
Abstract
Baer’s pochard (Aythya baeri) is a critically endangered species historically widespread throughout East Asia, whose population according to a recent estimate has decreased to between 150 and 700 individuals, and faces a long-term risk of extinction. However, the lack of a reference genome limits the study of conservation management and molecular biology of this species. We therefore report the first high-quality genome assembly of Baer’s pochard. The genome has a total length of 1.14 Gb with a scaffold N50 of 85,749,954 bp and a contig N50 of 29,098,202 bp. We anchored 97.88% of the scaffold sequences onto 35 chromosomes based on the Hi-C data. BUSCO assessment indicated that 97.00% of the highly conserved Aves genes were completely present in the genome assembly. Furthermore, a total of 157.06 Mb of repetitive sequences were identified and 18,581 protein-coding genes were predicted in the genome, of which 99.00% were functionally annotated. This genome will be useful for understanding Baer’s pochard genetic diversity and facilitate the conservation planning of this species.
Similar content being viewed by others

Background & Summary
Baer’s pochard is a migratory duck belonging to the order Anseriformes, family Anatidae, and genus Aythya, whose closest relative and sister species is the ferruginous duck1. Baer’s pochard has typical sexual dimorphism. Males have white or light-yellow irises (Fig. 1), whereas females have dark brown irises. Females also have reddish brown spots at the base of the beak2,3, and are smaller in size. This species was once widespread in East and South Asia, but is currently predominantly only in China4,5 due to over-exploitation and habitat loss, which have caused a severe and global population decline over the past decades6,7. Baer’s pochard was classified as endangered by the International Union for Conservation of Nature (IUCN) in 2008, then as Critically Endangered in 2012, and in 2021 was included in the China Red Data Book of Endangered Animals. According to a recent estimate by the IUCN, its population has only 150–700 mature individuals8, and faces a long-term risk of extinction. Moreover, although there has been an increasing number of avian genome assemblies in recent years9, many non-model species including Baer’s pochard still lack genome resources.

An adult male Baer’s pochard.(Qiang Li).
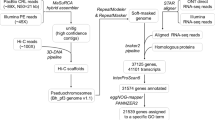
In order to provide genome-scale insights into a near-extinction species and promote conservation planning for it, we constructed the first high-quality Baer’s pochard chromosome-level reference genome using Illumina paired-end sequencing, Oxford Nanopore sequencing, and Hi-C technology. The genome had an assembly size of 1.14 Gb with a scaffold N50 of 85,749,954 bp and a contig N50 of 29,098,202 bp. These scaffolds were further clustered and ordered into 35 pseudo-chromosomes based on the Hi-C data, representing 97.88% of the assembled sequences. The genome contained 13.72% repeat sequences and 1,721 noncoding RNAs. A total of 18,581 protein-coding genes were predicted in the genome, of which 99.00% were functionally annotated. Searches for complete Aves BUSCO (Benchmarking Universal Single-Copy Ortholog) gene groups showed that 97.00% of BUSCO genes were complete, suggesting a high level of genome completeness. This genome provides a valuable genomics resource for studying the conservation genomics of critically endangered species to help recover their population size.
Methods
Ethics statement
All animal handling and experimental procedures were approved by the Qufu Normal University Biomedical Ethics Committee (approval number: 2022001).
Sample and sequencing
Baer’s pochard tissue for whole-genome sequencing was obtained from a dead individual that had strayed into a fishing net in Shandong (China). The muscle tissue that we collected was stored at −80 °C and used for genomic DNA extraction, genomic DNA sequencing. Nine additional transcriptomic samples (heart, kidney, lung, spleen, liver, craw, gallbladder, blood, and muscle) were collected from the same individual and stored at −80 °C until RNA were extracted for transcriptome sequencing. Paired-end libraries of genomic DNA (gDNA) were prepared using Illumina TruSeq Nano DNA Library Prep kits. The integrity and quality of the extracted DNA were checked using agarose gel electrophoresis and a Qubit Fluorometer. One library with an insertion size of 350 bp was constructed and sequenced using the Illumina HiSeq platform to enable genome survey and base-level correction. A total of 60.34 Gb (coverage of 49.69×) of 150-bp paired-end reads were generated. Purified DNA was then prepared for sequencing with the genomic sequencing kit SQK-LSK109 (Oxford Nanopore Technologies, Oxford, UK) following the provided protocol, and single-molecule real-time sequencing of long reads was conducted using the PromethION platform (ONT, Oxford, UK). Approximately 136.50 Gb of data was obtained (coverage of 112.42×). The Hi-C library was constructed using muscle tissue from the same Baer’s pochard individual and sequenced using the Illumina PE150 platform. A total of 125.64 Gb of 150-bp paired-end reads were obtained, which covered ~103.48× of the genome (Table 1). Finally, RNA was extracted from the nine transcriptomic samples and used for library construction, and RNA-Seq reads were generated for genome annotation using the Illumina NovaSeq 6000 platform. A total of 67.93 Gb of 150-bp paired-end reads were obtained after adapter trimming and quality filtering (Table 2).
Genome assembly
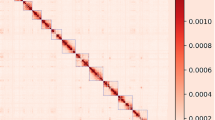
We used a combination of Nanopore long reads, Illumina short reads, and chromatin conformation capture (Hi-C) to generate chromosome-level reference genomes. The genome size and heterozygosity level of the Baer’s pochard were determined using Illumina short reads based on the k-mers spectrum10. The genome size was estimated to be approximately 1,214.25 Mb, and the heterozygosity rate of the genome is 0.38% (Table 3). NextDenovo (https://github.com/Nextomics) used Nanopore long reads for the initial scaffolding assemblies. However, long reads have low quality scores, and thus NextPolish11— which uses quality-controlled Illumina short reads, was employed to improve the assembled genome. These steps yielded the final Baer’s pochard genome with a total length of 1.14 Gb, which was mostly consistent with the k-mer-based estimation including 228 contigs with N50 = 29,098,202 bp, and the overall GC content of the genome was 41.94% (Table 4). We had obtained 125.64 Gb of Hi-C sequencing data to generate this chromosomal-level assembled genome. We first used HiCUP12 to map and process the reads obtained from the Hi-C library, then the Hi-C-corrected contigs were subjected to the ALLHiC pipeline13 for partition, orientation and ordering. A total of 135 scaffolds could be mapped to 35 chromosomes with lengths ranging from 1.77 Mb to 208.01 Mb, which covered 97.88% of the whole genome. Finally, we obtained the first chromosome-level high-quality Baer’s pochard assembly (1.14 Gb) with a scaffold N50 length of 85.75 Mb (Table 5 and Fig. 2). The genome size, scaffold N50 length, and GC content of Aythya baeri is similar to that of Aythya fuligula (RefSeq assembly access: GCF_009819795.1), a member of the same genus, but its contigN50 length is much longer than that of Aythya fuligula (Table 6). This indicates that the genome of Aythya baeri has high assembly quality.

Heat map of Hi-C assembly of the Baer’s pochard.
We used the Core Eukaryotic Genes Mapping Approach (CEGMA v2.5)14 and Benchmarking Universal Single-Copy Orthologs (BUSCO v4.1.2)15 methods to evaluate the completeness of genome assembly. A single-copy ortholog set was searched against the assembled genome of Baer’s pochard using BUSCO tool, of the 8,338 single-copy orthologs in the avian lineage (aves_odb10), approximately 97.00% were present in this assembly (Table 7). We took the conserved genes (248 genes) of six eukaryotic model organisms to form the core gene library, of which the CEGMA evaluation showed 95.97% was successfully assembled (Table 8).
Annotation of genomic repeat sequences
We annotated the Baer’s pochard whole-genome repeat sequences based on homology alignment and de novo predictions. RepeatModeler (v1.0.8)16, RepeatScout (v1.0.5)17 and LTR_FINDER (v1.0.7)18 were used to build a de novo repetitive element database. Tandem repeats were extracted using TRF19 via ab initio prediction. Homolog prediction was performed using the Repbase database20 whilst employing the RepeatMasker (v4.0.5) software21 to extract repeat regions (Table 9). According to these analyses, approximately 1,571 Mb of repeat sequences were revealed, which accounted for 13.72% of the whole genome; thus, the content of repeat sequence in A. baeri genome is slightly higher than that in the A. fuligul genome (13.00%). Among the repeat elements, long interspersed nuclear elements (LINEs) account for 8.80% of the genome, short interspersed nuclear elements (SINEs) for 0.01%, long terminal repeats (LTRs) for 4.13% and DNA transposons for 0.15% (Table 10).
Annotation of gene structure
We combined three approaches to predict protein-coding genes, including homologous comparison, ab initio prediction, and RNA-Seq-assisted prediction. For homologous comparison, the reference protein sequences of five bird species— the tufted duck (Aythya fuligula), mallard (Anas platyrhynchos), mute swan (Cygnus olor), red junglefowl (Gallus gallus), and ruddy duck (Oxyura jamaicensis), were sourced from the Ensembl database (release 91), and aligned to the Baer’s pochard genome using TBlastN (v2.2.26; E-value ≤ 1e-5)22. The potential gene structures were predicted using Genewise (v2.4.1)23. For ab initio analysis based gene prediction, we used Augustus (v3.2.3)24, Geneid (v1.4)25, Genescan (v1.0)26, GlimmerHMM (v3.04)27 and SNAP28 with appropriate parameters to perform de novo predictions. To optimize the genome annotation, RNA-Seq reads from nine different tissues were assembled de novo using Trinity (v2.1.1)29, and TopHat (v2.0.11)30 was used to align RNA-seq reads to the Baer’s pochard genome sequences. Cufflink software was then employed to determine potential gene structures. We used EvidenceModeler (EVM,v1.1.1) and PASA (Program to Assemble Spliced Alignment) to integrate all the results generated from the three aforementioned methods and create a non-redundant reference gene set31 composed of 18,581 genes, with an average CDS lengths of 1,600.42 bp, average exon and intron lengths were 169.04 bp and 2,763.57 bp, respectively (Table 11).
We also predicted 432 tRNAs using the program tRNAscan-SE32. We identified 664 ncRNAs, including 342 miRNAs and 322 snRNAs, by searching against the Rfam database with default parameters using Infernal33. For rRNAs that were highly conserved, we chose related species’ rRNA sequences as references and predicted 161 rRNA sequences using Blast34 (Table 12).
Functional annotation of protein-coding genes
We functionally annotated the predicted proteins in the Baer’s pochard genome according to homologous searches against six databases: SwissProt35, InterPro36, Pfam37, Kyoto Encyclopedia of Genes and Genomes (KEGG)38, Gene Ontology (GO)39, and Nr (http://www.ncbi.nlm.nih.gov/protein). Respectively, 82.39%, 98.90%, 76.00%, 77.40%, 91.90%, and 85.30% of genes matched the database entries (Fig. 3). In summary, 18,401 genes (99.00%) were successfully annotated by gene function and conserved protein motifs (Table 13).

Functional annotation statistics. Venn diagram illustrating the distribution of high-score matches of the functional annotation in the Baer’s pochard genome against six public databases.
Synteny analysis using the Tufted duck genome
We conducted whole-genome synteny analysis between the Tufted duck (GCA_009819795.1) and the Baer’s pochard genomes using MUMmer40. The whole-genome alignment between the tufted duck and the Baer’s pochard genomes was visualized using RectChr (BGI-shenzhen/RectChr), as shown in Fig. 4. The results showed the overall high consistency of the tufted duck and the Baer’s pochard genomes.

Circos plot of the synteny analysis between the tufted duck and the Baer’s pochard genome.
Data Records
The Nanopore, Illumina, and Hi-C sequencing data used for genome assembly were deposited in the NCBI Sequence Read Archive database with accession numbers SRR1756878541, SRR1751855342, and SRR1750990543. The transcriptomic sequencing data were stored under accession numbers SRR1743318244 and SRR1749702345-SRR17497030. The assembled genome was deposited in the NCBI assembly with the accession number JAKRSJ00000000046. The annotation results of repeated sequences, gene structure and functional prediction were deposited in the Figshare database47.
Technical Validation
The integrity of the extracted DNA was checked by agarose gel electrophoresis, and the main band was found to be approximately 45 Kb long. The concentration of DNA was determined using a Qubit fluorometer (Thermo Fisher Scientific, USA) with an absorbance of approximately 1.80 at 260/280.
We used the sequence identity method to evaluate the completeness of the genome assembly, selected small fragment library reads, and used BWA software (http://bio-bwa.sourceforge.net/) to align them with the assembled genome. The alignment rate of all small fragment reads to the genome was approximately 99.71%, and the coverage rate was approximately 99.45%, indicating consistency between the reads and assembled genome.
SNPs were identified using Samtools (v0.1.19), resulting in the identification of 3,162,696 SNPs, including 3,157,033 heterozygous SNPs and 5,663 homozygous SNPs. The proportion of homozygous SNPs was 0.000502%, indicating the high accuracy of this assembly.
Code availability
All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software. No specific code has been developed for this study.
References
Livezey, B. C. A phylogenetic analysis of modern pochards (Anatidae: Aythyini). The Auk 113, 74–93 (1996).
Kear, J. Ducks, Geese, and Swans. (Ducks, Geese, and Swans, 2005).
Mackinnon, J. & Phillipps, K. A Field Guide to the Birds of China. Colonial Waterbirds 18, 841–843 (2000).
Chowdhury, S. U., Lees, A. C. & Thompson, P. M. Status and distribution of the endangered Baer’s Pochard Aythya baeri in Bangladesh. Forktail 28, 57–61 (2012).
Wang, X., Barter, M., Cao, L., Lei, J. & Fox, A. D. Serious contractions in wintering distribution and decline in abundance of Baer’s Pochard Aythya baeri. Bird Conservation International 22 (2012).
Hearn, R. A species in serious trouble: Baer’s Pochard Aythya baeri is heading for extinction in the wild. (2013).
Hearn, R. The troubled Baer’s Pochard Aythya baeri: cause for a little optimism? (2015).
Misch, E. A. & Hawn, T. R. Toll-like receptor polymorphisms and susceptibility to human disease. Clinical ence 114, 347–360 (2008).
Feng, S., Stiller, J., Deng, Y., Armstrong, J. & Zhang, G. Dense sampling of bird diversity increases power of comparative genomics. Nature 587, 252–257 (2020).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quantitative Biology 35, 62–67 (2013).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics, 7 (2019).
Steven, W. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000res 4, 1310 (2015).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants 5 (2019).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Panagiotis, I., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 3210–3212.
Smit, A. & Hubley, R. RepeatModeler Open-1.0 (2008).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, 351–358 (2005).
Zhao, X. & Hao, W. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–268 (2007).
Gary, B. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research, 573–580 (1999).
Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current protocols in human genetics 25 (2009).
Schffer, A. A., Richa, A., Yu, Y. K., Michael, G. E. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biology,4,1(2006-12-07) 4, 41 (2006).
Birney, E. GeneWise and Genomewise. Genome Research 14, 988 (2004).
Mario, S. & Burkhard, M. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research 33, W465–467 (2005).
Parra, G., Blanco, E. & Guigó, R. GeneID in Drosophila. Genome Research 10, 511–515 (2000).
Burge, C. Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology 268 (1997).
Majoros, W., Pertea, M. & Salzberg, S. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Ian, K. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Haas, B. J. et al. transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols.
Kim, D., Pertea, G., Trapnell, C., Pimentel, H. & Kelley, R. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14 (2013).
Haas, B. J., Salzberg, S. L., Wei, Z. & Pertea, M. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research (2019).
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A. & Bateman, A. Rfam: Annotating Non-Coding RNAs in Complete Genomes. Nucleic Acids Research 33, D121–124 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. (1990).
Amos, B. & Rolf, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research, 45 (2000).
InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Research, D190–D199 (2017).
Jaina, M. et al. Pfam: The protein families database in 2021. Nucleic Acids Research.
Minoru, K., Yoko, S., Masayuki, K., Miho, F. & Mao, T. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research, D457–D462 (2016).
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D. & Cherry, J. M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics 25, 25–29 (2000).
Kurtz, S., Phillippy, A., Delcher, A. L. & Smoot, M. Versatile and open software for comparing large genomes. Genome Biology 5 (2004).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR17568785 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR17518533 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR17509905 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR17433182 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR17497023 (2021).
GenBank https://identifiers.org/ncbi/insdc:JAKRSJ0000000000 (2021).
Zhang, L. Whole genome sequencing of the Baer’s pochard (Aythya baeri). figshare. https://doi.org/10.6084/m9.figshare.21971360 (2023).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (32070405, 32270444, 32200407,32200349, and 32000291).
Author information
Authors and Affiliations
Contributions
Lei Zhang, Xiaodong Gao, Tian Xia, and Hong Zhang designed the study. Xiufeng Yang and Guolei Sun collected samples. Zhao and Liu extracted DNA. Xiaodong Gao and Tian Xia performed the research and analyzed the data. Lei Zhang drafted the manuscript for publication. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Gao, X., Xia, T. et al. Chromosome-level genome assembly of the critically endangered Baer’s pochard (Aythya baeri). Sci Data 10, 176 (2023). https://doi.org/10.1038/s41597-023-02063-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02063-9
