
Abstract
Species from the genus Talaromyces produce useful biomass-degrading enzymes and secondary metabolites. However, these enzymes and secondary metabolites are still poorly understood and have not been explored in depth because of a lack of comprehensive genetic information. Here, we report a 36.51-megabase genome assembly of Talaromyces pinophilus strain 1–95, with coverage of nine scaffolds of eight chromosomes with telomeric repeats at their ends and circular mitochondrial DNA. In total, 13,472 protein-coding genes were predicted. Of these, 803 were annotated to encode enzymes that act on carbohydrates, including 39 cellulose-degrading and 24 starch-degrading enzymes. In addition, 68 secondary metabolism gene clusters were identified, mainly including T1 polyketide synthase genes and nonribosomal peptide synthase genes. Comparative genomic analyses revealed that T. pinophilus 1–95 harbors more biomass-degrading enzymes and secondary metabolites than other related filamentous fungi. The prediction of the T. pinophilus 1–95 secretome indicated that approximately 50% of the biomass-degrading enzymes are secreted into the extracellular environment. These results expanded our genetic knowledge of the biomass-degrading enzyme system of T. pinophilus and its biosynthesis of secondary metabolites, facilitating the cultivation of T. pinophilus for high production of useful products.
Similar content being viewed by others

Introduction
Talaromyces pinophilus, formerly designated Penicillium pinophilum, is a fungus that produces biomass-degrading enzymes such as α-amylase1, cellulase2, endoglucanase3, xylanase2, laccase4 and α-galactosidase2. In one study, a blended enzyme cocktail produced by T. pinophilus and Chrysoporthe cubensis improved the efficiency of hydrolysis of glucan and xylan in sugarcane bagasse for glucose and xylose production, compared with enzymes from a single strain2. A relatively high level of β-glucosidase activity is observed in T. pinophilus under solid state fermentation5. Therefore, T. pinophilus is considered a potential alternative to Trichoderma reesei for cellulase production and efficient biomass hydrolysis. T. pinophilus produces a variety of medically useful metabolites such as 3-O-methylfunicone, which is used to inhibit mesothelioma cell motility6, and talaromycolides 1–3, 5 and 11, which inhibit the growth of the human pathogen methicillin-resistant Staphylococcus aureus 7.
The fungal strain T. pinophilus 1–95 was isolated from the soil of a dried, ploughed field in Wuzhou, China. This strain produces a highly efficient, calcium-independent α-amylase1. Application of calcium-independent α-amylase in starch conversion avoids problems caused by addition of calcium ions8. Additionally, we found that T. pinophilus 1–95 produces 1.21 ± 0.30 U/mL of filter-paper cellulase, 10.72 ± 0.74 U/mL of carboxymethylcellulose cellulase, 0.71 ± 0.02 U/mL of p-nitrophenyl-β-cellobioside cellulase, 0.27 ± 0.01 U/mL of p-nitrophenyl-β-glucopyranoside cellulase and 41.93 ± 2.84 U/mL of xylanase activities in submerged flask cultivation (data not shown). However, a comprehensive understanding of the biomass-degrading enzyme system in this fungus is still lacking.
We describe the de novo whole-genome assembly of T. pinophilus strain 1–95, a nearly complete genome sequence of a high biomass-degrading enzyme-producing species in the genus Talaromyces. Carbohydrate-active enzyme (CAZyme) genes and secondary metabolism gene clusters were observed in the sequenced genome. Comparative genomic analysis suggested that T. pinophilus harbors more biomass-degrading enzymes and secondary metabolites than other related filamentous fungi. In addition, the predicted secretory protein patterns of T. pinophilus 1–95 were analyzed.
Results
Genome sequencing, assembly and annotation
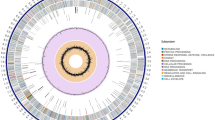
Genome sequencing of the fungal strain T. pinophilus 1–95 (CGMCC No. 2645), isolated from soil in a dried, ploughed field in Wuzhou, China, was performed using a combination of single molecule real-time (SMRT) DNA sequencing and next generation sequencing technology. A high-quality genome sequence of T. pinophilus 1–95 was generated on the PacBio RS II platform. Approximately 1.94 Gb of clean subreads, with sequences from a single pass of a polymerase on a single strand of an insert within a SMRTbell template and no adapter sequences with an N50 size of 10,045 bp and average length 8,102 bp were generated. Additionally, a paired-end (PE) library with a 500-bp average insert size was constructed using the Illumina HiSeq 4000 platform, and 3.88 Gb clean, short-sequence PE reads were generated with a length of 125 bp. Reads were used to correct wrong bases in the assembled genome sequence on the PacBio RS II platform. Finally, a 36.51-Mb genome of T. pinophilus 1–95 was generated with 159-fold coverage. This size was in accordance with the estimated genome size of 28–36 Mb for three Talaromyces species9,10,11. The genome was covered by nine scaffolds, including eight large scaffolds (accession number CP017344-CP017351 in GenBank) without gaps (Fig. 1) and a smaller circular scaffold (accession number CP017352 in GenBank). The N50 and N90 sizes of the scaffolds were, respectively, 4.80 Mb and 2.99 Mb (Table 1). Telomeric repeats (primarily 5ʹ-TTAGGG-3′) were found at both ends of all large scaffolds except for scaffold number 2, for which a telomeric repeat was only found at one end, possibly due to incompleteness of the scaffold sequence data. We inferred that the T. pinophilus 1–95 genome consisted of eight chromosomes (Fig. 1). Additionally, a smaller circular scaffold of 31.73 kb was assembled as a mitochondrial genome (Table 1). Several sequenced Penicillium and Aspergillus species such as P. oxalicum 114-212 and Aspergillus niger ATCC 101513 are also predicted to have eight chromosomes. The overall GC content of the T. pinophilus 1–95 genome was 46.23%. The GC content was 50.08% for the coding sequences and 24.84% for the mitochondrial genome. Other general features of the T. pinophilus 1–95 genome are in Table 1.

Circular map of genomic features of the T. pinophilus 1–95 genome. (a) Location of predicted genes. Numbers represent predicted chromosomes. (b) Schematic representation of genomic characteristics of T. pinophilus pseudogenome (Mb scale). (c) Gene density represented by number of genes in 100-kb nonoverlapping windows. (d) Gene density of genes annotated by the GO database in each 100-kb window. (e) Exon positions of protein-coding genes (circle). Red, exon number (>10) in a gene; green, one exon included. (f) Intron positions of protein-coding genes (circle). Red, exon number (>10) in a gene; green, no intron included. (g) The tRNA density represented by number of tRNAs in 100-kb nonoverlapping windows.
In total, 13,472 protein-coding genes were predicted from the genome assembly using five ab initio gene prediction programs: Augustus (http://bioinf.uni-greifswald.de/augustus/), GeneMark-ES (http://exon.gatech.edu/GeneMark/), Genewise (http://www.ebi.ac.uk/Tools/psa/genewise/), SNAP14 and an unsupervised learning system program Glean version 1 (https://sourceforge.net/projects/glean-gene/). The number of coding genes was significantly higher than other filamentous fungi that produce biomass-degrading enzymes (see Supplementary Table S1). Of the T. pinophilus coding genes, 8162 (60.58%), were annotated in the Gene Ontology (GO) database (http://geneontology.org/), 12,828 (95.21%) in the UniProt database (http://www.uniprot.org/), 12,946 (96.09%) in the NCBI non-redundant (NR) protein sequences database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/) and 4437 (32.93%) in the Clusters of Orthologous Groups of proteins database (http://www.ncbi.nlm.nih.gov/COG/). A total of 6817 (50.6%) genes belonging to 331 pathways were also annotated in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.kegg.jp/) (see Supplementary Fig. S1). The coding regions of the predicted genes constituted almost 53.37% of the genome, with an average length of 1446.5 bp. BUSCO15 and CEGMA16 were used to evaluate the integrity of the genome assembly and prediction of gene sets. A BUSCO set for fungi comprising 1438 single-copy ortholog genes from more than 100 fungal strains was used to evaluate the genome assembly and gene sets of T. pinophilus 1–95. More than 98% of the orthologous genes matched with genes in the genome and gene sets of T. pinophilus 1–95 (see Supplementary Table S2). Using CEGMA, 238 of 248 core eukaryotic genes for fungi were completely identified by evaluating the genome assembly, and 427 of 437 eukaryotic clusters of orthologous groups were identified with an overlap rate > 0.5 when predicted gene sets were assessed (see Supplementary Table S1). These results indicated that our genome assembly and prediction of gene sets for T. pinophilus 1–95 were of high quality and confidence.
Overall genome and proteome comparison
The genome size and number of protein-encoding genes of T. pinophilus 1–95 were comparable to other sequenced filamentous fungi (see Supplementary Table S1). A phylogenetic tree constructed based on 2082 single-copy orthologs (see Supplementary Dataset S1) indicated that T. pinophilus 1–95 was most closely related to T. cellulolyticus Y-94 (Fig. 2a). Analyzing the top hits of a BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) search of all-vs.-all findings showed that T. pinophilus 1–95 and T. cellulolyticus Y-94 shared 10,260 orthologous proteins, accounting for 76.16% of the total proteome, with an average amino acid sequence identity of 98.28%. In contrast, low identity was observed to other sequenced Talaromyces species17, Trichoderma sp.18, Penicillium sp.19, 20 and Aspergillus sp.13, 21, 22, ranging from 70% to 88% (see Supplementary Table S3).

Comparative genomic analysis of T. pinophilus and other fungal species. (a) Maximum-likelihood phylogenetic tree of T. pinophilus and 10 fungal species. (b) Comparative analysis of carbohydrate-active enzyme (CAZyme) numbers. GH, glycoside hydrolase; AA, auxiliary activity; GT, glycosyl transferase; CE, carbohydrate esterase; CBM, carbohydrate-binding module; PL, polysaccharide lyase. (c) Comparative analysis of secondary metabolite gene cluster numbers. In, Indole and Indole-Nrps; La, Lantipeptide; Nr, Nrps, Nrps-Indole, Nrps-T1pks and Nrps-Terpene; Ph, Phosphonate; Si, Siderophore; T1, T1pks, T1pks-Indole and T1pks-Nrps; T3, T3pks; Te, Terpene-Nrps, Terpene-Nrps-Indole and Terpene-T1pks; Ot, Others; To, total number of secondary metabolite gene clusters. Vertical axes in (b) and (c) correspond to fungal species in (a).
The predicted proteome of T. pinophilus 1–95 was 23.84% larger than the proteome of T. cellulolyticus Y-949, whereas the genome size of 1–95 was similar to Y-94 (see Supplementary Table S1). The reason for this finding was that the genome sequencing and assembly of T. pinophilus 1–95 were of high quality (see Supplementary Table S2), partly due to the lack of gaps in the scaffolds. A comparative analysis of proteins annotated by the GO database between T. pinophilus 1–95 and T. cellulolyticus Y-94 showed functional differences between the proteins mostly for “biological regulation”, “cellular process” and “metabolic process” in the biological process category and “binding”, “catalytic activity” and “nucleic acid binding transcription factor activity” in the molecular function category (see Supplementary Fig. S2).
We used data from the PE library with an average insert size of 500 bp from T. pinophilus 1–95 to map the entire genome sequence of T. cellulolyticus Y-947. The PE reads covered 35 scaffolds with 32.06 Mb of sequence from the T. cellulolyticus Y-94 genome, resulting in 88% average coverage and 83-times average depth (see Supplementary Table S4). We found 489 genes from T. cellulolyticus Y-94 contained insertion-deletion mutations when mapping PE reads from the T. pinophilus 1–95 to the genome of T. cellulolyticus Y-94; 257 of these occurred in coding sequence regions. Only 33 of the mutated genes showed no similarity with genes in the T. pinophilus 1–95 proteome, and approximately half were annotated by the GO database as related to “oxidation-reduction”, “binding” and “catalytic activity”.
Biomass degrading machinery in T. pinophilus 1–95
Among the 13,472 unique proteins of T. pinophilus 1–95, 803 were annotated as encoding CAZymes using carbohydrate-active enzyme annotations from dbCAN23. The putative CAZymes included 72 families of glycoside hydrolases (GHs), 35 families of glycosyl transferases (GTs), 13 families of carbohydrate esterases (CEs), 10 families of auxiliary activities (AAs), 5 families of polysaccharide lyases (PLs) genes and 19 families of carbohydrate-binding modules (CBMs) (Fig. 2b). Further analysis revealed that 156 CAZymes were predicted to be plant cell wall-degrading enzymes (CWDEs), specifically 42 cellulolytic enzymes, 97 hemicellulases and 17 pectinases (Fig. 3; see Supplementary Table S5).

Carbohydrate-active enzyme (CAZymes) genes in T. pinophilus 1–95 including plant cell wall-degrading enzymes (CWDEs) and starch-degrading enzymes (SDEs). Blue gene IDs, secreted CWDEs and SDEs. (a) Heatmap of different enzyme types. (b,c) Orange, starch-degrading enzymes; purple, plant CWDEs. (d) Colors represent different CAZyme families.
Among the cellulolytic enzymes, 2 cellobiohydrolases (CBHs, EC 3.2.1.91), 8 β-1,4-endoglucanases (EGs, EC 3.2.1.4) and 29 β-glucosidases (BGLs, EC 3.2.1.21) were included as cellulases. Of these, the known cellulases included Cel7A-2 (TP09412), Cel5A (TP03457), Cel5B (TP07499), Cel7B (TP08514) and Bgl3A (TP09042). Compared with known filamentous fungi used for cellulase production, i.e., T. cellulolyticus Y-94 and P. oxalicum HP7-1, larger numbers of BGLs were classified into GH families 1 and 3 in the T. pinophilus 1–95 genome (see Supplementary Table S5). We examined lytic polysaccharide monooxygenases (LPMO) that catalyze the initial oxidative cleavage of recalcitrant cellulose, resulting in the slow release of oxidized oligosaccharides into solution. LPMO-cellulase synergy is beneficial for degradation of large and highly resistant crystalline cellulose24, 25. Only one, TP03971, an ortholog of Cel61A from P. oxalicum HP7–1, was identified, which was fewer than in P. oxalicum HP7–1 (see Supplementary Table S5).
The T. pinophilus 1–95 genome was rich in hemicellulose-degrading enzymes (97 genes) assigned into 29 predicted CAZyme families; this finding was compared to 77 genes in T. cellulolyticus Y-94 and 80 genes in P. oxalicum HP7–1, which includes Xyn11A (TP00436) and Xyn10A (TP06900), encoding the important β-1,4-endoxylanases. This result supports the high xylanase activity of T. pinophilus 1–95. The predicted hemicellulases in T. pinophilus 1–95 were divided into 19 types by substrate specificities. The large differences among T. pinophilus 1–95, T. cellulolyticus Y-94 and P. oxalicum HP7–1 broadly covered most of the listed hemicellulase types (see Supplementary Table S5). For instance, T. pinophilus 1–95 possessed more β-D-xylosidases (EC 3.2.1.37), acetyl xylan esterases (EC 3.1.1.72) and feruloyl esterases (EC 3.1.1.73) than the two others. β-D-xylosidases hydrolyze xylobiose or linear xylooligosaccharides to the monomer xylose. Acetyl xylan esterases liberate acetic acid esterifying position 2 and 3 on mono- and di-O-acetylated β-1,4-linked D-xylopyranosyl residues in xylan chains. Feruloyl esterases liberate trans-ferulic acid from 5-O-feruloylated L-arabinofuranosyl residues. These enzymes facilitate the hydrolysis of hemicellulose26. In contrast, the T. pinophilus 1–95 genome possessed a similar number of pectin-degrading enzymes as T. cellulolyticus Y-94 and P. oxalicum HP7–1 (see Supplementary Table S5).
T. pinophilus 1–95 produces a highly efficient, calcium-independent α-amylase1. Application of this enzyme in starch conversion might avoid the drawbacks of calcium ion addition8. Among 803 CAZymes, 24 were involved in starch degradation, while 5 to 21 were found in the other 10 investigated filamentous fungi (see Supplementary Table S6). These 24 enzymes were composed of 5 α-amylases (EC 3.2.1.1), which mainly break internal α-1,4-glucosidic linkages and some branched α-1,6-glycosidic linkages from the inner starch chain; 13 α-glucosidases (EC 3.2.1.20), which break α-1,4-linkages in mainly maltose and short maltooligosaccharides to release glucose at nonreducing ends27; 5 glucoamylases (EC 3.2.1.3), which mainly hydrolyze α-1,4-glucosidic linkages from the nonreducing ends of starch chains with the release of β-D-glucose28; and a 1,4-α-glucan branching enzyme (EC 2.4.1.18) that cleave α-1,4 glucosidic linkages of glucan chain, and then transfer the cut end to 6-position of glucose residue within the cleaved or another glucan chain, resulting the generation of an α-1,6 glucosidic linkage27, 29. Comparative analysis indicated that T. pinophilus 1–95 had more α-glucosidases and glucoamylases than the other investigated fungi (see Supplementary Table S6), supporting that it has a high capacity for starch hydrolysis1. Proteomic mass spectrometry and proteome prediction analyses indicated that TP04014 may encode a highly efficient, calcium-independent α-amylase, as reported in Xian et al.1, but this result needs to be further confirmed experimentally.
Transcription factor prediction
Transcription factors (TFs) are essential for modulating diverse biological processes by regulating gene expression. In total, 943 TFs were found in the predicted proteome of T. pinophilus 1–95. The largest family was proteins (716 members) containing zinc-finger structures, such as the Zn2Cys6 type, the C2H2 type and CCHC type, followed by the winged helix repressor DNA-binding family (97 members) (Fig. 4). TFs are used in genetic engineering to improve biomass-degrading enzyme yields. We carried out an orthology search of known TFs involved in regulation of lignocellulolytic genes in filamentous fungi against the translated proteins in the T. pinophilus 1–95 genome using an identity of 40% and an E-value threshold of 1E-10. Orthologs of most known TFs were identified in T. pinophilus 1–95, including the carbon catabolite repressor CreA (TP09972), the cellulase transcription activator CLR-2 (TP10486), the starch degradation regulator AmyR (TP09286) and the xylan degradation regulator XlnR (TP02627) (Table 2). Several related proteins include ACEII, Xpp1, ClrC and BglR, had no ortholog in T. pinophilus 1–95.

Top 10 types of predicted transcription factors in T. pinophilus 1–95.
A repertoire of secondary metabolism gene clusters
We found that T. pinophilus 1–95 had a wealth of secondary metabolites using the AntiSMASH web service30. A total of 68 secondary metabolism gene clusters harboring 401 putative genes were identified. The predicted products of 52 secondary metabolism gene clusters were classified into 8 different types: 28 T1 polyketide synthase (T1PKS) gene clusters, 9 non-ribosomal peptide synthase (NRPS) gene clusters, 9 terpene gene clusters, 2 Nrps-T1pks gene clusters, 1 phosphonate gene cluster, 1 T1pks-Indole gene cluster, 1 T1pks-Nrps gene cluster and 1 Terpene-T1pks gene cluster; the remaining 16 gene clusters synthesized other unknown secondary metabolites (Fig. 2c; see Supplementary Table S7). When compared with known gene clusters for secondary metabolites, eight were predicted to produce emericellin, pestheic acid, azanigerone and azaphilone (data not shown).
The transporter and secretory system
Transporters are important in microbial growth and reproduction because they assist microbes in the uptake of nutrients and energy from the surrounding environment. In total, 1,238 genes encoding putative transporters belonging to seven superfamilies were identified in the T. pinophilus 1–95 genome (see Supplementary Dataset S2). Of these, members of the electrochemical potential-driven transporters (EPTs) were the most abundant, accounting for 54.7%, followed by primary active transporters (18.3%). Among the EPTs, 431 members of a major facilitator superfamily were selected; these are involved in the transport of substances including sugar, drugs and peptides. Of these EPTs, TP13272 and TP06909, respectively, were orthologs of the cellodextrin transporters Cdt-C and Cdt-D, which are crucial for induction of cellulase gene expression by insoluble cellulose31.
Many proteins are commonly secreted into extracellular regions, including enzymes involved in biomass degradation. A comprehensive pipeline was designed to predict the T. pinophilus 1–95 secretome. T. pinophilus 1–95 potentially secreted 1,203 extracellular proteins (8.9% of predicted total proteins), comprising 831 classical and 372 nonclassical secretory proteins (see Supplementary Dataset S3). Using KEGG annotation, 546 putatively secreted proteins were assigned, indicating that these abundant secretory proteins were mainly involved in metabolism, especially carbohydrate metabolism and xenobiotic biodegradation and metabolism (see Supplementary Fig. S3). We found that 323 of these, including 35 nonclassical secreted proteins, were present in the CAZyme database, accounting for 26.8% of the total secretome. The repertoire of secreted CAZymes consisted of 54 GH families, 18 GT families, 12 CE families, 8 AA families, 4 PL families and 13 CBM families. The most common GH family, comprising 188 enzymes, contributed to 58.2% of total secreted CAZymes, followed by the AA families (14.9%). The most prevalent GH CAZyme classes were GH7, GH3, GH5, GH10-13 and GH31, which represent cellulases, endoxylanases and amylases, all of which are required for biomass degradation. The most abundant CBM family was CBM1, accounting for 43.8% of total CBMs, which are known to bind to crystalline cellulose and aid in its enzymatic hydrolysis32.
Further analysis indicated that 82 of 323 predicted secretory CAZymes including two nonclassical proteins and six non-CAZymes were identified as CWDEs. Of these, 18 cellulases consisting of two CBHs, seven EGs and nine BGLs were investigated. These cellulases included the major CWDEs for cellulose degradation Cel7A-2 (TP09412), Cel7B (TP08415), Cel5A (TP13457), Cel5B (TP07499), Cel5C (TP08784), Cel45A (TP06957) and Bgl3A (TP09042). We also identified 62 hemicellulose-degrading enzymes and 6 pectin-degrading enzymes, including eight β-1,4-endoxylanases, nine acetyl xylan esterases, four α-galactosidases, 10 α-L-arabinofuranosidases, five α-L-fucosidases, one endo-1,4-β-mannanase and four feruloyl esterases, as well as two pectin esterases, two tannases, one pectate lyase and one pectin lyase (see Supplementary Dataset S3). In addition to CWDEs, 10 starch-degrading enzymes were found in the predicted secretome of T. pinophilus 1–95: four α-amylases, one glucoamylase and five α-glucosidases (see Supplementary Dataset S3).
Discussion
A systematic genetic investigation of filamentous fungi would contribute to genetic engineering of more diverse and productive industrial microbial strains for improving cellulolytic enzyme production. We sequenced, assembled and analyzed the entire T. pinophilus 1–95 genome in detail. T. pinophilus is a promising filamentous fungus for the industrial production of biomass-degrading enzymes. This study describes the nearly complete genome sequence of a member of the genus Talaromyces. The total assembled genome size was 36.51 Mb, which was within the range of filamentous fungi that produce cellulolytic enzymes, including Penicillium, Aspergillus, Trichoderma and Neurospora species.
Comparative genome analysis indicated that the most closely related species to T. pinophilus 1–95 was T. cellulolyticus Y-94. T. cellulolyticus Y-94 was identified as T. pinophilus based on only an internal transcribed spacer sequence33. It was proposed as the new species T. cellulolyticus in the genus based on morphological and phenotypic differences from T. pinophilus 34. The reported genome sequence of Y-94 is a draft with a number of gaps9. In this study, a nearly complete genome sequence of T. pinophilus 1–95 was obtained. The number of protein-encoding genes in T. pinophilus was higher than in other investigated fungal strains. This result may be because of the presence of more genes or the result of high-quality SMRT DNA sequencing technology and a different methodology for gene prediction. In particular, a large inventory of CAZymes was found, including CWDEs and starch-degrading enzymes. This result supported the high capacity of this species to degrade biomass, comparable to T. cellulolyticus Y-9435 and P. oxalicum HP7-120. Of note, T. pinophilus 1–95 possessed the most BGLs (29 coding genes), glucoamylases (5 coding genes) and α-glucosidases (13 coding genes) among species we compared. BGLs are important for releasing inhibition of cellulase activity36. Furthermore, the predicted secretome of T. pinophilus 1–95 showed that approximately 50% of CWDEs and starch-degrading enzymes were secreted into the extracellular region, including major cellulases, hemicellulases and amylases. This result indicated a promising application of T. pinophilus in biorefining. These results also demonstrated that T. pinophilus 1–95 is more excellent cellular machinery for biomass-degrading enzymes than that of P. oxalicum HP7–1 and T. cellulolyticus Y-94 as previously observed20, 35, meriting further study.
Comparative analysis to 10 filamentous fungi from four genera, Talaromyces, Trichoderma, Penicillium and Aspergillus, indicated that T. pinophilus 1–95 possessed the most secondary metabolism gene clusters except for A. niger and A. oryzae. T. pinophilus 1–95 had more T1PKs than Trichoderma sp., Penicillium sp. and Aspergillus sp., and fewer NRPS than Aspergillus species. These results suggested that T. pinophilus 1–95 has potential for producing bioactive secondary metabolites. Although thus far, several bioactive secondary metabolites have been extracted and characterized from T. pinophilus 6, 7, according to the genomic data, additional secondary metabolites could be generated using this species.
In summary, this study provided a nearly complete genome sequence for the genus Talaromyces. The result provided new insights for a comprehensive understanding of the biomass-degrading enzyme system of Talaromyces at the genome level. Detailed comparative genomic analysis revealed a complex biomass-degrading enzyme system in T. pinophilus, indicating its promising application in biomass biorefineries. This study provides a genome-sequence basis for developing strategies that use T. pinophilus as a microbial cell factory for production of high-value enzymes and secondary metabolites.
Materials and Methods
Culture conditions and genomic DNA extraction
T. pinophilus 1–95 was isolated from soil in a dried, ploughed paddy field in Wuzhou, China1 and was deposited at the China General Microbiological Culture Collection Center (CGMCC) under accession number CGMCC No. 2645. Total DNA extraction from mycelia was performed using a phenol-chloroform method with some modifications37. Mycelia were ground in liquid nitrogen and put in 1 mL lysate reagent (40 mM Tris-HCl, 20 mM sodium acetate, 10 mM ethylenediaminetetraacetic acid, and 1% sodium dodecyl sulfate, pH 8.0) per 100 mg mycelia powder. Genomic DNA was collected by centrifugation at 11,300 × g for 10 min.
Genome sequencing and assembly
The T. pinophilus strain 1–95 genome was sequenced using a PacBio RS II platform and Illumina HiSeq 4000 platform at the Beijing Genomics Institute (BGI, Shenzhen, China). Four SMRT cells zero-mode waveguide arrays of sequencing, were used by the PacBio platform to generate the subreads set. PacBio subreads (length < 1 kb) were removed. The program Pbdagcon (https://github.com/PacificBiosciences/pbdagcon) was used for self-correction. Draft genomic unitigs, which are uncontested groups of fragments, were assembled using the Celera Assembler38 against a high-quality corrected circular consensus sequence subreads set. Order, distance and orientation of unitigs and combined scaffolds were generated using software SSPACE39. An upgraded draft genome was obtained after filling or reducing as many captured gaps as possible using software PBJelly40. To improve the accuracy of the genome sequences, GATK (https://www.broadinstitute.org/gatk/) and SOAP tool packages (SOAP2, SOAPsnp, SOAPindel)41, 42 were used to make single-base corrections.
A DNA library of 500 bp inserts was constructed and PE sequenced. For generated HiSeq reads, Q20, representing the probability of the incorrectness of the corresponding base call, was detected. If Q20 reads accounted for less than 60%, they were discarded. Software Pilon43 used reasonable PE sequence data from Illumina libraries to perform comprehensive variant detection and improve genome assembly.
Gene detection and functional annotation
Protein-coding genes in the T. pinophilus 1–95 genome were predicted independently with the gene prediction programs Augustus (http://bioinf.uni-greifswald.de/augustus/), GeneMark-ES (http://exon.gatech.edu/GeneMark/), Genewise (http://www.ebi.ac.uk/Tools/psa/genewise/), SNAP14, and the unsupervised learning system program Glean (https://sourceforge.net/projects/glean-gene/) version 1. Augustus and SNAP, using default parameters, were trained on gene models for A. oryzae, P. oxalicum, T. marneffei and T. stipitatus (see Supplementary Table S1). Programs GeneWise and GeneMark-ES were used to obtain different gene sets and worked in a self-training manner. Finally, all prediction gene sets were integrated by Glean.
For functional annotation of translated proteins in the T. pinophilus 1–95 genome, a BLASTp version 2.2.28+ search against the NCBI NR database (update 05, 2015) and Swiss-Prot and TrEMBL databases (http://www.mrc-lmb.cam.ac.uk/genomes/madanm/pres/swiss2.htm) (update 01, 2016) and KEGG database (http://www.kegg.jp/) version 76, were used to assign general protein function profiles. We used cut-off E-value ≤ 1e-5, overlap 0.4 and identity 30. InterProScan5 (http://www.ebi.ac.uk/interpro/), stand-alone version 55, and GO (http://geneontology.org/) were also used to annotate the predicted proteome. TFs were predicted based on InterPro IDs in the Fungal Transcription Factor Database (http://ftfd.snu.ac.kr/). The hmmsearch program in the HMMER 3.1b2 package (http://hmmer.org/), was used to search all predicted proteomes with the family-specific hidden Markov model profiles of CAZymes from the dbCAN database26. Primary results were processed with an E-value threshold of 1E-7. Protein kinases and phosphatases were detected using hmmsearch based on the Eukaryotic Kinase and Phosphatase Database (http://ekpd.biocuckoo.org/). Membrane transport proteins were classified and identified by a BLASTp search against the transport classification database44, with E-value threshold 1E-10, overlap 0.4 and identity of 30. AntiSMASH30 was used to annotate secondary metabolism gene clusters.
Phylogenetic analysis
An all-against-all pairwise BLASTp similarity search was performed using proteomes from 11 filamentous fungi (see Supplementary Table S2) with E-value cutoff 1E-7, according to the method described previously45. We selected 2,082 single-copy genes from 118,099 genes in 11 fungal genomes. MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) version 3.7 with default parameters was used to perform multiple sequence alignment of single-copy genes. A neighbor-joining tree was calculated using TreeBeST46 with bootstrapping set to 100. The phylogenetic tree was visualized using SVGKit (http://svgkit.sourceforge.net/) and PERL scripts.
Secretome prediction
The total set of 13,472 proteins of T. pinophilus strain 1–95 was analyzed using Secretome P47 v1.0 and SignalP48 v4.0 for ab initio prediction of classical and nonclassical secretory proteins, except for 11 proteins with more than 4,000 amino acid residues. All proteins containing signal peptides were selected, with proteins without signal peptides chosen as candidates in cases of neural network score ≥ 0.6. All selected proteins were analyzed by TargetP49 v1.1 with localization = secretory pathway and RC≤ 4 as screening criteria. Protein sets were scrutinized for the presence of transmembrane domains using TMHMM v2.0 (http://www.cbs.dtu.dk/services/TMHMM/) and for glycosylphosphatidyl inositol anchors use web server PredGPI (http://gpcr.biocomp.unibo.it/predgpi/). Software tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/) version 1.3 was used for transfer RNA prediction using the T. pinophilus 1–95 genome with option C and other default parameters.
References
Xian, L., Wang, F., Luo, X., Feng, Y. L. & Feng, J. X. Purification and characterization of a highly efficient calcium-independent alpha-amylase from Talaromyces pinophilus 1–95. PLoS One 10, e0121531 (2015).
Visser, E. M., Falkoski, D. L., de Almeida, M. N., Maitan-Alfenas, G. P. & Guimarães, V. M. Production and application of an enzyme blend from Chrysoporthe cubensis and Penicillium pinophilum with potential for hydrolysis of sugarcane bagasse. Bioresour Technol 144, 587–594 (2013).
Pol, D., Laxman, R. S. & Rao, M. Purification and biochemical characterization of endoglucanase from Penicillium pinophilum MS 20. Indian J Biochem Biophys 49, 189–194 (2012).
Dhakar, K., Jain, R., Tamta, S. & Pandey, A. Prolonged laccase production by a cold and pH tolerant strain of Penicillium pinophilum (MCC 1049) isolated from a low temperature environment. Enzyme Res 2014, 120708 (2014).
Elnaggar, E. A., Haroun, S. A., Oweis, E. A. & Sherief, A. A. Identification of newly isolated Talaromyces pinophilus and statistical optimization of glucosidase production under solid-state fermentation. Prep Biochem Biotech 45, 712–729 (2014).
Buommino, E. et al. Cell-growth and migration inhibition of human mesothelioma cells induced by 3-O-Methylfunicone from Penicillium pinophilum and cisplatin. Invest New Drug 30, 1343–1351 (2012).
Zhai, M. M. et al. Talaromycolides A-C, novel phenyl-substituted phthalides isolated from the green chinese onion-derived fungus Talaromyces pinophilus AF-02. J Agr Food Chem 63, 9558–9564 (2015).
Haki, G. D. & Rakshit, S. K. Developments in industrially important thermostable enzymes: a review. Bioresour Technol 89, 17–34 (2003).
Fujii, T., Koike, H., Sawayama, S., Yano, S. & Inoue, H. Draft genome sequence of Talaromyces cellulolyticus strain Y-94, a source of lignocellulosic biomass-degrading enzymes. Genome Announc 3, e00014–15 (2015).
Hu, L. et al. Draft genome sequence of Talaromyces verruculosus (“Penicillium verruculosum”) strain TS63-9, a fungus with great potential for industrial production of polysaccharide-degrading enzymes. J Biotechnol 219, 5–6 (2016).
Schafhauser, T. et al. Draft genome sequence of Talaromyces islandicus (“Penicillium islandicum”) WF-38-12, a neglected mold with significant biotechnological potential. J Biotechnol 211, 101–102 (2015).
Liu, G. et al. Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens. PLoS One 8, e55185 (2013).
Andersen, M. R. et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21, 885–897 (2011).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 1–9 (2004).
Sim, A. F. O., Waterhouse, M. R., Ioannidis, P., Kriventseva, V. E. & Zdobnov, M. E. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Parra, G., Bradnam, K., Ning, Z., Keane, T. & Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Res 37, 289–297 (2009).
Nierman, W. C., Fedorovaabrams, N. D. & Andrianopoulos, A. Genome sequence of the AIDS-associated pathogen Penicillium marneffei (ATCC18224) and its Near taxonomic relative Talaromyces stipitatus (ATCC10500). Genome Announc 3 (2015).
Martinez, D. et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26, 553–560 (2008).
Specht, T., Dahlmann, T. A., Zadra, I., Kürnsteiner, H. & Kück, U. Complete sequencing and chromosome-scale genome assembly of the industrial progenitor strain P2niaD18 from the penicillin producer Penicillium chrysogenum. Genome Announc 2, e00577–14 (2014).
Zhao, S. et al. Comparative genomic, transcriptomic and secretomic profiling of Penicillium oxalicum HP7-1 and its cellulase and xylanase hyper-producing mutant EU2106, and identification of two novel regulatory genes of cellulase and xylanase gene expression. Biotechnol Biofuel 9, 203 (2016).
Galagan, J. E. et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438, 1105–1115 (2005).
Zhao, G. et al. Draft genome sequence of Aspergillus oryzae strain 3.042. Eukaryot Cell 11, 1178–1178 (2012).
Yin, Y. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40, W451 (2012).
Eibinger, M. et al. Cellulose surface degradation by lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J Biol Chem 289, 35929–35938 (2014).
Kracher, D. et al. Extracellular electron tranfer systems fuel cellulose oxidative degradation. Science 352, 1098–1101 (2016).
Biely, P., Singh, S. & Puchart, V. Towards enzymatic breakdown of complex plant xylan structures: state of the art. Biotechnol Adv 34, 1260–1274 (2016).
Møller, M. S. & Svensson, B. Structural biology of starch-degrading enzymes and their regulation. Curr Opin Struc Biol 40, 33–42 (2016).
Marín-Navarro, J. & Polaina, J. Glucoamylases: structural and biotechnological aspects. Appl Microbiol Biotechnol 89, 1267–1273 (2011).
Li, W. W. et al. Retrogradation behavior of corn starch treated with 1,4-α-glucan branching enzyme. Food Chem 203, 308–313 (2016).
Weber, T. et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43, 237–243 (2015).
Li, J. et al. Cellodextrin transporters play important roles in cellulase induction in the cellulolytic fungus Penicillium oxalicum. Appl. Microbiol Biotechnol 97, 10479–10488 (2013).
Duan, C. J., Feng, Y. L., Cao, Q. L., Huang, M. Y. & Feng, J. X. Identification of a novel family of carbohydrate-binding modules with broad ligand specificity. Sci Rep 6, 19392 (2016).
Houbraken, J., de Vries, R. P. & Samson, R. A. Modern taxonomy of biotechnologically important Aspergillus and Penicillium species (ed. Sima Sariaslani, S. & Gadd, G. M.) 86, 199-249 (Elsevier, 2014).
Fujii, T., Hoshino, T., Inoue, H. & Yano, S. Taxonomic revision of the cellulose-degrading fungus Acremonium cellulolyticus nomen nudum to Talaromyces based on phylogenetic analysis. FEMS Microbiol Lett 351, 32–41 (2014).
Inoue, H., Decker, S. R., Taylor, L. E. 2nd, Yano, S. & Sawayama, S. Identification and charaterization of core cellulolytic enzymes from Talaromyces cellulolyticus (formerly Acremonium cellulolyticus) critical for hydrolysis of lignocellulosic biomass. Biotechnol Biofuel 7, 151 (2014).
Gusakov, A. V. Alternatives to Trichoderma reesei in biofuel production. Trends Biotechnol 29, 419–425 (2011).
Sun, X., Liu, Z., Qu, Y. & Li, X. The effects of wheat bran composition on the production of biomass-hydrolyzing enzymes by Penicillium decumbens. Appl Biochem Biotechnol 146, 119–128 (2008).
Myers, E. W. et al. A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000).
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 211 (2014).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7, e47768–e47768 (2012).
Li, R. et al. SNP detection for massively parallel whole-genome resequencing. Genome Res 19, 545–552 (2009).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Jr, S. M., Reddy, V. S., Tamang, D. G. & Västermark, A. The transporter classification database. Nucleic Acids Res 42, 251–258 (2014).
Kim, E. B. et al. Genome seqeucing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011).
Vilella, A. J. et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19, 327–335 (2009).
Bendtsen, J. D., Jensen, L. J., Blom, N., Von, H. G. & Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel Peds 17, 349–356 (2004).
Petersen, T. N., Brunak, S., Von, H. G. & Nielsen, H. SIGNALP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8, 785–786 (2011).
Emanuelsson, O., Nielsen, H., Brunak, S. & Heijne, G. V. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300, 1005–1016 (2000).
Aro, N., Saloheimo, A., Ilmen, M. & Penttila, M. ACEII, a novel transcriptional activator involved in regulation of cellulase and xylanase genes of Trichoderma reesei. J Biol Chem 276, 24309–24314 (2001).
Lockington, R. A., Rodbourn, L., Barnett, S., Carter, C. J. & Kelly, J. M. Regulation by carbon and nitrogen sources of a family of cellulases in Aspergillus nidulans. Fungal Genet Biol 37, 190–196 (2002).
Battagliae, E. et al. Regulation of pentose utilisation by AraR, but not XlnR, differs in Aspergillus nidulans and Aspergillus niger. Appl Microbiol Biotechnol 91, 387–397 (2011).
Li, Z. H. et al. Synergistic and dose-controlled regulation of cellulase gene expression in Penicillium oxalicum. PLoS Genet 11, e1005509 (2015).
Nitta, M. et al. A new Zn(II)(2)Cys(6)-type transcription factor BglR regulates beta-glucosidase expression in Trichoderma reesei. Fungal Genet Biol 49, 388–397 (2012).
Qin, Y. et al. Penicillium decumbens BrlA extensively regulates secondary metabolism and functionally associates with the expression of cellulase genes. Appl Microbiol Biotechnol 97, 10453–10467 (2013).
Coradetti, S. T. et al. Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. P Natl Acad Sci USA 109, 7397–7402 (2012).
Tilburn, J. et al. The Aspergillus PacC zinc finger transcription factor mediates regulation of both acid- and alkaline-expressed genes by ambient pH. EMBO J 14, 779–790 (1995).
Wang, B. et al. A transcriptomic analysis of Neurospora crassa using five major crop residues and the novel role of the sporulation regulator rca-1 in lignocellulase production. Biotechnol Biofuel 8, 21 (2015).
Xiong, Y., Sun, J. P. & Glass, N. L. VIB1, a link between glucose signaling and carbon catabolite repression, is essential for plant cell wall degradation by Neurospora crassa. PLoS Genet 10, e1004500 (2014).
Mach-Aigner, A. R., Grosstessner-Hain, K., Pocas-Fonseca, M. J., Mechtler, K. & Mach, R. L. From an electrophoretic mobility shift assay to isolated transcription factors: a fast genomic-proteomic approach. BMC Genomics 11, 644 (2010).
Acknowledgements
This work was financially supported by grants from the Guangxi BaGui Scholars Program Foundation (grant number 2011A001), the ‘One Hundred Person’ Project of Guangxi, and Excellent Teaching Program of Guangxi High Education-Program of Advantageous and Characteristic Specialty (Excellent Undergraduate Major). We thank the Beijing Genomics Institute (Shenzhen, China) for physical sequencing of Talaromyces pinophilus 1–95 genome and the software support.
Author information
Authors and Affiliations
Contributions
J.X.F. designed and supervised the research, involved in data analysis, and prepared the manuscript. S.Z. prepared the manuscript and involved in data analysis. C.X.L. sequenced and analyzed genome data. T.Z. and L.S.L. extracted genomic DNA and assayed the enzyme activity. L.X. and J.L.L. were involved in data analysis. All the authors have approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession Codes: This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LSFK00000000.1, and the 8 chromosomes and the mitochondrial genome have been assigned accession numbers CP017344-CP017352. The BioProject ID in GenBank is PRJNA310372.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, CX., Zhao, S., Zhang, T. et al. Genome sequencing and analysis of Talaromyces pinophilus provide insights into biotechnological applications. Sci Rep 7, 490 (2017). https://doi.org/10.1038/s41598-017-00567-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00567-0
This article is cited by
-
Systematic identification of CAZymes and transcription factors in the hypercellulolytic fungus Penicillium funiculosum NCIM1228 involved in lignocellulosic biomass degradation
Biotechnology for Biofuels and Bioproducts (2023)
-
Platinum Nanoparticles as Potent Anticancer and Antimicrobial Agent: Green Synthesis, Physical Characterization, and In-Vitro Biological Activity
Journal of Cluster Science (2023)
-
Solid-state co-culture fermentation of simulated food waste with filamentous fungi for production of bio-pigments
Applied Microbiology and Biotechnology (2022)
-
Fungal glycosyl hydrolases for sustainable plant biomass valorization: Talaromyces amestolkiae as a model fungus
International Microbiology (2021)
-
The transcription factor TpRfx1 is an essential regulator of amylase and cellulase gene expression in Talaromyces pinophilus
Biotechnology for Biofuels (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
