
Abstract
Tropical peatlands in South-East Asia are some of the most carbon-dense ecosystems in the world. Extensive repurposing of such peatlands for forestry and agriculture has resulted in substantial microbially-driven carbon emissions. However, we lack an understanding of the microorganisms and their metabolic pathways involved in carbon turnover. Here, we address this gap by reconstructing 764 sub-species-level genomes from peat microbiomes sampled from an oil palm plantation located on a peatland in Indonesia. The 764 genomes cluster into 333 microbial species (245 bacterial and 88 archaeal), of which, 47 are near-complete (completeness ≥90%, redundancy ≤5%, number of unique tRNAs ≥18) and 170 are substantially complete (completeness ≥70%, redundancy ≤10%). The capacity to respire amino acids, fatty acids, and polysaccharides was widespread in both bacterial and archaeal genomes. In contrast, the ability to sequester carbon was detected only in a few bacterial genomes. We expect our collection of reference genomes to help fill some of the existing knowledge gaps about microbial diversity and carbon metabolism in tropical peatlands.
Similar content being viewed by others


Background & Summary
Peatlands occupy only 3% of the total land area but contain 600–650 Gt C1,2,3, nearly double the biomass stored in the world’s tropical rainforests4. Peatland ecosystems thus have a significant impact on global carbon storage, greenhouse gas emissions, and climate regulation. Although peatlands are widely distributed across the earth, some of the most carbon-dense (80–120 Gt C) peatlands exist in the tropics due to the sheer depth of peat deposits that occur within a relatively small area (0.4 million km2)5. Peatlands in South-East Asia (SEA) account for 56% of those that occur in the tropics and store nearly 77% of carbon locked in tropical peatlands globally2. Since 1990, tropical peatlands in SEA have been extensively repurposed (20–40% of land area) for forestry (pulpwood plantations) and agriculture (oil palm plantations)6, resulting in a substantial loss of sequestered carbon through peat oxidation (2.5 Gt C between 1990–2015)5. Peat oxidation is largely driven by microorganisms and thus our ability to predict carbon outcomes relies on identifying those with the capacity to sequester or emit carbon. In context to the data presented in this study, microorganisms refer to bacteria and archaea only.
Microorganisms in SEA tropical peat microbiomes have largely been identified and studied using marker-based approaches7,8,9,10,11. While such studies have helped uncover the high microbial diversity9,10,11 that exists within SEA tropical peatlands and its association with environmental variables7,8,9,10,11, they do not provide direct insights into microbial carbon metabolism. Identifying and understanding the role of specific microorganisms that turnover carbon in the community requires reference genomes which link microbial identity with metabolic capacity. However, genomically resolving such communities is highly challenging owing to their sheer complexity. So far, the carbon-processing potential of tropical peat microbiomes remains either genomically unresolved or poorly resolved at best, but, recent studies in temperate peatlands have shown that the functional potential of peat microbiomes can be genomically resolved using metagenomics approaches12,13.
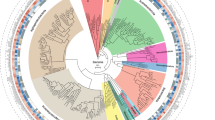
Here, we deeply sequenced peat metagenomes proximal to and distant from oil palm trees in an oil palm plantation. Oil palm plantations on peatlands are of particular interest as they are hotspots of microbially-driven carbon emissions. Using assembly-based approaches, we reconstructed 764 sub-species level (99% gANI) metagenome-assembled genomes (MAGs) with a completeness ≥50% and redundancy ≤10% which cluster into 333 species-level (95% gANI) MAGs (245 bacterial and 88 archaeal) (Fig. 1). Of these, 38 bacterial and 13 archaeal genomes are near-complete (completeness ≥90%, redundancy ≤5%, number of unique tRNAs ≥18), while an additional 207 bacterial and 91 archaeal genomes are substantially complete (completeness ≥70%, redundancy ≤10%). The MAGs have a median size of 3.23 Mbp (range: 0.43–10.91 Mbp), median N50 of 6.34 kbp (range: 3.3–104.84 kbp) and encode a total of 2,530,130 protein-coding genes. The sub-species-level collection spans 14 different bacterial and archaeal phyla with a majority (53.1%) belonging to the phyla Acidobacteriota and Thermoplasmatota, both of which occur widely in peatlands and in acidic soils14,15. Within these phyla, the MAGs provide maximum phylogenetic gain for the orders UBA7540 (13 genomes; phylogenetic gain: 33%) and UBA184 (56 genomes; phylogenetic gain: 76.72%). To our knowledge, this catalogue represents the largest collection of microbial genomes from a tropical peat ecosystem.

Maximum likelihood tree of bacterial species-level MAGs. The phylogenetic tree was constructed using a concatenated set of 120 conserved bacterial marker genes. Concentric rings (moving outward) represent genome completeness and redundancy. The bar plot represents the size of the MAG in Mbp.
Carbon-processing potential of MAGs was determined using a comprehensive marker-gene-based approach which integrates gene functional annotations from multiple databases such as KEGG16, dbCAN17, PFAM18, and TIGRFAM19. The ability to respire a broad spectrum of carbon substrates such as amino acids, polysaccharides, and fatty acids was widespread across both bacterial and archaeal species but the capacity to fix carbon was detected only in a few genomes (Fig. 2). Fermentative pathways which produce alcohols and organic acids such as ethanol and acetate, as well as hydrogen and carbon dioxide were also prevalent. None of the archaeal genomes encode for pathways to convert fermentative end-products into methane, however, the capacity to oxidise methane was detected in a small fraction of MAGs (34 MAGs; 10.2%) from the phyla Acidobacteriota, Actinobacteriota, Protebacteria, Desulfobacterota_B, Thermoplasmata, Thermoproteota, and Micrarchaeota. In contrast, the capacity to oxidise non-methane trace gases such as methanol (93 MAGs; 27.9%), ethanol (69 MAGs; 20.7%), hydrogen (103 MAGs; 30.9%), and carbon monoxide (177 MAGs; 53.2%) was detected in several MAGs. Interestingly, 38 of the 93 MAGs, capable of oxidising methanol belong to the phylum Acidobacteriota, members of which are not typically linked to methanol consumption.

Genome-resolved carbon-processing potential of bacterial and archaeal species-level MAGs. Heatmap showing the presence (black) or absence (white) of key carbon-processing pathways across MAGs within phyla containing a minimum of ten genomes with a completeness ≥70% and redundancy ≤10%. Phylum names are shown either below or next to the heatmap slice corresponding to MAGs from the particular phylum. MAGs within each phylum are clustered based on the occurrence frequency of different pathways. Carbon-processing pathways are grouped and color-coded on the left for visual clarity.
Overall, we expect our genomes database and metagenomes to be widely useful as a reference for metatranscriptomic experiments, comparative studies, and genome-guided isolation efforts. Availability of statistics describing the prevalence of carbon-processing functions across microbial populations will help fill existing knowledge gaps about their diversity, distribution, and metabolism. This data is particularly timely as carbon emissions from repurposed tropical peatlands continue to accelerate at an unprecedented rate, posing a grave threat to our climate.
Methods
Sample collection
Peat samples proximal to (0.5–1 m) and distant from (≥5 m) oil palm trees were collected as part of a time-stamped fertilizer intervention experiment in an oil palm plantation located in Jambi, Indonesia (103°49ʹ 32.23ʺ E, 1°40ʹ58.24ʺ S). The plantation was considered young as age of drainage was ≤10 years8. The sampling location, local weather conditions and peat physiochemical parameters have been previously described8. The mineral fertilizer intervention involved a single application of NPK (16:16:16; P as P2O5, and K as K2O; 1.6–1.8 kg palm−1) and urea (0.5–1 kg palm−1) following local practices20,21. Peat samples were collected from four oil palm trees across two time-points before (days 1 – 2015-01-14 – and 4) and four time-points after (days 6, 7, 10, and 14) fertilizer application. All peat samples were collected from a depth 0–20 cm using an auger and flash frozen in liquid nitrogen on site.
Metagenome sequencing and assembly
Genomic DNA was extracted from all samples using the Zymo Research Soil MidiPrep kit (Zymo Research, CA, USA). Shotgun DNA libraries were prepared from a total of 36 samples using the TruSeq DNA library preparation kit (Illumina, San Diego, CA, USA) with 2 × 250 bp chemistry, and sequenced on the Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) at SCELSE (https://www.scelse.sg), Nanyang Technological University, Singapore. We generated a total of 133.7 Gbp of raw sequence data, with each sample, containing on an average, 3.8 Gbp.
Raw sequence reads were processed using Cutadapt v3.422 with parameters: --error-rate 0.2, --minimum-length 75, --no-indels to remove Illumina sequence adapters. Low-quality regions from adapter-free reads were trimmed using bbmap v38.96 (https://sourceforge.net/projects/bbmap/) with parameters: trimq = 20, qtrim = rl, minlen = 75. Overall, 121.6 Gbp of sequence data were retained after quality filtering.
Samples were then assembled de-novo both individually and co-assembled in groups using MEGAHIT v1.2.723 with parameters: --k-min 27, --k-max 197, --k-step 10 on 48-core compute nodes with 2T RAM. Co-assemblies of proximal and distant peat samples were performed separately by first pooling samples from each time-point and then from all the time-points. Assemblies were length-filtered to retain only contigs ≥1 kbp and renamed using the rename.sh script (bbmap v38.96) with parameters: minscaf = 1000. This resulted in a total of 10.35 million contigs (equivalent to 21.05 Gbp) with a median N50 of 1.96 kbp (range: 1.55–3.27 kbp). Read containment and across-sample contig coverage was estimated by cross-mapping each assembly against quality-filtered reads from all the samples using Bowtie2 v2.4.524 with parameters: -no-unal, -X 1000, SAMtools v1.625, and the jgi_summarize_bam_contig_depths script (METABAT2 v1.2.926). Summaries of individual samples, assemblies, and assembled contigs ≥1 kbp are available on figshare27 in the files “jopf_sample_data.csv”, “jopf_assembly_summary.csv”, “jopf_single_sample_assemblies.tar.gz” and “jopf_co_assemblies.tar.gz” respectively.
Genome binning
Genome bins were recovered from each assembly using METABAT2 v1.2.926, CONCOCT v1.1.028, and MaxBin2 v2.2.729 with parameters: -min_contig_length 2500, all of which use a combination of differential coverage and tetranucleotide frequency information. Bins obtained using the three binning algorithms were then pooled and processed using DASTool v1.1.230 with parameters: --score_threshold 0, --write_bin_evals 1, --search_engine diamond, and --write_bins 1 to achieve a unified set of non-redundant bins. This resulted in a total of 1,535 genome bins with a median size of 3.49 Mbp (range: 0.46–17.88 Mbp) and a median N50 of 6.76 kbp (range: 3.04–134.04 kbp).
Genome refinement and dereplication
Genome bins were first refined using refineM v0.1.231 with parameters: --cov_corr 0.8. Contigs were removed (a) if either their GC content or tetranucleotide frequency exceeded reference-based thresholds (98th percentile) or (b) if across-sample coverage correlation was <80%. Bins were further refined using reference-based approaches implemented in MAGpurify v2.1.232. Contigs were removed if they contained taxonomically-discordant marker genes, known contaminants, lacked a concordant marker gene or if they aligned poorly to conspecific genomes (when available) from the IGGdb database32. Refined bins with completeness ≥50% and redundancy ≤10% were designated as MAGs. Species and sub-species-level collections were generated by dereplicating the MAGs using dRep v3.4.033 with parameters: -sa 0.95/0.99 --S_algorithm fastANI.
Genome quality assessment and taxonomic classification
MAG statistics were estimated using CheckM v1.1.334 with parameters: lineage_wf, -t 24 --pplacer_threads 1, --tab_table and are summarised in Fig. 3. Transfer RNA (tRNA) gene sequences were predicted using tRNAScan-SE v2.0.935 using kingdom-specific HMM models. Taxonomic annotation was performed using GTDB-Tk v2.1.136,37,38,39,40,41,42,43,44,45 and the Genome Taxonomy Database r20746. MAGs were classified as near-complete31,47 if they had completeness ≥90%, redundancy ≤5%, and ≥18 unique tRNAs, or as medium-quality otherwise. MAG statistics and taxonomic labels for the species and sub-species-level collections are available on figshare27 in the file “jopf_mag_quality_summary.csv”.

Genome statistics for the 764 sub-species-level MAGs. Histograms (from left to right, starting from the top) show genome completeness, redundancy, genome size, number of contigs, contig N50, mean contig length, length of the longest contig, and the number of tRNAs corresponding to the 20 standard amino acids identified in each MAG.
Estimating phylogenetic diversity and gain
Phylogenetic diversity and gain were estimated by constructing kingdom-specific maximum likelihood trees integrating species-level MAGs from this study and reference genomes from GTDB r20746. Phylogenetic trees were constructed using GTDB-Tk v2.1.136,37,38,39,40,41,42,43,44,45 with parameters: de_novo_wf, --bacteria/--archaea, and --outgroup_taxon p__Patescibacteria/p__Altiarchaeota. Relative taxon phylogenetic diversity and phylogenetic gain were computed using GenomeTreeTk v0.1.6 (https://github.com/dparks1134/GenomeTreeTk) with parameters: pd_clade.
De-novo trees comprising only MAGs from this study were constructed using GTDB-Tk v2.1.136,37,38,39,40,41,42,43,44,45 with parameters: de_novo_wf, --bacteria/--archaea, --skip_gtdb_refs, and --outgroup_taxon p__Acidobacteriota/p__Thermoplasmatota. Trees were visualised and annotated using iTOL v648. Unrooted Archaeal/Bacterial trees with and without GTDB reference genomes are available on figshare27 in the files “jopf_bacteria_with_gtdb_r207_refs_unrooted.tree”, “jopf_archaea_with_gtdb_r207_refs_unrooted.tree”, “jopf_bacteria_unrooted.tree”, and “jopf_archaea_unrooted.tree”.
Functional annotation
Carbon-processing potential of the MAGs was estimated using METABOLIC v4.049 which integrates functional annotations from KEGG16, dbCAN217, PFAM18, TIGRFAM19, and custom HMMs for specific metabolic functions. Metabolic pathways were considered present if the MAG contained at least one associated marker gene or absent otherwise. Presence/absence of carbon-processing pathways in MAGs is available on figshare27 in the file “jopf_carbon_processing_pathways.csv”.
Technical Validation
MAGs reported in this study only consist of those that met the medium quality threshold or above as defined in Bowers et al.51.
Usage Notes
Users/researchers should independently assess the accuracy of genes, contigs, and functional assignments for genomes of interest prior to downstream analysis.
Code availability
Open-source software packages were used to process data and generate data products. Software versions and non-default parameters are specified where required.
References
Dargie, G. C. et al. Age, extent and carbon storage of the central Congo Basin peatland complex. Nature 542, 86–90 (2017).
Page, S. E., Rieley, J. O. & Banks, C. J. Global and regional importance of the tropical peatland carbon pool. Global Change Biology 17, 798–818 (2011).
Yu, Z., Loisel, J., Brosseau, D. P., Beilman, D. W. & Hunt, S. J. Global peatland dynamics since the Last Glacial Maximum. Geophysical Research Letters 37 (2010).
Page, S. E. & Baird, A. J. Peatlands and Global Change: Response and Resilience. Annual Review of Environment and Resources 41, 35–57 (2016).
Miettinen, J., Hooijer, A., Vernimmen, R., Liew, S. C. & Page, S. E. From carbon sink to carbon source: extensive peat oxidation in insular Southeast Asia since 1990. Environmental Research Letters 12, 024014–024014 (2017).
Miettinen, J. et al. Extent of industrial plantations on Southeast Asian peatlands in 2010 with analysis of historical expansion and future projections. GCB Bioenergy 4, 908–918 (2012).
Jackson, C. R., Liew, K. C. & Yule, C. M. Structural and functional changes with depth in microbial communities in a tropical malaysian peat swamp forest. Microbial Ecology 57, 402–412 (2009).
Mishra, S. et al. Microbial and metabolic profiling reveal strong influence of water table and land-use patterns on classification of degraded tropical peatlands. Biogeosciences 11, 1727–1741 (2014).
Too, C. C., Keller, A., Sickel, W., Lee, S. M. & Yule, C. M. Microbial Community Structure in a Malaysian Tropical Peat Swamp Forest: The Influence of Tree Species and Depth. Frontiers in Microbiology 9, 2859–2859 (2018).
Dom, S. P. et al. Linking prokaryotic community composition to carbon biogeochemical cycling across a tropical peat dome in Sarawak, Malaysia. Scientific Reports 2021 11:1 11, 1–19 (2021).
Tripathi, B. M. et al. Distinctive tropical forest variants have unique soil microbial communities, but not always low microbial diversity. Frontiers in Microbiology 7, 376–376 (2016).
Woodcroft, B. J. et al. Genome-centric view of carbon processing in thawing permafrost. Nature 2018 560:7716 560, 49–54 (2018).
St. James, A. R., Yavitt, J. B., Zinder, S. H. & Richardson, R. E. Linking microbial Sphagnum degradation and acetate mineralization in acidic peat bogs: from global insights to a genome-centric case study. The ISME Journal 2020 15:1 15, 293–303 (2020).
Belova, S. E. et al. Hydrolytic capabilities as a key to environmental success: Chitinolytic and cellulolytic acidobacteriafrom acidic sub-arctic soils and boreal peatlands. Frontiers in Microbiology 9, 2775–2775 (2018).
Sheridan, P. O., Meng, Y., Williams, T. A. & Gubry-Rangin, C. Recovery of Lutacidiplasmatales archaeal order genomes suggests convergent evolution in Thermoplasmatota. Nature Communications 2022 13:1 13, 1–13 (2022).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28, 27–30 (2000).
Zhang, H. et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Research 46, W95–W101 (2018).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Research 49, D412–D419 (2021).
Haft, D. H., Selengut, J. D. & White, O. The TIGRFAMs database of protein families. Nucleic Acids Research 31, 371–373 (2003).
Woittiez, L. S. et al. Fertiliser application practices and nutrient deficiencies in smallholder oil palm plantations in Indonesia. Experimental Agriculture 55, 543–559 (2019).
Comeau, L.-P. et al. How do the heterotrophic and the total soil respiration of an oil palm plantation on peat respond to nitrogen fertilizer application? Geoderma 268, 41–51 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
Li, D. et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 2012 9:4 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kang, D. D. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, e7359–e7359 (2019).
Bandla, A., Mukhopadhyay, S., Sridhar Sudarshan, A. & Swarup, S. Genome-resolved carbon processing potential of tropical peat microbiomes from an oil palm plantation. figshare https://doi.org/10.6084/m9.figshare.20744788 (2023).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nature Methods 2014 11:11 11, 1144–1146 (2014).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Sieber, C. M. K. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nature Microbiology 2018 3:7 3, 836–843 (2018).
Parks, D. H. et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature Microbiology 2, 1533–1542 (2017).
Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 2019 568:7753 568, 505–510 (2019).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. The ISME Journal 2017 11:12 11, 2864–2868 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research 25, 1043–1055 (2015).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Research 49, 9077–9096 (2021).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Hyatt, D. et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 1–11 (2010).
Eddy, S. R. Accelerated Profile HMM Searches. PLOS Computational Biology 7, e1002195–e1002195 (2011).
Matsen, F. A., Kodner, R. B. & Armbrust, E. V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC bioinformatics 11, 538–538 (2010).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature Communications 2018 9:1 9, 1–8 (2018).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE 5, e9490–e9490 (2010).
Ondov, B. D. et al. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biology 17, 1–14 (2016).
Sukumaran, J. & Holder, M. T. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Harris, C. R. et al. Array programming with NumPy. Nature 2020 585:7825 585, 357–362 (2020).
Costa-Luis, C. D. et al. tqdm: A fast, Extensible Progress Bar for Python and CLI. (2022).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Research 50, D785–D794 (2022).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nature Biotechnology 39, 105–114 (2021).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research 49, W293–W296 (2021).
Zhou, Z. et al. METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 10, 1–22 (2022).
Bandla, A., Mukhopadhyay, S., Mishra, S., Sridhar Sudarshan, A. & Swarup, S. Peat metagenomes from an oil palm plantation in Jambi, Indonesia. NCBI BioProject https://identifiers.org/bioproject:PRJNA883528 (2023).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology 35, 725–731 (2017).
Acknowledgements
This work was co-funded by the National Research Foundation, Prime Minister’s Office, Singapore and the Economic Development Board, Singapore as part of the Singapore Peking Oxford Research Enterprise (SPORE) program (SPORE, COY-15-EWI-RCFSA/N197-1), Singapore-Delft Water Alliance program (R264-001-011-490) and the Integrated Tropical Peatlands Research Program (A-0001192-00-00). The computational work for this study was fully performed using resources available at the National Supercomputing Centre, Singapore (https://www.nscc.sg).
Author information
Authors and Affiliations
Contributions
A.B., S. Mishra and S.S. conceived this study. S. Mishra designed the field experiment, collected and processed samples. A.B. designed the methodology, performed the computational analysis, and generated the figures, tables, datasets, and data products. A.S. and S. Mukhopadhyay performed parts of the analysis under A.B.’s supervision. A.B., S. Mukhopadhyay, and S.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bandla, A., Mukhopadhyay, S., Mishra, S. et al. Genome-resolved carbon processing potential of tropical peat microbiomes from an oil palm plantation. Sci Data 10, 373 (2023). https://doi.org/10.1038/s41597-023-02267-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02267-z
