
Abstract
During Schizosaccharomyces pombe meiotic prophase, homologous chromosomes are co-aligned by linear elements (LinEs) analogous to the axial elements of the synaptonemal complex (SC) in other organisms. LinE proteins also promote the formation of meiotic DNA double-strand breaks (DSBs), the precursors of cross-overs. Rec10 is required for essentially all DSBs and recombination, and three others (Rec25, Rec27, and Mug20) are protein determinants of DSB hotspots – they bind DSB hotspots with high specificity and are required for DSB formation there. These four LinE proteins co-localize in the nucleus in an interdependent way, suggesting they form a complex. We used random mutagenesis to uncover recombination-deficient missense mutants with novel properties. Some missense mutations changed essential residues conserved among Schizosaccharomyces species. DSB formation, gene conversion, and crossing-over were coordinately reduced in the mutants tested. Based on our mutant analysis, we revised the rec27 open reading frame: the new start codon is in the previously annotated first intron. Genetic and fluorescence-microscopy assays indicated that the Rec10 N- and C-terminal regions have complex interactions with Rec25. These mutants are a valuable resource to elucidate further how LinE proteins and the related SCs of other species regulate meiotic DSB formation to form crossovers crucial for meiosis.
Similar content being viewed by others

Introduction
Meiosis is a special process in which diploid cells undergo one round of DNA replication followed by two consecutive rounds of chromosome segregation to generate haploid gametes. In the first meiotic division, homologous chromosomes physically associate with each other via crossing-over during prophase, resulting in the reciprocal exchange of chromosome portions, and then segregate. This unique feature of meiosis – high-levels of homologous recombination – is critical for sexually reproducing species and is regulated in multiple ways. Homologous recombination is initiated by DNA double-strand breaks (DSBs) formed by the conserved protein Spo11 (called Rec12 in fission yeast, studied here)1. These programmed DSBs are not evenly distributed throughout the genome: the frequency of breakage is much higher than average at widely-spaced loci called DSB hotspots. Here, we describe novel mutants of proteins critical for determining DSB hotspots. These proteins also comprise a structure important for the pairing of homologs described below.
During meiotic prophase in most eukaryotes studied, a tripartite chromosomal structure called the synaptonemal complex (SC) is observed under the electron microscope [reviewed in ref. 2]. The SC consists of a central element and two parallel lateral elements to which chromatin loops are attached, thereby aligning and holding together the two homologous chromosomes. Axial elements, formed shortly after replication, become the lateral elements of the SC later in meiosis. The ultra-structure of the SC is highly conserved, but the proteins composing it are highly diverged among species.
In meiotic prophase of the fission yeast Schizosaccharomyces pombe, linear elements (LinEs), but not full-length SC, are detected by electron microscopy (EM) of spread-out nuclear contents; LinEs appear similar to lateral elements of the SC of other species but do not extend end-to-end on the chromosomes in such EM analyses3, 4. Like the SC, LinE formation requires a meiosis-specific sister chromatid cohesin complex containing Rec8 and Rec11 subunits, and phosphorylation of one of them (Rec11) by a casein kinase 1 ortholog5, 6. Rec8 forms filamentous structures with the chromosomes aligned in parallel from one end to the other during the horsetail stage of meiosis as observed in live cells by super-resolution structured illumination microscopy (SIM)7. This observation reveals a structural similarity of S. pombe meiotic chromosomes and those of other species. The time-course of LinE development, like that of the SC, is coordinated with other meiotic prophase events – LinEs form after replication but before recombination is complete3, 8, 9. Although LinEs differ somewhat from the SC in structure or stability, both SC and LinE proteins are required for high levels of homolog pairing and meiotic recombination10,11,12,13,14.
Four S. pombe proteins have been identified as LinE components – Rec10, Rec25, Rec27 and Mug20. The morphology of immunostained Rec10 is similar to that of LinEs observed by EM, suggesting Rec10 is a primary structural component4. In the C-terminal truncation mutant rec10-155, LinEs are not detectable by EM13, 15. Rec25, Rec27 and Mug20 are small proteins (125–151 amino acids) predicted to form coiled-coil domains, which are also found in SC proteins. In rec25Δ or rec27Δ cells, no Rec10 immunostaining signal, and therefore no LinEs, can be detected by fluorescence microscopy11. In the absence of Mug20, LinEs fail to develop to full-length12 or to form distinct nuclear foci16. Furthermore, these four LinE components interdependently co-localize in meiotic nuclei of live cells; i.e., focus-formation of any one depends on each of the others tested11, 16. Rec10 physically interacts with the other three proteins, as detected by mass spectrometry of immuno-precipitates, and directly with Rec25, as detected in yeast two-hybrid assays12, 17. Thus, these four proteins appear to function as a physical complex (designated the LinE complex), although there is no direct biochemical evidence for such a complex. Two other proteins, Hop1 and Mek1, also localize to LinEs and their localization depends on Rec109.
LinE proteins are crucial for meiotic recombination. Like elimination of Rec12 or any of its partner proteins, deletion of rec10 abolishes meiotically induced homologous recombination in all intervals tested and DSB-formation genome-wide16, 18, 19. Rec10 interacts with Rec15, a Rec12 partner protein, in two-hybrid assays20 and appears to activate Rec12 for DSB formation. Rec25, Rec27 and Mug20 are meiotic DSB hotspot determinants – they bind specifically to nearly all hotspots and are essential for most hotspot DSBs16. In contrast to deletion of rec10, deletion of rec25, rec27 or mug20 reduces recombination frequencies strongly in some intervals (100-fold or more) but only modestly in others (as little as 2-fold)11, 12, 21. Thus, Rec10 acts in DSB formation genome-wide, whereas Rec25, Rec27, and Mug20 seem to function primarily or exclusively at DSB hotspots.
Although LinE proteins play a fundamental role in DSB formation, their mechanistic role is still unclear. Reduced recombination frequency could be a consequence of reduced DSB formation, but the available data do not exclude an additional function of LinE proteins in DSB repair after break formation. Some rec10 non-deletion mutants (rec10-109, rec10-144 and rec10-155) were used to study LinE development and function in recombination and homologous chromosome pairing4, 13, 15, 22, but no rec25, rec27 and mug20 alleles other than deletions have been reported, to our knowledge. Non-deletion mutants are especially useful to study multi-functional proteins, such as LinE proteins involved in DSB formation, homolog pairing and recombination. Here, we identified multiple missense and nonsense alleles of rec25, rec27, mug20 and rec10 and investigated recombination and DSB formation in these mutants. We revised the rec27 open reading frame and the deduced sequence of the Rec27 protein. We also investigated Rec25-GFP focus-formation in multiple rec10 missense mutants. Our results demonstrate the importance of certain conserved amino acids, lend further insight into LinE formation and function, and provide material useful for further investigations. Our study of LinE proteins provides a new perspective on the still-unclear relationship between the SC (axial elements), DSB formation, and recombination.
Results
A screen for novel LinE protein mutants deficient in meiotic recombination
We designed a genetic screen to isolate new rec25, rec27, and mug20 mutations affecting meiotic recombination by modifying a screen for recombination-deficient (Rec−) mutants described previously23 (Fig. 1). We used a diploid strain heteroallelic for mat (h +/h −) and for two non-complementing ade6 mutations (ade6-3049/ade6-M26). Upon starvation, these cells undergo meiosis and recombine to ade + at high frequency due to the ade6-3049 and ade6-M26 recombination hotspots19. ade6-3049 and ade6-M26 are also DSB hotspots; each is a single base-pair change mutation of ade6 + that creates a binding site for the transcription factor Atf1-Pcr124, 25. Binding of Atf1-Pcr1, as well as LinE proteins, is required for DSB formation there16, 26. Chromatin structural changes accompanying this binding appear to facilitate, by an unknown mechanism, DSB formation by Rec12 and its putative partner proteins27,28,29. A LinE gene, such as rec25, was deleted on each chromosome but was present on a derivative of the low copy plasmid pFY2030 as a mutagenized library. After sporulation, spores were spotted on YEA plates without supplementary adenine, on which non-recombinant (Ade−) spores form red colonies, while recombinant (Ade+) spores form white colonies; this produces a red lawn with some white (Ade+) papillae. When the diploid cells express a recombination-deficient (Rec−) LinE mutant protein from the plasmid, the frequency of white papillae is reduced.

Schematic of the screen to isolate LinE mutants. A library of plasmids containing a randomly (PCR) mutagenized rec25, rec27 or mug20 gene was transferred to diploid cells homozygous for rec25Δ, rec27Δ or mug20Δ; linE* means rec25, rec27 or mug20. After sporulation on EMM2+Ade plates, Ade+ recombinant frequencies were estimated by spot test (bottom right), in which recombination-deficient mutants produce a low frequency of white (Ade+) papillae on a background of red (Ade−) cells.
We used mutagenic PCR to form a library of plasmids with mutations restricted to the LinE protein gene. We sequenced the LinE protein gene from randomly chosen plasmids (9 for rec25, 10 for rec27, and 8 for mug20) and found 0.27 base-pair changes per rec25 gene (0.12/kb), 2.5 base-pair changes per rec27 gene (1.2/kb), and 0.65 base-pair changes per mug20 gene (0.36/kb). We transformed an appropriate diploid strain with these libraries and screened ~400 transformants for rec25 or rec27 and ~200 transformants for mug20. We identified 43, 32, and 15 Rec− candidates (i.e., with reduced frequency of white papillae) of rec25, rec27, and mug20, respectively. Among these candidates, 24, 17, and 6 were sequenced (Suppl. Figs S1–S3). Missense mutants, especially those changed in conserved amino acids, were chosen for further study. For all subsequent experiments reported here, the mutations were put onto the chromosome at the endogenous location, to ensure stability and appropriate gene expression.
Four amino acids essential for Rec25 activity
We analyzed four missense mutations of rec25 – rec25-234 (N36D, L137P), rec25-235 (N63I), rec25-236 (K124I), and rec25-237 (L90P) – with substitutions of amino acids conserved in at least four of the five examined Schizosaccharomyces species (N36 is not conserved; Suppl. Fig. S4A). Each of these mutations reduced recombinant frequencies, both ade6 intragenic (gene conversion) and ade6 – arg1 intergenic (crossing-over), by factors not significantly different from that of the rec25 deletion (null) mutation (Fig. 2A; Suppl. Table S3; by unpaired t-test p > 0.07, except for rec25-235 gene conversion in the ade6-M26 x ade6-52 cross with p = 0.04). ade6 gene conversion was reduced by factors of 30–50, and ade6 – arg1 crossing-over by factors of 16–28, indicating the importance of Rec25, and these residues specifically, for recombination.

Meiotic recombination of newly isolated LinE mutants. Meiotic gene conversion at ade6 (two allele pairs) and crossing-over between ade6 and arg1 in LinE mutants were measured as described in Methods. Data (mean ± SEM) are from Supplementary Table S3; n ≥ 3 for each mutant. NS (not significant; P > 0.05), **(P < 0.01), and ***(P < 0.001) indicate significance of the difference from wild type by unpaired t-test.
Revised rec27 ORF starts within the annotated first intron
The rec27 open reading frame (ORF) in the public database (http://www.pombase.org/) has ten sequential As (adenine) in the first exon close to the designated ATG start codon. We sequenced the rec27 + ORF of two not-closely-related wild-type strains (GP13 and GP20) and found that each had ten sequential As, as expected. We found, however, that two isolates from our screen had only nine As (rec27-248) or eleven As (rec27-249) but were as recombination-proficient (Rec+) as another isolate with ten As, which we designate wild type (rec27 +) (Fig. 3A). These potential frameshift mutations would result in truncated Rec27 proteins only 20 and 22 amino acids long, yet they had wild-type recombination phenotype.

Revised Rec27 ORF starts within the first formerly designated intron. (A) Meiotic recombination between ade6-M26 and ade6-52 in rec27 mutants. Data (mean ± SEM) are from Supplementary Table S3; n = 4 for each strain. NS (non-significant) and * (P < 0.05) indicate significance of the difference from rec27 + by unpaired t-test. (B) Map of revised rec27 ORF (lower line) compared to the previous annotation (upper line) (http://www.pombase.org/spombe/result/SPBC577.05c). Pink boxes are exons.
This unexpected observation raises the question, where does rec27 translation start? Recent RNA-seq data show abundant transcripts covering only part of the former first intron, followed by a short exon and then the rest of the gene as designated in the database31. We fortunately recovered a mutation in the first annotated intron [rec27-246 (M1L, N50I)] which exhibited the mutant null phenotype and changed a Met codon to a Leu codon. This Met codon, but not another Met codon toward the 5′ end of this intron, is covered by the RNA-seq data. We conclude, based on these data, that rec27-246 changed the true start codon ATG (Met) to TTG (Leu) in what we conclude is the wild-type first exon (Table 1 and Fig. 3). Using these data, we revised the rec27 ORF such that rec27 translation starts within the former first intron at nucleotide (nt) 156 (relative to the previously designated ATG). Thus, the newly designated first exon extends from nt 156 to 196 and the new first intron extends from nt 197 to 247 (Fig. 3B). As a result, the predicted Rec27 protein has nine amino acids less than the annotated Rec27 protein and 12 amino acids different at the N-terminus. As expected, the revised Rec27 protein aligned better with other Schizosaccharomyces species Rec27 and C. elegans SYP-2 proteins, which were previously shown to share sequence similarity (Fowler et al.16; Suppl. Fig. S4B). The nucleotide changes and amino acid changes of the rec27 mutants listed in Table 1 and Suppl. Fig. S2 were defined based on the revised ORF.
Four rec27 missense mutations with recombination-reduced or null phenotype
Three rec27 missense mutants – rec27-238 (K28E), rec27-240 (K36E), and rec27-241 (Q119K) – were modestly or strongly Rec− in meiosis. The meiotic gene conversion at ade6 was reduced only 2- to 7-fold in rec27-238 and rec27-240 compared to that in wild-type cells. Crossing-over in the ade6 – arg1 interval was reduced by a factor of 1.4 for rec27-238 and by a factor of 3 for rec27-240 (Fig. 2B; Suppl. Table S3). rec27-241, however, was strongly Rec−: it reduced both gene conversion and crossover frequencies to rec27Δ levels (Fig. 2B; Suppl. Table S3). rec27-238 and rec27-240 mutations changed K (lysine) to E (glutamic acid) at conserved sites. Interestingly, the mutation with the greatest defect (Q119) is not among the conserved sites of Schizosaccharomyces species; it is the seventh amino acid from the C-terminus and is apparently missing in two other related species (Table 1; Suppl. Fig. S4B).
Since the Rec− mutants rec27-238 (K28E) and rec27-240 (K36E) altered conserved K (lysine) to E (glutamic acid), we used site-directed mutagenesis to similarly alter five other conserved K or R (arginine) sites to E (R32E, K52E, K57E, K65E and K108E) (Table 1). rec27-239 (R32E), but not the other mutations – rec27-242 (K52E), rec27-243 (K57E), rec27-244 (K65E) and rec27-245 (K108E) – modestly reduced meiotic recombination (Fig. 2B; Suppl. Fig. S5; Suppl. Table S3). The amino acids altered in the three Rec− mutations, K28E, R32E and K36E, are conserved and clustered, suggesting this region is part of a functional domain.
Two mug20 missense mutations with recombination-reduced or null phenotype
In two mug20 missense mutants – mug20-250 (L52P) and mug20-251 (V78E) – gene conversion and crossing-over were reduced. Both types of recombination were modestly reduced in mug20-251 (1.5 to 3-fold) but were reduced to the level of mug20Δ in mug20-250 (Fig. 2C; Suppl. Table S3). The amino acids changed in these two mutants (V78 and L52) are not conserved in Schizosaccharomyces species other than the closest related species S. kambucha (Suppl. Fig. S4C).
Analysis of eight new and previously isolated rec10 mutations
Rec10 is essential for meiotic recombination: deletion of rec10 decreases recombinant frequencies about 1000-fold, to the levels in mutants lacking Rec12, the protein with the active site for DSB formation (Table 2)19. Here we analyzed seven rec10 mutants (rec10-109, rec10-116, rec10-133, rec10-134, rec10-136, rec10-144 and rec10-155) previously described but not sequenced (except for rec10-144 and rec10-155)11, 19, 22, 23, 32, 33 and one newly created mutant (rec10-216).
The first rec10 mutant isolated, rec10-109, had two nearby amino acids changed in its conserved N-terminal quarter – V176I and G178D (Suppl. Fig. S4D; Table 1). Gene conversion at ade6 was reduced by a factor of 100, but, remarkably, crossing-over in the lys3 – ura1 or ura1 – met5 intervals on another chromosome was not affected, compared to wild-type cells (Table 2). This observation is consistent with the previous results with rec10-109 which indicated that Rec10 is a region-specific activator of meiotic recombination15, 23, 33.
rec10-216 (R184F, D185T) was newly generated by random mutagenesis of two N-terminal codons for amino acids conserved in all Schizosaccharomyces species (Suppl. Fig. S4D) and close to the sites mutated in rec10-109. rec10-216 reduced gene conversion and ade6 – arg1 crossing-over nearly to the level of rec10Δ (Table 2). rec10-116 (W575*), rec10-133 (Q729*), rec10-134 (E271K, R434H), and rec10-136 (E309K, W603*) were previously isolated as nitrosoguanidine-induced Rec− mutants32, and three contain non-sense mutations (designated by *). rec10-155 is an insertion of S. cerevisiae LEU2 into the coding sequence34 and results in Rec10 lacking the C-terminal 101 amino acids with ten additional amino acids encoded by the insertion11 (Suppl. Fig. S6). Two mutations – rec10-116 (W575*) and rec10-155 (E691I and frame-shift) – decreased ade6 gene conversion to the level of rec10Δ. Crossovers, however, were decreased substantially less: < 11-fold reduction in these mutants but > 50-fold reduction in rec10Δ 11, 19 (Table 2). rec10-144 (G727E) changes an amino acid in a conserved region that is removed by the rec10-155 truncation (E691I and frame-shift). Both rec10-144 and rec10-155 reduced recombination similarly for crossing-over, but rec10-144 had less effect on gene conversion at ade6 (Table 2).
We conducted complementation analyses by testing N-terminal mutations rec10-109 (V176I, G178D) and rec10-216 (R184F, D185T) with the C-terminal truncation mutation rec10-155 (E691I and frame-shift) and the missense mutation rec10-144 (G727E). Both rec10-109 and rec10-216 partially complemented the missense mutation rec10-144, consistent with previous reports of rec10-109 and rec10-144 complementation22, 32, but failed to complement the truncation mutation rec10-155 (Table 2). Deletion of rec25 in the N-terminal mutant rec10-109 reduced the recombinant frequency to the rec10Δ level at ade6 and also significantly reduced crossing-over (p < 0.01 by chi-square test). But deletion of rec25 in the C-terminal rec10-155 truncation or even in the missense mutation rec10-144 did not have this strong additive affect (Table 2). These results suggest that Rec25 interacts with the C-terminal region of Rec10 and agrees with the absence of LinEs in the rec10-155 C-terminal truncation mutant15.
DSBs are reduced or eliminated in parallel with recombination reduction in the newly isolated LinE mutants
The deficiency of recombination observed in LinE mutants could result from failure either to form or to repair DSBs. DSBs are abolished genome-wide in rec10Δ mutants and strongly reduced at DSB hotspots in rec27Δ; at the few hotspots tested, DSBs are also strongly reduced in rec25Δ 16, 19, 21. We therefore assayed DSB formation at the ade6-3049 hotspot in representative novel LinE mutants. Cells were induced to enter meiosis synchronously using the pat1-114 temperature-sensitive repressor of meiosis in the rad50S background to accumulate unrepaired broken DNA18. There were no detectable DSBs in strong Rec− mutants – rec27-241 (Q119K), rec25-235 (N63I) and mug20-250 (L52P) (Figs 4 and S7). In weaker Rec− mutants – rec27-238 (K28E), rec27-240 (K36E) and mug20-251 (V78E) – DSBs at ade6-3049 accumulated to 22%, 10% and 16%, respectively, of total DNA, compared to 30% in wild-type cells (mean percentage of broken DNA at 5 hr and 6 hr). Overall, DSBs were reduced in parallel with the reduction of recombinant frequencies (Figs 2 and 4).

Meiotic DSBs of LinE mutants. LinE mutants were meiotically induced; DNA was extracted at the indicated times, digested with BsrG1 and analyzed for DSBs at the ade6-3049 hotspot by Southern blot hybridization. Unbroken DNA (10.1 kb) migrates at the top of the blots. Meiotically broken DNA (3–4 kb) migrates as multiple bands near the middle of the blots. The percentage of total radioactivity in the DSB position was quantified with ImageQuant and is shown below each lane and in the graph on the right. Data are individual values or (for wild-type and rec27-238 cells) the range for two independent experiments. Full-length blots are presented in Suppl. Figure S7.
Rec25-GFP forms nuclear foci in a rec10 C-terminus mutant but not in an N-terminus mutant
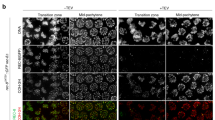
The formation of nuclear foci by Rec25-GFP, Rec27-GFP, Mug20-GFP and immunostained-Rec10 in nuclear spreads depends on the presence of each of the other tested LinE proteins, suggesting that these four proteins form an active complex11, 12, 16. To test whether mutations in the largest protein, Rec10, interrupt formation of this putative complex, we investigated Rec25-GFP nuclear focus-formation in three rec10 mutants. rec10-109 (V176I, G178D) and rec10-216 (R184F, D185T) alter the N-terminal region, and rec10-144 (G727E) alters the C-terminal region. In rec10-109 cells, Rec25-GFP fluorescence became visible in the nucleus about 1.5 hr after meiotic induction, similarly as in wild-type cells, but the Rec25-GFP foci were scarce, faint or not distinct, even at 4 hr after meiotic induction (Fig. 5). In rec10-216 cells, Rec25-GFP entered the nucleus with similar kinetics, but the signal was less concentrated (weaker, and without distinct foci) than in wild-type cells, especially at early time points, and nuclear accumulation was not clear until later in meiosis (Fig. 5). This suggests that in rec10-216 mutant cells Rec25-GFP protein does not enter the nucleus efficiently. Flow cytometry showed that DNA replication in rec10-109 and rec10-216 cells was normal, indicating that the retarded Rec25-GFP accumulation and focus formation in the nucleus was not caused by delayed meiotic progression (data not shown); indeed, both mutants entered meiosis I with normal timing compared to control cells (Fig. 5). In rec10-144 cells, Rec25-GFP appeared in the nucleus at the same time as in wild-type cells, and with similar intensity, but only a few foci were formed on top of a pan-nuclear signal not observed in the control at times when discrete Rec25-GFP foci were present (Fig. 5).

Rec25-GFP localization in rec10 missense mutants. Cells containing Rec25-GFP and the indicated rec10 mutations were induced for meiosis and fixed for fluorescence microscopy. The upper panels show Rec25-GFP, while the lower panels show DAPI-stained DNA. The figures show only the time points when GFP signal was detected (from 1.5 hr to 4 hr) after meiotic induction.
Discussion
Missense mutations are especially useful to study a protein’s function, particularly that of multi-functional proteins such as the S. pombe LinE proteins. We report here eleven newly isolated missense mutations of LinE proteins – four rec25 mutants, four rec27 mutants, two mug20 mutants and one rec10 mutant – and their levels of meiotic recombination, DSB formation and putative complex formation to understand further how these proteins act in meiosis. Additionally, we revised the rec27 ORF.
LinE components share some similarities with SC proteins found in other species. Rec10 has limited amino-acid sequence similarity to Red1, an S. cerevisiae axial element protein; the similarity is limited to ~10% of the protein near the C-terminus that in both proteins is predicted to form a coiled-coil4 (Suppl. Fig. S4D). Red1 appears to play a role in the bias toward DSB repair with the homolog rather than with the sister chromatid35, 36. Rec27 and Mug20 share amino-acid sequence similarity with Caenorhabditis SC proteins SYP-2 and DDL-1, respectively16. Rec25, Rec27, and Mug20 are also predicted to be coiled-coil proteins, a feature of many SC components from numerous organisms. In addition, LinE components are highly conserved among Schizosaccharomyces species. For example, the S. pombe and S. kambucha genomes share 99.5% nucleotide sequence identity37, and their Rec25, Rec27, and Mug20 proteins are 100% identical, and Rec10 98% identical (Suppl. Fig. S4). Among all annotated orthologs in the genomes of three other Schizosaccharomyces species, the mean amino-acid sequence identities, compared to those of S. pombe, range from 66% for S. octosporus to 55% for S. japonicus 37. Among the four individual LinE proteins, amino-acid sequence identities, compared to those of S. pombe, range from 40% for Mug20 of S. cryophilus to 25% for Rec10 of S. japonicus. Although these LinE proteins are somewhat less conserved than proteins in general, as is typical for meiotic proteins, the conclusions with S. pombe mutants, described here, are likely to extend to the other Schizosaccharomyces species and perhaps to more distant species such as C. elegans as well.
In the rec25, rec27 and mug20 mutants studied here, there is co-ordinate reduction or loss of DSBs and recombination frequency. Meiotic recombination is initiated by DSBs, whose formation requires multiple proteins. LinE proteins Rec10, Rec25, Rec27 and Mug20 form a putative LinE complex and are required for most DSBs at hotspots, to which they bind with high specificity16, 19, 21. Since the viable spore yields of the LinE mutants studied here were not strongly reduced (unpublished data), the observed recombination-deficiency of these mutants is likely a consequence of subdued complex formation or impaired DSB formation rather than inefficient DSB repair. Missense LinE mutants are a useful resource to address these aspects of DSB regulation and to identify protein domains involved in these processes.
The LinE mutants we identified reduced recombination to different levels: four rec25 mutants – rec25-234 (N36D, L137P), rec25-235 (N63I), rec25-236 (K124I), and rec25-237 (L90P), one rec27 mutant – rec27-241 (Q119K), and one mug20 mutant – mug20-250 (L52P) had the null phenotype, indicating that these amino acids are critical for each protein’s function, although some of these amino acids are not conserved among Schizosaccharomyces species (Suppl. Fig. S4). As noted above, three rec27 mutants – rec27-238, rec27-239 and rec27-240 – clustered in a small N-terminal region of Rec27 and reduced recombination frequencies moderately; similar reductions were observed in mug20-251.
Since the recombination-deficiencies in these LinE mutants spanned a range from null to nearly wild-type, we measured DSBs at the ade6-3049 hotspot in these mutants. DSBs in mutants with strongly reduced recombination – rec25-235 (N63I), rec27-241 (Q119K) and mug20-250 (L52P) – were reduced to background level. In other mutants with reduced but not abolished recombination – rec27-238 (K28E), rec27-240 (K36E) and mug20-251 (V78E) – DSBs were correspondingly reduced but not abolished (Figs 4 and S7). The detectable DSBs in these mutants suggest that the putative Rec10-Rec25-Rec27-Mug20 complex formed and accumulated in the nucleus, but the complex’s binding-efficiency to hotspots may be inefficient and thereby reduce but not eliminate DSB formation. At the DSB hotspots ade6-3049 and mbs1, DNA breaks are repaired 3- or 4-times more frequently with the sister chromatid than with the homolog38,39,40. We infer that in the rec27-238, rec27-240 and mug20-251 mutants DSBs are repaired with the same bias as in wild-type cells, since DSB and recombinant frequencies are reduced in parallel.
A KR cluster near the N-terminus of Rec27 is important for its function. Three rec27 mutants changed K (lysine) or R (arginine) to E (glutamic acid): rec27-238 (K28E), rec27-239 (R32E), and rec27-240 (K36E) are each separated by three amino acids and clustered near the N-terminus of Rec27. They partially reduced recombination (both gene conversion and crossing-over) (Fig. 2B; Suppl. Table S3) and partially reduced DSBs at ade6-3049 (Figs 4 and S7). These three amino acids are identical in the Rec27 proteins of five Schizosaccharomyces species and may be part of a functional domain to perform the protein’s function.
Our data indicate complex interactions of the N- and C-terminal domains of Rec10. Rec25-GFP, Rec27-GFP and Mug20-GFP interdependently co-localize at about 15 distinct foci in meiotic nuclei; nuclear accumulation of the proteins and formation of foci depends on Rec10 11, 16. In the absence of one or another LinE protein, there is no observed GFP accumulation in the nucleus, even though where tested the proteins’ abundance in the cells is similar to that in wild-type cells11. The nuclear foci of LinE proteins are unlikely to be single protein molecules visible in the microscope we used. Instead, these foci may represent multiple distant regions on a chromosome bound by LinE proteins and forming a cluster that locally regulates DSB formation (Fowler et al., unpublished data).
Rec10, but not the other LinE proteins, has a predicted nuclear localization signal (NLS) in the middle of the protein (K494-S503) (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi). In mitotic cells, ectopically expressed Rec10 strongly localizes in the nucleus, while Rec25 and Mug20 are distributed evenly over the cytoplasm and nucleus41. Rec25-Rec27-Mug20-Rec10 may form a complex and then be brought into the nucleus by Rec10. This view is consistent with Rec10, but not the other LinE proteins, having an obvious NLS. Alternatively, the different components may independently enter the nucleus and LinE complex formation may cause nuclear retention. Since Rec25, Rec27, and Mug20 are small (125–151 amino acids), they may freely diffuse into the nucleus but not accumulate there without association with Rec10.
The rec10 mutations we used to investigate Rec25-GFP focus-formation did not occur in this predicted NLS. In these rec10 mutants Rec25-GFP accumulated in the nucleus, although in the case of the N-terminal mutant rec10-216 (R184F, D185T) less efficiently and with some delay (Fig. 5). This suggests that LinE complex formation or nuclear import is defective in this mutant, which affects two amino acids located in a region of Rec10 (H183-L191) identical in five Schizosaccharomyces species (Fig. S4D). The deficiency in localization of the LinE complex agrees with the recombination deficiency of the rec10-216 mutant, which shows a strong reduction in recombination (gene conversion and crossing-over) nearly to the levels of rec10Δ (Table 2).
In another N-terminal mutant rec10-109 (V176I, G178D), LinE formation assayed in chromosome spreads is considerably reduced4. In intact rec10-109 cells, we observed that Rec25-GFP signal was concentrated in the nucleus with normal kinetics, suggesting that LinE complex formation and nuclear import or retention are normal but the signal did not evolve into dot-like foci as distinct as those in rec10 + cells (Fig. 5). Gene conversion at the hotspot ade6-M26 is reduced in rec10-109 by a factor > 40, but, remarkably, crossing-over is not significantly reduced in two other intervals on another chromosome (Table 2)23, 32, 34. Genome-wide analysis showed that most of the DSBs at most hotspots, including ade6-3049, are strongly reduced but not eliminated in rec10-109; at some hotspots, however, DSBs are still prominent16. The defect in LinE focus formation agrees with the genome-wide analysis of DSBs in this mutant. The LinE complex’s binding to hotspots may be defective in the rec10-109 mutant, and the recombination still observed in rec10-109 mutants may originate mostly from DSBs outside of hotspots16.
A different Rec25-GFP signal was observed in intact cells of the C-terminal rec10-144 (G727E) mutant. A few distinct foci formed in the nucleus although later than, and not as efficiently as, in wild-type cells. The Rec25-GFP nuclear foci in rec10-144 may be limited to a set of hotspot clusters noted above; alternatively, binding of the LinE complex to hotspots may be unstable, hampering normal levels of DSB formation at the hotspot.
In the rec10-109 (V176I, G178D) mutant, the residual gene conversion at the hotspot ade6-M26 and crossing-over in the intervals tested (lys3 – ura1 and ura1 – met5) was Rec25-dependent: deletion of rec25 in rec10-109 reduced gene conversion to the rec10Δ level and crossing over to the rec25Δ level (Table 2). This result indicates that there is still some LinE-dependent recombination activity in rec10-109. Perhaps LinE complexes still load onto some hotspots but at such a low level that they cannot be as readily detected by Rec25-GFP fluorescence as in rec10 + cells; alternatively, hotspot clustering may be defective (see above). Unlike the N-terminal mutant rec10-109, the C-terminal mutant rec10-155 (E691I and frame-shift) did not manifest this genetic interaction with rec25Δ: the recombinant frequencies in the double mutant rec10-155 rec25Δ were the same as that in each single mutant, which are similar (Table 2). This suggests that Rec25 interacts with the Rec10 C-terminus, and that this interaction is lost in the truncated Rec10-155 protein. The similar recombination phenotypes of the C-terminal rec10-144 (G727E) and rec25Δ mutants and the mild effect of rec25Δ in rec10-144 background are also consistent with Rec25 interacting with this region of Rec10.
Our study shows that rec10 mutations altering either the N-terminal region (rec10-109; V176I, G178D) or the C-terminal region (rec10-144; G727E) are defective in LinE focus formation. This result and the intragenic complementation of N-terminal and C-terminal missense rec10 mutations (Table 2)22, 32 suggest that Rec10 protein is self-interacting. There may be domains provided from different Rec10 molecules in a dimeric or higher order complex. In addition, the C-terminal region appears to interact with Rec25 for DSB formation. Two adjacent residues (R184, D185) in the N-terminal region may have a role in LinE complex formation or nuclear import. In summary, the rec10 mutants studied here differ from rec10 + with respect to their interactions with Rec25, assayed both genetically and microscopically, but no clear pattern has yet emerged to explain these alterations.
The eleven novel missense mutations of LinE proteins reported here will help shed more light on LinE complex organization, regulation, and functions of the SC in DSB formation to form meiotic crossovers.
Materials and Methods
S. pombe strains, growth media, and genetic methods
Suppl. Table S1 lists the S. pombe strains used in this study. Media for cell growth and methods for meiotic crosses and random spore analysis were described42.
S. pombe plasmids, oligonucleotides, and site-directed mutant construction
rec25-256::ura4 +, rec27-257::ura4 + and mug20-258::ura4 + were constructed using the method described43. Briefly, the ura4 + ORF (open reading frame) was amplified from plasmid pFY2030 using primers OL3266 and OL3267 (Suppl. Table S2) with 80 bp of homology to DNA flanking the rec25 ORF, or OL3268 and OL3269 with 80 bp of homology to DNA flanking the mug20 ORF. For rec27-257::ura4 +construction, a 1.8 kb DNA fragment containing ura4 + 44 with ~250 bp of homology to DNA flanking each side of the rec27 ORF, as formerly annotated, was generated in two steps by DNA polymerase chain-reactions (PCRs): first, ~250 bp DNA fragments upstream or downstream of the former rec27 ORF were amplified in PCRs using OL3441 with OL3438, or OL3439 with OL3440, respectively, and wild-type (strain GP13) genomic DNA. Then, the purified PCR products together with pFY20 as template were used to amplify the 1.8 kb ura4 + by a secondary PCR using primers OL3442 and OL3443. Since the PCR products from the first step overlap with the 5′ and 3′ ends of ura4 + on plasmid pFY20, the second step PCR generated ura4 + with ~250 bp of homology to regions flanking rec27. The PCR products containing ura4 + and homology to regions flanking rec25, rec27 and mug20 were used to transform strain GP4915 (ura4-D18) to uracil prototrophy. Integration at the rec25 locus was verified by a PCR using primers OL326 and OL3258; OL326 and OL3260 for mug20; and OL3255 and OL3256 for rec27.
Plasmids pLM06, pLM08 and pLM09 with wild-type rec25 + , rec27 + and mug20 +, respectively, were constructed as follows. The rec25 + gene with 297 bp of 5′ and 1118 bp of 3′ flanking DNA was amplified from wild-type (strain GP13) genomic DNA using OL3250 and OL3251 as primers, and the PCR fragment was cloned into pFY20 at the XmaI site to produce plasmid pLM06. The rec27 + gene with 292 bp of 5′ and 314 bp of 3′ flanking the rec27 ORF, as formerly annotated, was PCR-amplified using OL3248 and OL3249 as primers and cloned into pFY20 at the KpnI site to produce pLM08. The mug20 + gene with 291 bp of 5′ and 597 bp of 3′ flanking DNA was PCR-amplified using OL3253 and OL3254 as primers and cloned into pFY20 at the KasI site to produce pLM09.
The mutations rec27-239, rec27-242, rec27-243, rec27-244, and rec27-245 were introduced into plasmid pLM08 by mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent catalog # 210518). Primers used to introduce point mutations were: OL3399 and OL3400 for rec27-239; OL3401 and OL3402 for rec27-242; OL3403 and OL3404 for rec27-243; OL3405 and OL3406 for rec27-244; and OL3407 and OL3408 for rec27-245. After sequencing to confirm the mutations, the mutated rec27 ORFs with flanking homologous sequences were released from the plasmids by KpnI digestion and integrated into the chromosome at the rec27 locus by transformation of strain GP8513 (rec27-257::ura4 +) to 5-fluorouracil (FOA)-resistance. Integration at the rec27 locus was identified by a PCR using primers OL3255 and OL3256.
rec10-216 was generated as follows: primers OL2502 and OL2503, each of which contains six random nucleotides in place of the wild-type codons for R184 and D185, were used to generate randomly mutagenized R184, D185 mutations in rec10. Plasmid pYL17634, bearing the wild-type rec10 gene with ~550 bp of 5′ and ~270 bp of 3′ flanking DNA, was used as a template. After PCR using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent catalog # 210518) and DpnI digestion to remove the template plasmid DNA, the mutant plasmids were introduced into ultracompetent Escherichia coli XL10-Gold cells {∆(mcrA)183 ∆(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac Hte [F′ proAB lacI q Z∆M15 Tn10 (TetR) Amy CamR)]} (Agilent) by transformation to ampicillin-resistance. Plasmids were isolated from seven isolated colonies and sequenced using OL1192; three were mutant: R184V, D185*; R184F, D185T; and R184I, D185S. (*Indicates a non-sense mutation.) Strain GP6993 (rec10-175::kanMX6) was transformed with the mutated plasmids and crossed with GP6994 (rec10-175::kanMX6) to assay intergenic and intragenic recombination. The missense mutation R184F, D185T (rec10-216) showed strong recombination deficiency, equal to that of the nonsense mutation R184V, D185* (rec10-261) (data not shown). The rec10-216 ORF with flanking homologous sequences was released from the plasmid by PvuII and NsiI digestion and integrated into the chromosome at the rec10 locus by transformation of strain GP7301 (rec10-260::ura4 +) to 5-fluorouracil (FOA)-resistance. Integration at the rec10 locus was verified by a PCR using primers OL1778 and OL1779. GP7301 (rec10-260::ura4 +) was constructed by transformation of strain GP6994 to Ura+ using a 1.8 kb ura4 + PCR fragment with 80 bp of homology to DNA flanking the rec10 ORF [amplified from pFY20 which contains ura4 + 30 using OL2722 and OL2723 as primers]. Integration at the rec10 locus was verified by PCR using primers OL1778 and OL1779.
rec25, rec27 and mug20 mutant library construction
Random mutations were introduced into each LinE ORF using the Genemorph II EZclone Domain Mutagenesis Kit (Agilent catalog # 200552). Oligos OL3257 and OL3258 for rec25, OL3255 and OL3256 for rec27, and OL3259 and OL3260 for mug20 were used to synthesize gene fragments containing random mutations using pLM06, pLM08 or pLM09, respectively, as templates. After denaturation and annealing to pLM06, pLM08 or pLM09, the mutated PCR products served as “mega-primers” for a PCR with a specialized high-fidelity enzyme mix in the kit. After DpnI digestion to remove the template plasmid DNA, the mutant plasmids were introduced into ultracompetent E. coli XL10-Gold cells by transformation to ampicillin-resistance. DNA from separate pools of ~6500 transformants for rec25, ~4000 transformants for rec27, and ~2800 transformants for mug20 generated the rec25, rec27 and mug20 mutant libraries. This DNA was used to transform diploid strains GP8370 (rec25Δ), GP8367 (rec27Δ), and GP8484 (mug20Δ) to uracil prototrophy by selection on EMM2 + Ade plates incubated at 32 °C for 5–6 days to allow growth into visible colonies, mating, and sporulation. Single colonies were picked into 75 μl of sterile water in a sterile 96-well plate, and 4 μl of the cell suspension was spotted onto NBA + Ade “keeper” plates. Then, 25 μl of glusulase (1:50 dilution in water) was added to each well. After overnight incubation at 32 °C 100 μl of 60% ethanol was added to each well and held at room temperature for 30 min. The plates were centrifuged at 4000 rpm (2880× g) for 4 min in an Eppendorf centrifuge model 5810; the cells were washed twice with 125 μl of sterile water and suspended in 125 μl of sterile water. Aliquots of 4 μl of the spore suspensions were spotted on YEA plates and incubated for 4 days at 32 °C to assay the Ade+ recombinant frequencies (Fig. 1).
The rec27 mutant genes were put into the chromosome as for the site-directed rec27 mutant genes described above. The rec25 mutant genes with flanking DNA were released by KpnI and XmaI digestion and used to transform GP8210 (rec25-256::ura4 +) to FOA-resistance; a PCR using OL3257 and OL3258 as primers identified homologous replacements. The mug20 mutant genes were released by KasI digestion and similarly transferred to strain GP8211 (mug20-258::ura4 +) and identified by a PCR using OL3259 and OL3260 as primers.
Assay for meiotic DSBs
Meiotic induction and DNA analysis were described by45, 46. DNA in agarose plugs was digested with BsrG1 overnight at 37 °C, subjected to electrophoresis through 0.8% agarose, and transferred to a nylon membrane (Zeta-probe GT membrane, Biorad). DNA broken at the ade6-3049 DSB hotspot was detected by hybridization to a 1 kb probe from the right end of the 10.1 kb BsrGI fragment. The probe was a PCR product amplified from wild-type genomic DNA using OL3693 and OL3694 as primers and labeled with [α-32P] dCTP (3000 Ci/mmol). Signals were detected using a Typhoon storage PhosphorImaging system (GE Healthcare).
Fluorescence microscopy
Diploid pat1-114 rec10 mutant cells producing Rec25 with GFP fused to its C-terminus (rec25-204::GFP) were induced for meiosis, and the GFP signals were examined as described16. Cells were fixed sequentially with 100% methanol and 100% acetone before and at different times after induction of meiosis. The cell suspension was spread on a poly-L-lysine coated slide, acetone allowed to evaporate, and mounting solution (Vectashield; Vector Laboratories, Inc.) supplemented with DAPI (2 μg/ml) added. Images are maximal projections of 9–11 sections, step size of 0.4 mm, to cover the whole cell (approx. 4 µm total). DNA images are a single focal plane, because out-of-focus DAPI fluorescence obscures the projection. Images were obtained with a Nikon Eclipse 90i microscope equipped with a 100/1.4 Oil Plan APO VC lens, a Hamamatsu ORCAER camera, and MetaMorph software (Molecular Devices).
References
Keeney, S., Lange, J. & Mohibullah, N. Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet. 48, 187–214 (2014).
Zickler, D. & Kleckner, N. Meiotic chromosomes: integrating structure and function. Annu Rev Genet. 33, 603–754 (1999).
Bähler, J., Wyler, T., Loidl, J. & Kohli, J. Unusual nuclear structures in meiotic prophase of fission yeast: a cytological analysis. J. Cell Biol. 121, 241–256 (1993).
Lorenz, A. et al. S. pombe meiotic linear elements contain proteins related to synaptonemal complex components. J. Cell Sci 117, 3343–3351 (2004).
Phadnis, N. et al. Casein kinase 1 and phosphorylation of cohesin subunit Rec11 (SA3) promote meiotic recombination through linear element formation. PLoS Genetics 11, e1005225, doi:10.1371/journal.pgen.1005225 (2015).
Sakuno, T. & Watanabe, Y. Phosphorylation of cohesin Rec11/SA3 by casein kinase 1 promotes homologous recombination by assembling the meiotic chromosome axis. Dev. cell 32, 220–230, doi:10.1016/j.devcel.2014.11.033 (2015).
Ding, D. Q. et al. Meiotic cohesin-based chromosome structure is essential for homologous chromosome pairing in Schizosaccharomyces pombe. Chromosoma 125, 205–214 (2016).
Lorenz, A., Estreicher, A., Kohli, J. & Loidl, J. Meiotic recombination proteins localize to linear elements in Schizosaccharomyces pombe. Chromosoma 115, 330–340 (2006).
Loidl, J. S. pombe linear elements: the modest cousins of synaptonemal complexes. Chromosoma 115, 260–271 (2006).
Rockmill, B. & Roeder, G. S. Meiosis in asynaptic yeast. Genetics 126, 563–574 (1990).
Davis, L., Rozalén, A. E., Moreno, S., Smith, G. R. & Martin-Castellanos, C. Rec25 and Rec27, novel components of meiotic linear elements, link cohesin to DNA breakage and recombination in fission yeast. Curr. Biol. 18, 849–854 (2008).
Estreicher, A., Lorenz, A. & Loidl, J. Mug20, a novel protein associated with linear elements in fission yeast meiosis. Curr. Genetics 58, 119–127, doi:10.1007/s00294-012-0369-3 (2012).
Molnar, M., Doll, E., Yamamoto, A., Hiraoka, Y. & Kohli, J. Linear element formation and their role in meiotic sister chromatid cohesion and chromosome pairing. J. Cell Sci 116, 1719–1731 (2003).
Tung, K. S. & Roeder, G. S. Meiotic chromosome morphology and behavior in zip1 mutants of Saccharomyces cerevisiae. Genetics 149, 817–832 (1998).
Wells, J. L., Pryce, D. W., Estreicher, A., Loidl, J. & McFarlane, R. J. Linear element-independent meiotic recombination in Schizosaccharomyces pombe. Genetics 174, 1105–1114, doi:10.1534/genetics.106.063818 (2006).
Fowler, K. R., Gutiérrez-Velasco, S., Martín-Castellanos, C. & Smith, G. R. Protein determinants of meiotic DNA break hotspots. Mol. Cell 49, 983–996 (2013).
Spirek, M. et al. SUMOylation is required for normal development of linear elements and wild-type meiotic recombination in Schizosaccharomyces pombe. Chromosoma 119, 59–72, doi:10.1007/s00412-009-0241-5 (2010).
Young, J. A., Schreckhise, R. W., Steiner, W. W. & Smith, G. R. Meiotic recombination remote from prominent DNA break sites in S. pombe. Mol. Cell 9, 253–263 (2002).
Ellermeier, C. & Smith, G. R. Cohesins are required for meiotic DNA breakage and recombination in Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 102, 10952–10957 (2005).
Miyoshi, T. et al. A central coupler for recombination initiation linking chromosome architecture to S phase checkpoint. Mol. Cell 47, 722–733, doi:10.1016/j.molcel.2012.06.023 (2012).
Martin-Castellanos, C. et al. A large-scale screen in S. pombe identifies seven novel genes required for critical meiotic events. Curr. Biol. 22, 2056–2062 (2005).
Pryce, D. W., Lorenz, A., Smirnova, J. B., Loidl, J. & McFarlane, R. J. Differential activation of M26-containing meiotic recombination hot spots in Schizosaccharomyces pombe. Genetics 170, 95–106, doi:10.1534/genetics.104.036301 (2005).
Ponticelli, A. S. & Smith, G. R. Meiotic recombination-deficient mutants of Schizosaccharomyces pombe. Genetics 123, 45–54 (1989).
Fox, M. E., Yamada, T., Ohta, K. & Smith, G. R. A family of CRE-related DNA sequences with meiotic recombination hotspot activity in Schizosaccharomyces pombe. Genetics 156, 59–68 (2000).
Schuchert, P., Langsford, M., Kaslin, E. & Kohli, J. A specific DNA sequence is required for high frequency of recombination in the ade6 gene of fission yeast. EMBO J 10, 2157–2163 (1991).
Steiner, W. W., Schreckhise, R. W. & Smith, G. R. Meiotic DNA breaks at the S. pombe recombination hotspot M26. Mol. Cell 9, 847–855 (2002).
Steiner, W. W. & Smith, G. R. Natural meiotic recombination hot spots in the Schizosaccharomyces pombe genome successfully predicted from the simple sequence motif M26. Mol. Cell Biol. 25, 9054–9062 (2005).
Wahls, W. P. & Davidson, M. K. Discrete DNA sites regulate global distribution of meiotic recombination. Trends Genet. 26, 202–208, doi:10.1016/j.tig.2010.02.003 (2010).
Fowler, K. R., Sasaki, M., Milman, N., Keeney, S. & Smith, G. R. Evolutionarily diverse determinants of meiotic DNA break and recombination landscapes across the genome. Genome Res. 24, 1650–1664 (2014).
Li, Y. F., Numata, M., Wahls, W. P. & Smith, G. R. Region-specific meiotic recombination in Schizosaccharomyces pombe: the rec11 gene. Mol. Microbiol. 23, 869–878 (1997).
Lee, N. N. et al. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155, 1061–1074 (2013).
DeVeaux, L. C., Hoagland, N. A. & Smith, G. R. Seventeen complementation groups of mutations decreasing meiotic recombination in Schizosaccharomyces pombe. Genetics 130, 251–262 (1992).
DeVeaux, L. C. & Smith, G. R. Region-specific activators of meiotic recombination in Schizosaccharomyces pombe. Genes Dev. 8, 203–210 (1994).
Lin, Y. & Smith, G. R. Molecular cloning of the meiosis-induced rec10 gene of Schizosaccharomyces pombe. Curr. Genetics 27, 440–446 (1995).
Schwacha, A. & Kleckner, N. Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 90, 1123–1135, doi:S0092-8674(00)80378-5 (1997).
Thompson, D. A. & Stahl, F. W. Genetic control of recombination partner preference in yeast meiosis. Isolation and characterization of mutants elevated for meiotic unequal sister-chromatid recombination. Genetics 153, 621–641 (1999).
Rhind, N. et al. Comparative functional genomics of the fission yeasts. Science 332, 930–936, doi:10.1126/science.1203357 (2011).
Hyppa, R. W. & Smith, G. R. Crossover invariance determined by partner choice for meiotic DNA break repair. Cell 142, 243–255, doi:10.1016/j.cell.2010.05.041 (2010).
Cromie, G. A. et al. A discrete class of intergenic DNA dictates meiotic DNA break hotspots in fission yeast. PLoS Genet. 3, e141 (2007).
Cromie, G. A. et al. Single Holliday junctions are intermediates of meiotic recombination. Cell 127, 1167–1178 (2006).
Matsuyama, A. et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nature Biotechnology 24, 841–847 (2006).
Smith, G. R. In Meiosis Methods in Molecular Biology (ed. S. Keeney) Ch. 6, 65–76 (Humana Press, 2009).
Bähler, J. et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 (1998).
Grimm, C., Bahler, J. & Kohli, J. M26 recombinational hotspot and physical conversion tract analysis in the ade6 gene of Schizosaccharomyces pombe. Genetics 135, 41–51 (1994).
Cervantes, M. D., Farah, J. A. & Smith, G. R. Meiotic DNA breaks associated with recombination in S. pombe. Mol. Cell 5, 883–888 (2000).
Hyppa, R. W. & Smith, G. R. In Meiosis Methods in Molecular Biology (ed. S. Keeney) Ch. 15, 235–252 (Humana Press, 2009).
Acknowledgements
We thank Susana Gutiérrez-Velasco for technical assistance, and Sue Amundsen and Mridula Nambiar for helpful comments on the manuscript. This research was supported by grant R01 GM 032194 and R35 GM118120 from the National Institute of General Medical Sciences to G.R.S., and by Spanish Ministry of Science and Innovation (MICINN) grant FEDER-BFU2010-14954 and Spanish Ministry of Economy and Competitiveness (MINECO) grant FEDER-BFU2013-45182-P to C.M.-C.
Author information
Authors and Affiliations
Contributions
All authors participated in the design of the study, analyzed the data, and wrote and reviewed the manuscript text. L.M., K.R.F., and C.M.C. performed the experiments and prepared the figures.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, L., Fowler, K.R., Martín-Castellanos, C. et al. Functional organization of protein determinants of meiotic DNA break hotspots. Sci Rep 7, 1393 (2017). https://doi.org/10.1038/s41598-017-00742-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00742-3
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
