found that about 30% of the known RP genes showed a conserved feature because very few non-synonymous variants could be detected either in RPs or in controls, including PRPF3, TOPORS, PRCD, ZNF513, RP2, PDE6G, DHX38, ARL6, CRX, IDH3B, LRAT, SNRNP200, IMPDH1, PRPF6, PRPF31, GUCA1B, ARL2BP, PRPF8, NRL, and CLRN1. In our in-house control dataset, the average count of non-synonymous variants of the RP genes was about 73 (3.9 variants per 100 sample); however, the RP genes listed above showed less than three non-synonymous variant counts in all samples (0.15 variants per 100 sampled). These results suggest that these genes may play very basic roles in human health in addition to their roles in the causation of RP.
Mutations in other retinal disease genes
RP is one of the most commonly inherited retinopathy diseases and is highly heterogeneous. Mutations in other retinopathy genes might also cause the RP phenotype. Thus, we searched for mutations in another 161 retinopathy-related genes in the RetNet database. We detected eight genes that showed mutations in 11 sporadic samples (Table 3, Fig. 1). Mutations in seven genes (KCNV2, HMCN1, CYP4V2, COL11A1, CAPN5, CACNA1F, and ADAMTS18) typically cause only simple symptoms of RP, while patients with mutations in IFT140 showed RP plus cataract (onset at 7 years of age). Three mutations in CYP4V2 were detected in three samples (p.Trp340*, c.1091-2A > G27 (splicing), and p.Lys376Glu). RP-064 and RP-131 were detected as having the same compound heterozygous mutations (p.Trp340* and c.1091-2A > G)27. CYP4V2 gene encodes a polypeptide of cytochrome P450 hemethiolate protein superfamily involved in oxidizing substrates in the metabolic pathway28. Mutations in this gene can also cause Bietti crystalline corneoretinal dystrophy in the recessive inheritance model29. Typical features of this disease include crystals scattered over the fundus, degeneration of the retina, and sclerosis of the choroid vessels, ultimately causing night blindness and vision loss30. Although the same mutations were detected in the present study in sporadic patients RP-064 and RP-131, both of whom were 25 years of age, different phenotypes were observed. Patient RP-064 was diagnosed with a syndrome causing lens opacity in both eyes. This patient developed the mottled appearance of the RPE caused by bone spicule formation, a waxy appearance of the optic nerve, and thin-walled blood vessels in the retina in both eyes. However, patient RP-131 showed only bone spicule formation in the retinas of both eyes, similar to another patient, RP-074, who had a CYP4V2 p.Lys376Glu mutation. No syndrome was observed in either of these patients. Collectively, mutations in these genes may cause the heterogeneity in the clinical phenotypes of eye diseases.
Discussion
RP is the term given to a set of hereditary retinal diseases that feature degeneration of rod and conephotoreceptors2; these diseases typically present with poor night vision (due to rod dysfunction) in early or middle life. The condition of RP patients progresses to the loss of the mid-peripheral field of vision, which gradually extends and causes a small central island of vision due to the preservation of macular cones2. RP is one of the two main causes of blindness in those aged 20 to 6431. RP is a highly heterogeneous genetic disease. So far, mutations in 77 genes that can cause RP have been found in the RetNet database, and the number is increasing. In our WES data of 98 small RP families, we detected mutations in the genes known for RP in 40 (40.8%) families. It appears that the detected mutation rate might be higher among the larger pedigrees. In the present study, we detected mutations in known RP genes in 59% of the dominant inheritance families, 42% of the recessive families, 80% of the X-linked families, and 31% of the sporadic small families. This mutation detection rate is consistent with previous research on a Chinese RP cohort32. Most of the genes that cause inherited PR degeneration contribute to a small fraction of cases. Consistent with previous reports, the most common single genes that were found to cause RP in this study are USH2A 2, RPGR 33, ABCA4 34, and RP1 35. The frequency of the mutations that were detected in RHO was lower than in Caucasians (8–25%)2, 4, 36, but was similar to reports in Japanese (∼1%)37 and Indians (∼1%)38. Other genes caused only a small proportion of cases. The genetic diversity of RP presents challenges for clinical therapy, but molecular diagnosis would provide valuable information in gene replacement therapy39, 40.
In sporadic small families, which account for the vast majority of RP patients, it is difficult to confirm mutations, either for known RP genes or for the discovery of new RP genes, because co-segregation analysis cannot be performed. The possible reasons for the lower mutation rate detection of sporadic patients may include the fact that many genes responsible for sporadic RP have not yet been identified41, or the fact that non-genetic factors such as environmental phenocopies may easily occur in the case of sporadic RP36, 42. In our arRP families with detected known RP gene mutations in China, half were homozygotes, and the rest were compound heterozygous mutations. In sporadic patients, most were compound heterozygous mutations, which may be related to rarity of the phenomenon of close-relative marriages among the Han Chinese in China.
In this study, we used the WES method to examine the genetic etiology of RPs. We identified 57potential mutations in known RP genes, among which 12 are recurrent mutations and 45 are mutations first reported in this study. It is difficult to confirm a new missense mutation to be pathogenetic without in vivo and in vitro functional studies such as knock-in in animal models, a task that is beyond the scope of this study. Here, a multistep bioinformatics analysis strategy was utilized to systematically identify the putative pathogenic mutations for each of the 98 families (see method section). Our results extended the mutation spectrum of known RP genes, which will be beneficial for molecular RP diagnosis in the future. Our experience in demonstrating the high diagnostic yield by WES, coupled with its role in discovering etiologies, supports the use of WES in retinal disease practices. Although our diagnostic evaluation is a forward genetics approach, we hypothesize that it will be pivotal for future use in personalized genomic medicine and individualized RP treatments.
References
Hamel, C. Retinitis pigmentosa. Orphanet J Rare Dis 1, 40 (2006).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Hu, D. N. Prevalence and mode of inheritance of major geneticeye diseases in China. Journal of Medical Genetics. 24, 584–588 (1987).
Ferrari, S. et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 12, 238–249 (2011).
Sun, H., Smallwood, P. M. & Nathans, J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet 26, 242–246 (2000).
Yang, Z. et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest 118, 2908–2916 (2008).
Ali, R. R. et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet 25, 306–310 (2000).
van Huet, R. A. et al. Clinical characteristics of rod and cone photoreceptor dystrophies in patients with mutations in the C8orf37 gene. Invest Ophthalmol Vis Sci 54, 4683–4690 (2013).
Lu, F. et al. A novel PRPF31 mutation in a large Chinese family with autosomal dominant retinitis pigmentosa and macular degeneration. PLoS One 8, e78274 (2013).
Wang, X. et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. Journal of medical genetic (2013).
Srivastava, S. et al. Clinical whole exome sequencing in child neurology practice. Annals of neurology 76, 473–483 (2014).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249 (2010).
Rivera, A. et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. American journal of human genetics 67, 800–813 (2000).
Lewis, R. A. et al. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. American journal of human genetics 64, 422–434 (1999).
Fukui, T. et al. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Investigative ophthalmology & visual science 43, 2819–2824 (2002).
Stenirri, S. et al. Are microarrays useful in the screening of ABCA4 mutations in Italian patients affected by macular degenerations? Clinical chemistry and laboratory medicine: CCLM/FESCC 46, 1250–1255 (2008).
Downs, K. et al. Molecular testing for hereditary retinal disease as part of clinical care. Archives of ophthalmology 125, 252–258 (2007).
Seong, M. W. et al. Molecular characterization of Leber congenital amaurosis in Koreans. Molecular vision 14, 1429–1436 (2008).
Audo, I. et al. EYS is a major gene for rod-cone dystrophies in France. Hum Mutat 31, E1406–1435 (2010).
Zernant, J. et al. Genotyping microarray (disease chip) for Leber congenital amaurosis: detection of modifier alleles. Investigative ophthalmology & visual science 46, 3052–3059 (2005).
Bowne, S. J. et al. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Human molecular genetics 11, 559–568 (2002).
Sohocki, M. M. et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Human mutation 17, 42–51 (2001).
Dreyer, B. et al. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat 29, 451 (2008).
Nakanishi, H. et al. Identification of 11 novel mutations in USH2A among Japanese patients with Usher syndrome type 2. Clin Genet 76, 383–391 (2009).
Xu, W. et al. Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis 17, 1537–1552 (2011).
Wang, Y. et al. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with retinitis pigmentosa. PLoS One 7, e33673 (2012).
Mackay, D. S. & Halford, S. Focus on molecules: cytochrome P450 family 4, subfamily V, polypeptide 2 (CYP4V2). Exp Eye Res 102, 111–112 (2012).
Li, A. et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet 74, 817–826 (2004).
Okialda, K. A., Stover, N. B., Weleber, R. G. & Kelly, E. J. In GeneReviews(R) eds Pagon, R. A. et al. (1993).
Buch, H. et al. Prevalence and causes of visual impairment and blindness among 9980 Scandinavian adults: the Copenhagen City Eye Study. Ophthalmology 111, 53–61 (2004).
Xu, Y. et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum Genet 133, 1255–1271 (2014).
Breuer, D. K. et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet 70, 1545–1554 (2002).
Maugeri, A. et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 67, 960–966 (2000).
Wang, D. Y. et al. Gene mutations in retinitis pigmentosa and their clinical implications. Clin Chim Acta 351, 5–16 (2005).
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M. & Bhattacharya, S. S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet 11, 273–284 (2010).
Fujiki, K. et al. Missense mutation of rhodopsin gene codon 15 found in Japanese autosomal dominant retinitis pigmentosa. Jpn J Hum Genet 40, 271–277 (1995).
Gandra, M. et al. Retinitis pigmentosa: mutation analysis of RHO, PRPF31, RP1, and IMPDH1 genes in patients from India. Mol Vis 14, 1105–1113 (2008).
Smith, A. J., Bainbridge, J. W. & Ali, R. R. Prospects for retinal gene replacement therapy. Trends Genet 25, 156–165 (2009).
Chung, D. C., Lee, V. & Maguire, A. M. Recent advances in ocular gene therapy. Curr Opin Ophthalmol 20, 377–381 (2009).
Xu, Y. et al. Mutation analysis in 129 genes associated with other forms of retinal dystrophy in 157 families with retinitis pigmentosa based on exome sequencing. Mol Vis 21, 477–486 (2015).
Boughman, J. A. & Fishman, G. A. A genetic analysis of retinitis pigmentosa. Br J Ophthalmol 67, 449–454 (1983).
Acknowledgements
We would like to thank all the RP patients and their families for participating in this study. This research project was supported by: National key scientific research program (2016YFC0905200, Z.Y.), the National Natural Science Foundation of China (81170883and 81430008 (Z.Y.), 81300802 and 81670895 (L.H.), 81271048 (J. Y.), 81570848 (C.Q) and 81100693 (C.Q.); the Department of Science and Technology of Sichuan Province, China (2014SZ0169, 2015SZ0052 (Z.Y.), 2015JZ0004 (C.Q.), 2015JQO057 (L.H.), 2017JQ0024 (L.H.), 2016HH0072 (L.H.), 2015SZ0060 (Y.L.) and 2013JY0195 (L.H.).
Author information
Authors and Affiliations
Contributions
Z.Y. designed the study. P.Z., Q.Z., X.H., C.Q., S.M. and Z.Y. recruited the participants. L.H., Y.M., C.T., J.Y., Y.L., F.L. and Z.Y. performed the genotyping. L.H. and Z.Y. performed the statistical analysis. L.H. wrote the initial draft, with edits from Z.Y. All authors critically revised and gave final approval of this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, L., Zhang, Q., Huang, X. et al. Mutation screening in genes known to be responsible for Retinitis Pigmentosa in 98 Small Han Chinese Families. Sci Rep 7, 1948 (2017). https://doi.org/10.1038/s41598-017-00963-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00963-6
This article is cited by
-
Inherited retinal disorders: a genotype–phenotype correlation in an Indian cohort and the importance of genetic testing and genetic counselling
Graefe's Archive for Clinical and Experimental Ophthalmology (2023)
-
Whole-exome sequencing in 168 Korean patients with inherited retinal degeneration
BMC Medical Genomics (2021)
-
Retinitis pigmentosa is associated with shifts in the gut microbiome
Scientific Reports (2021)
-
Multi-level evidence of an allelic hierarchy of USH2A variants in hearing, auditory processing and speech/language outcomes
Communications Biology (2020)
-
EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
